

Abstract
Prediction of side-chain conformations is an important component of several biological modeling applications. In this work, we have developed and tested an advanced Monte Carlo sampling strategy for predicting side-chain conformations. Our method is based on a cooperative rearrangement of atoms that belong to a group of neighboring side-chains. This rearrangement is accomplished by deleting groups of atoms from the side-chains in a particular region, and regrowing them with the generation of trial positions that depends on both a rotamer library and a molecular mechanics potential function. This method allows us to incorporate flexibility about the rotamers in the library and explore phase space in a continuous fashion about the primary rotamers. We have tested our algorithm on a set of 76 proteins using the all-atom AMBER99 force field and electrostatics that are governed by a distance-dependent dielectric function. When the tolerance for correct prediction of the dihedral angles is a <20° deviation from the native state, our prediction accuracies for χ1 are 83.3% and for χ1 and χ2 are 65.4%. The accuracies of our predictions are comparable to the best results in the literature that often used Hamiltonians that have been specifically optimized for side-chain packing. We believe that the continuous exploration of phase space enables our method to overcome limitations inherent with using discrete rotamers as trials.
Keywords: rotamer library, configurational-bias sampling, side-chain packing, Monte Carlo methods
Accurate prediction of side-chain conformations is an important undertaking in a variety of biomolecular simulations (Vasquez 1996). Correct positioning of the amino acid side-chains is an important refinement step in investigating protein–protein interactions (Gray et al. 2003; Zacharias 2003; Wang et al. 2005) for protein docking applications. A rational drug design protocol involves side-chain packing of the active site to investigate potential ligand–protein interactions (Claussen et al. 2001). Design problems that involve introducing mutations in proteins to modify their stability or selection of sequence to achieve a specific fold require optimization of the identity and conformations of side-chains (Desjarlais and Handel 1995; Dahiyat and Mayo 1997; Gordon et al. 2003; Havranek and Harbury 2003; Kraemer-Pecore et al. 2003; Kuhlman et al. 2003; Looger et al. 2003). Investigation of protein folding using homology modeling or ab initio methods requires repacking the side-chains to obtain optimal low-energy structures (Holm and Sander 1992; Bower et al. 1997; Huang et al. 1998).
The topic of accurate prediction of side-chain conformations in proteins has been addressed in a number of detailed investigations (Holm and Sander 1992, Mendes et al. 1999; Xiang and Honig 2001; Jacobson et al. 2002a; Liang and Grishin 2002; Canutescu et al. 2003; Peterson et al. 2004). From the point of view of methodology, these investigations have focused on the development of sampling techniques as well as on adaptation of force fields to enhance their ability to discriminate between native and non-native conformations. Combinatorial sampling techniques such as dead-end elimination (Desmet et al. 1992; Lasters et al. 1995; De Maeyer et al. 1997; Gordon and Mayo 1998; Looger and Hellinga 2001), Branch-and-Terminate (Gordon and Mayo 1999), and graph theory-based methods (Canutescu et al. 2003) are guaranteed to find the global energy minimum on convergence. However, they require Hamiltonians that are of a pairwise additive nature between side-chains. Sampling methods based on a Monte Carlo–type algorithm do not have such a restriction on the nature of the potential function, but they are not guaranteed to converge to the global energy minimum. However, a careful implementation of Monte Carlo methods can be expected to reach conformations that are close to the global energy minimum. Examples of Monte Carlo–type techniques include simulated-annealing algorithms (Hwang and Liao 1995; Liang and Grishin 2002), Monte Carlo moves in dihedral space (Dunbrack and Karplus 1993), and rotamer substitution protocols (Xiang and Honig 2001; Jacobson et al. 2002a; Gray et al. 2003). Self-consistent mean-field theory (Mendes et al. 1999) with flexible rotamers has also been used successfully for predicting side-chain conformations. These studies have also contributed to our understanding of whether commonly used force fields are applicable for the more restricted problem of side-chain placement given a fixed backbone conformation (Petrella et al. 1998; Xiang and Honig 2001; Jacobson et al. 2002b; Liang and Grishin 2002). As a result, several modifications have been suggested to existing force fields to make them amenable to the above task. These modifications include addition of hydrogen-bonding potentials (Peterson et al. 2004; Gray et al. 2003), surface- and volume-overlap terms (Liang and Grishin 2002; Peterson et al. 2004), and knowledge-based terms (Liang and Grishin 2002; Gray et al. 2003; Peterson et al. 2004) derived from a statistical analysis of the Protein Data Bank (PDB). Additionally, the relative contributions of nonbonded interactions such as van der Waals (vdW) and electrostatics have been reweighted (Fernandez-Recio et al. 2002; Liang and Grishin 2002; Gray et al. 2003; Peterson et al. 2004) in order to obtain predictions closer to the native state. It has often been argued that the unmodified Hamiltonians may not be suitable for side-chain predictions due to their inability to accurately account for solvation effects, electrostatics, and hydrogen bonding. Even in the case of Hamiltonians, which contain potential functions to account for the above-mentioned effects, the contributions from the different terms may not be weighted correctly to obtain accurate results. Nearly all modern-day prediction algorithms rely on rotamer libraries (Tuffery et al. 1991; Dunbrack and Cohen 1997; Xiang and Honig 2001) to reduce the expanse of conformational space that needs to be explored for side-chain placement. Rotamer libraries list the primary side-chain dihedral angles by residue type based on a statistical analysis of the Protein Data Bank. Hamiltonians in several investigations (Liang and Grishin 2002; Gray et al. 2003; Peterson et al. 2004) employ a potential energy term based on the backbone-dependent frequency of occurrence of a rotamer. An important conclusion from those studies (Liang and Grishin 2002; Peterson et al. 2004) is that significant overall improvement in the prediction quality for the surface residues results from the inclusion of this term. Such a result is not surprising since the frequency of occurrence of a rotamer can be viewed as a free-energy term that incorporates both the energetic and entropic terms arising from dispersion interactions and electrostatic and solvation effects. The latter two are expected to play an increasingly important role in the prediction of surface residues. Thus, the inclusion of this term can alleviate some of the shortcomings of the Hamiltonian in accounting for these effects. In addition to the Hamiltonian used for the prediction, it is imperative for the sampling methodology to be sophisticated in order to reliably sample the resulting energy landscape. A simplistic sampling technique will be unable to surmount the energy barriers in a reasonable span of time, thereby possibly trapping the system in a local minimum far from the global minimum for the potential function. Rotamer libraries seek to alleviate the sampling problem by guiding the sampling technique to a region of conformational space that is likely to be populated. However, since the rotamer libraries are discrete in nature, whereas the side-chains conformations are not, researchers are increasingly moving toward incorporating flexibility in the rotamer libraries by minimization in torsional space (Wang et al. 2005), addition of rotamers around primary rotamers (Gray et al. 2003; Peterson et al. 2004), introduction of flexibility in the angles for a side-chain, and incorporation of rotamer ensembles using continuous flexibility about dihedral angles (Mendes et al. 1999).
There are certain shortcomings with using discrete libraries of rotamer conformations and knowledge-based Hamiltonians when the goal is to rigorously sample an ensemble average property of the system. For example, the presence of a conformation that deviates significantly from a primary rotamer, though rare, is a possible conformation during a molecular dynamics simulation. Monte Carlo searches based on discrete rotamer libraries, however finely and widely they may be discretized around the primary rotamers, cannot be employed to supplement the sampling in conjunction with a molecular dynamics method, since the detailed balance condition (Frenkel and Smit 1996) cannot be satisfied. Briefly, this condition states that in order for a Monte Carlo algorithm to rigorously sample the distribution in the ensemble of interest, the rates of forward and reverse transition between every pair of linked states must be equal. While the primary goal of side-chain prediction methods is to obtain a probable low-energy conformer, we believe that advanced Monte Carlo methods have the potential to effectively supplement molecular dynamics methods. In this paper, we formulate and test an advanced Monte Carlo sampling protocol based on configurational-bias sampling methods. This type of sampling methodology has been used successfully to equilibrate dense polymer melts (Siepmann and Frenkel 1992; Escobedo and de Pablo 1994; Jain and de Pablo 2002b) and liquids (Cracknell et al. 1990), calculate chemical potentials (Kumar et al. 1991), estimate free energies (Jain and de Pablo 2002a), simulate peptides (Wu and Deem 1999), and study protein–protein association (Fernandez-Recio et al. 2002). Our implementation of this scheme for the purpose of side-chain placement involves performing successive patch refinements on neighboring groups of residues. By performing cooperative moves on groups of side-chains as opposed to individual side-chains, we attempt to achieve rapid exploration of the relevant conformational space. We develop an adaptive methodology that relies on introducing variations in the existing primary rotamers as dictated by the surroundings of a residue in the protein. This is achieved by an approach where a side-chain is built unit by unit starting from the backbone, with conformations that are biased toward, but not restricted to, the angles in the rotamer library. In this work, we restrict ourselves to the search for low-energy conformers, though with additional book-keeping this approach can be extended to perform rigorous sampling in the canonical ensemble at a temperature of interest. We test our algorithm on a set of 76 high-resolution protein structures using the AMBER99 all-atom force field (Cornell et al. 1995) with a simple distance-dependent dielectric function for the electrostatic interactions. When the tolerance for correct prediction is 20° within the native dihedral angle values, our results indicate that our method is able to correctly predict the side-chain conformations with an accuracy that is comparable to the existing methods in the literature that often use Hamiltonians that have been specifically optimized for this problem. For a tolerance of 40°, the results from our method are slightly worse than the best prediction accuracies in the literature (Peterson et al. 2004), and this loss of accuracy can be largely attributed to the absence of a knowledge-based rotamer frequency term in the Hamiltonian.
Results and Discussion
Simulation methodology
In this work, configurational-bias sampling methodology was utilized for incorporating flexibility into existing rotamer libraries. Our protocol for implementing this method is inspired by the configurational-bias method used for polymers. In the case for polymers, several repeat units from one or several chains in a particular region are deleted. The deleted units are then grown successively, by biasing their new positions with respect to a potential function and topological constraints (Escobedo and Chen 2000; Siepmann and Wick 2000). While the complete Hamiltonian can be used, it is often computationally more feasible to use a cheaper Hamiltonian for the growth step (Frenkel and Smit 1996; Escobedo and Chen 2000). After all the deleted units are regrown, the move is accepted or rejected on the basis of the detailed balance condition (Frenkel and Smit 1996) using the target Hamiltonian. This ensures correct sampling in the ensemble of choice. This method is able to successfully equilibrate dense systems due to the cooperative rearrangements that are proposed during each step. These drastic rearrangements enable the system to escape local minima that could not otherwise be easily overcome using simpler moves. We extend this method to side-chain packing using the Dunbrack rotamer library (Dunbrack and Cohen 1997). A brief overview of the procedure is given below. A pseudocode for the protocol can be found in Materials and Methods. For each patch refinement, a flexible residue is selected along with N − 1 neighboring residues, for a patch size of N residues. The regrowth process for each residue in the patch is started from either χ1 or χ2 (except for valine, which due to the flexible polar hydrogens on serine and threonine, is the only residue without a χ2). Trial moves are generated by rotating the residue about the flexible dihedrals according to the dihedral angle values in the rotamer library with additional continuous flexibility about these values. For each trial move, a cheap Hamiltonian is evaluated for the group of atoms whose positions are affected only by rotation about that particular dihedral. For example, for phenylalanine, for the choice of χ1, the unit comprises Cγ and the hydrogens on Cβ, since the positions of these atoms are not influenced by subsequent dihedrals. For χ2, the unit comprises the phenyl ring except for Cγ. The insertion of the group is biased on the basis of the nonbonded Lennard-Jones energy and the torsional energy about the rotatable bonds associated with the group. A trial, i, is picked out from several proposals, using the following probability distribution,
 |
where k denotes the index over the trials, m denotes the unit being grown, βprop corresponds to the temperature used for move proposal, and pmi is the probability of selecting trial i for unit m. This process is repeated until all the deleted units are regrown. The correct acceptance probability, pacc, for the above move is
 |
where βacc corresponds to the temperature used for the acceptance of moves, ΔUfull is the change in the potential energy of the system using the Hamiltonian of interest, m ∈ [1 N] represents all the units being grown during the patch refinement, and superscripts F and R represent the probabilities in the forward and the reverse moves, respectively. Thus, to use the rigorous acceptance criterion, the reverse move must be performed to generate the original patch and determine the corresponding probabilities. As mentioned earlier, in this work our primary goal is to test whether the sampling methodology detailed above performs successfully in the context of the side-chain prediction problem by identifying a low-energy conformer that is close to the energy minimum of the prescribed Hamiltonian. Therefore, for reasons of computational efficiency, we do not implement the above acceptance criterion but instead employ the usual Metropolis criterion (Metropolis et al. 1953) that would result from setting the probabilities in Equation 2 to one. At the end of the move, a comparison is made between the rotamers of the newly generated patch and those belonging to the patch before the deletion of units. If the rotamers are the same for every residue in the patch, a counter called the “persistence counter” is incremented for the residues that comprise this patch. If the move is accepted and the new patch has different rotamers from the previous configuration, the persistence counter is reset to zero for all the residues in the patch. As long as the persistence counter for a residue is below predetermined value lp, that residue can be the central or the first residue for a patch refinement. If the counter exceeds or equals that value, the patch size is set to 1; i.e., only that particular residue is regrown. We label this sampling technique SPRUCE (Successive Patch Refinement Using Configurational-Bias Exploration). The method discussed above was applied to 76 high-resolution proteins from the PDB. In all cases, alanine, glycine, proline, and the residues involved in disulfide bridges were kept rigid. Proline was not predicted since its cyclic nature does not lend itself to a regrowth process as explained above. The temperature for the growth process was set to a high value in order to propose a diverse set of configurations during the generation of trial moves. However, since the overall goal of this study is to generate a structure close to the energy minimum for the system, the acceptance temperature was set to a low value. The values of the various parameters are listed in Materials and Methods. The AMBER99 force field (Cornell et al. 1995) and charge set used were used along with the modifications suggested by Okur et al. (2003). A simple distance-dependent dielectric (DDD) was used to account for the solvent-screened electrostatic interactions. The nonbonded Lennard-Jones interactions were truncated at 10 Å. No truncation was employed for the electrostatic interactions. Consistent with the AMBER99 force field, the 1–4 interactions were scaled by 0.5 and 0.833 for the Coulombic and the Lennard-Jones, respectively. A neighbor list was employed to speed up the nonbonded calculations.
Prediction accuracies and comparisons with literature
The sampling methodology employed in our investigation differs significantly from that used in other studies. Our algorithm incorporates flexibility into the placement of a side-chain by sampling the surrounding dihedral space of the primary rotameric angles in a continuous fashion, as opposed to the discrete conformations from the rotamer library. Additionally, we stress that no modifications to the AMBER99 force field or atomistic parameters were made during the conformational search process.
Stated prediction accuracies use the convention [X Y]tol, where tol is the maximum deviation from the native value of any “correctly” predicted dihedral, X is prediction accuracy for χ1, and Y is the prediction accuracy for χ1 and χ2, i.e., χ1+2. Predictions for χ1+2 are considered correct only when both χ1 and χ2 are predicted correctly. Statistics were calculated for three different subsets of the 76 proteins, in order to aid comparison with other detailed investigations in the literature. Table 1 lists the PDB codes for the three sets. Overall prediction accuracies of [86.7 74.0]40 and [83.3 65.8]20 were obtained over the 65-protein test set with an overall root mean square deviation (RMSD) value of 1.39 Å. Compared with the results from the NCN algorithm (Peterson et al. 2004) using vdW interactions and uniform electrostatics with a dielectric of 80 (row 2 in Table 2 in Peterson et al. 2004), they represent an improvement of 1.5% in the accuracy of χ1+2 and 0.04 Å in the overall RMSD. However, the accuracy for χ1 was lower by 0.3%. It should be noted, however, that Peterson et al. (2004) employed a vdW contribution that was averaged over the heavy atoms in the side-chain. This resulted in an increase in the prediction accuracy compared with the full vdW potential, although the precise magnitude of the improvement was not stated. Using the optimized Hamiltonian with hydrogen bonding, rotamer overlap, and knowledge-based rotamer frequency terms, the predictions from the NCN algorithm are better than our predictions by 2.7% and 3.7% for χ1 and χ1+2, respectively. The overall RMSD is also better by 0.12 Å. The test set also includes the seven proteins that were used to optimize the coefficients for the rotamer frequency term in the Hamiltonian used by Peterson et al. (2004). Furthermore, the size of their rotamer library was nearly 50,000 rotamers. Without the rotamer frequency term, the accuracies obtained are [87.4 73.2]40 with an overall RMSD of 1.42 Å, which is comparable with our predictions. The same study also evaluated the Liang and Grishin (2002) simulated annealing approach (LGA) and the SCAP algorithm (Xiang and Honig 2001) on the 65-protein test set. In terms of overall accuracy, the NCN algorithm performed better than both LGA and SCAP. Using a tolerance of 20°, the best results from the NCN method are [83.2 64.9]20. These accuracies are almost similar to our prediction, which surprisingly indicates that our simpler Hamiltonian with SPRUCE performs at par compared with the optimized Hamiltonian. The study of Xiang and Honig (2001) used a vdW potential and distance-dependent dielectric term. Their test set comprised 33 proteins, of which 31 are in our current test set. Prediction accuracies of [84 67]20 were obtained for these proteins compared with [81 62]20 (Table 3, row b, using CHARMM library bond lengths and angles, in Xiang and Honig 2001). The overall RMSD value of 1.35 Å is also better by 0.32 Å. The self-consistent mean-field (SCFMT) method of Mendes et al. (1999) incorporated a flexible ensemble of states around the principal rotamer. However, the ligands were constrained to their original positions along with the backbone, which increases the prediction accuracies for these structures (Liang and Grishin 2002). Liang and Grishin evaluated the SCMFT method on 15 proteins in the absence of the prosthetic groups and obtained accuracies of [83.9 65.4]40 with an overall RMSD of 1.48 Å. The accuracy of their own LGA algorithm was [87.6 71.5]40 with an overall RMSD of 1.36 Å. Our results for the same 15 proteins are [86.3 72.0]40 with an RMSD of 1.39 Å. Thus, our results compare favorably with those obtained using LGA and SCFMT, for configurations without any constrained ligands. The accuracies using LGA over a set of 30 proteins (Liang and Grishin 2002) is higher than those reported in the previous comparison. However, 15 of those proteins also comprised the training set used for optimizing the Hamiltonian, which can skew the results toward higher prediction accuracies. The prediction accuracy from SPRUCE over the entire set of 76 proteins is [86.7 73.6]40 and [83.3 65.4]20 with an overall RMSD of 1.40 Å. Table 2 lists the results obtained for each protein investigated using SPRUCE.
Table 1.
Test sets used and corresponding computation times for assessing prediction accuracies with SPRUCE and for comparison with literature studies

Table 2.
Prediction accuracies for the 76 proteins investigated
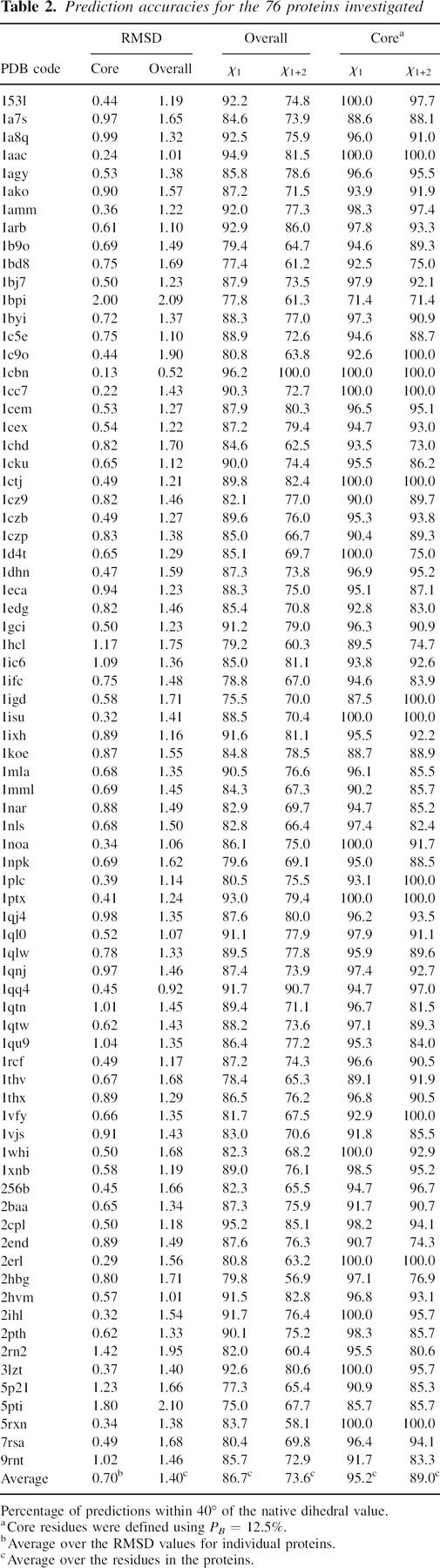
Table 3.
Prediction accuracies with SPRUCE as a function of PB

Incorporation of the knowledge-based backbone-dependent rotamer frequency term leads to the greatest improvement in the overall RMSD and prediction accuracy of the dihedrals. Interestingly, most of the improvement is concentrated in the prediction of the χ1+2 term for the surface residues. The NCN algorithm shows an improvement of 7.8% for χ1+2 for the surface residues on the inclusion of a rotamer frequency term, as opposed to an improvement of 0.9% for the core χ1+2 (Table 2, rows 1 and 7, in Peterson et al. 2004). For LGA, adding a backbone dependency to a surface and volume term increases the accuracy by 6.7% and 10.3% for χ1 and χ1+2, respectively (Table 2, rows 2 and 4, in Liang and Grishin 2002). There is considerable discussion regarding the influence of the neighboring environment on the dihedral angles for surface residues (Gelin and Karplus 1979; van Gunsteren and Berendsen 1984; Kossiakoff et al. 1992). In a study of the influence of the crystal environment (Jacobson et al. 2002a), it was found that when the same proteins crystallized with different unit-cell geometries, ∼75% of the χ1 values for surface side-chains were in the same dihedral bin, i.e., within 40° of each other. The corresponding value for χ1+2 was ∼60%. Thus, the crystal packing and environment can have a significant influence on the values of the dihedral angles, especially for surface residues. An important conclusion from the study was that the target for prediction accuracies for χ1 for core residues was >95.0%; for surface residues forming crystal contacts, >80.0%; and for surface residues not involved in crystal contacts, 60%–80%. In this work, as in the majority of studies investigating side-chain placement, a neglect of the crystal environment implies that no surface chains are involved in crystal contacts. Our prediction accuracy for χ1 for the surface residues is 80%. Thus, the quality of our predictions is consistent with the variations that can be expected on account of the neglect of the crystal environment.
Effect of burial
Core residues were defined on the basis of the percentage of the solvent-accessible surface area in the protein compared with the isolated amino acid in its native state. If this percentage was below a cutoff value, PB, the residues were designated as core residues. Increasing the value of PB leads to a larger fraction of residues being labeled as core residues. In order to study the influence of residue burial on the prediction accuracies of χ1 and χ1+2, we carried out post-processing at several different values of PB. Table 3 summarizes the prediction accuracies for the above-mentioned analysis. As expected, this analysis reveals a systematic decrease in the prediction accuracy as the fraction of residues considered as core residues increases. These accuracies are very similar to those reported in other investigations. In particular, for the fraction of core residues equaling 53.8%, our predictions are [93.6 86.7]40. The results compare favorably with the accuracies of [94.1 87.4]40, [93.7 84.6]40, and [91.4 84.0]40 reported by Peterson et al. (2004) using the NCN, LGA, and SCAP algorithms, respectively, with a fraction of core residues equaling 54.3%.
Accuracy by residue type
Table 4 lists the overall prediction accuracy and the prediction accuracy for core residues as a function of residue type. The fraction of residues of a particular type in the core is also listed. As expected, there is a clear preference for the polar residues at the surface of the protein, accompanied by a corresponding decrease in the overall prediction accuracy for these residues. In these cases, it is not surprising that the rotamer frequency terms enable the LGA (Liang and Grishin 2002; Peterson et al. 2004) and NCN (Peterson et al. 2004) algorithms to predict the dihedral angles for these residues to a higher accuracy (Table 7 in Peterson et al. 2004) than SPRUCE. However, in a comparison with the SCAP (Xiang and Honig 2001) investigation that employs only a DDD, we find that the predictions from SPRUCE are overall superior to those from SCAP over the 65-protein test set (Table 7 in Peterson et al. 2004). For the SCFMT study that also uses a DDD, we similarly find our results (data not shown) are superior over the 20-protein test set used by Mendes et al. (1999), except for tryptophan where SCFMT performs better. A significant contribution to the increased accuracy in the prediction of tryptophan by SCFMT can be attributed to the inclusion of flexible angles in generation of the ensemble of rotamers (Mendes et al. 1999). Proline was not predicted in this study. In studies conducted by Liang and Grishin (2002) and Peterson et al. (2004), it was seen that the prediction accuracy for proline was close to the overall prediction accuracies averaged over all residue types. Hence, subtracting the contributions of proline from the overall prediction quality in these studies changes the overall accuracies negligibly.
Table 4.
Prediction accuracies as a function of residue type

Conclusions
We have demonstrated the applicability of a novel sampling method SPRUCE for the prediction of side-chain conformations in proteins. In conjunction with the all-atom AMBER99 force field, we have shown that this sampling method is able to achieve high accuracies that are comparable with the other detailed investigations in the literature. This method can essentially sample a continuous range of dihedral angles around the primary rotameric angles. The continuous sampling has the advantage that it can adaptively admit conformations around rotamers that would otherwise result in clashes with the remainder of the protein. Significant deviations from the primary rotamers can occur at protein–protein interfaces where the use of discrete rotamers can lead to steric clashes, and thereby a rejection of near-native states (Wang et al. 2005). Such problems are often tackled by introducing additional rotamers around primary rotamers (Gray et al. 2003; Peterson et al. 2004), development of more detailed rotamer libraries (Xiang and Honig 2001), or scaling of vdW radii (Liang and Grishin 2002; Gray et al. 2003; Peterson et al. 2004). No scaling of the vdW radii or modifications to the Lennard-Jones potentials was found necessary for the tests carried out with SPRUCE. However, since continuous flexibility demands an increasing amount of conformational space to be sampled and eliminates possibility of pretabulation of residue energies, the computational time required for SPRUCE is modest and on the order of a day for a single run over the entire test set using one processor. This time is of the same order as in other investigations using Monte Carlo–based methods (Table 6 in Peterson et al. 2004). Note, however, that all calculations were done with the all-atom AMBER force field. Use of a united-atom force field as in other studies (Mendes et al. 1999; Xiang and Honig 2001; Peterson et al. 2004) would reduce the system size by ∼40% resulting in a speed up by a factor of ∼3. The execution times of SCWRL (Bower et al. 1997; Canutescu et al. 2003) are typically 2 orders of magnitude less than those of Monte Carlo–based methods, though the overall prediction accuracies are also lower (Mendes et al. 1999; Xiang and Honig 2001; Liang and Grishin 2002). Though we have used the AMBER99 force field in this study, we stress that this method is not restricted to Hamiltonians with particular types of interaction potentials. Indeed, the separation of the Hamiltonian into a move proposal and move acceptance part can enable the use of the aforementioned tuned Hamiltonians with this method. While such a study is outside the present scope of this work, we are currently working to incorporate other force fields in order to present a thorough comparison of their applicability for side-chain packing. The Hamiltonian in this work employs a simplistic DDD for electrostatic interactions. Despite the fact that DDD does not accurately account for solvation effects, we find that our prediction accuracies rival those from studies that incorporate such effects. It would indeed be worthwhile to incorporate more accurate solvation models such as Generalized Born (GB) within the framework of SPRUCE to study the resulting changes in prediction accuracies. Jacobson et al. (2002a) report that the use of GB models (Hawkins et al. 1996; Gallicchio et al. 2002; Onufriev et al. 2004) in conjunction with the OPLS united-atom force field performs significantly better than a DDD. A primary motivation for the development of SPRUCE was to enable Monte Carlo methods based on rotamer libraries to supplement sampling from molecular dynamics simulations where a continuous range of conformations is accessible. SPRUCE can also be used to construct trial moves as a part of a larger Monte Carlo procedure such as the Biased Probability Monte Carlo Conformational search (Abagyan and Totrov 1994). We believe that SPRUCE offers an elegant methodology to introduce continuous sampling in the conformational space accessible to amino acid side-chains.
Materials and methods
Force field
The AMBER force field (Cornell et al. 1995) and charge set with modifications suggested by Okur et al. (2003) was used throughout this work. No attempt was made to include potential terms to account for the effects of hydrogen bonding, surface-area burial, rotamer frequency, or steric overlap. No linearization of the Lennard-Jones potential function or scaling of vdW radii was performed. In this sense, our work can be considered to use an ab initio force field with no additional terms that are trained to the problem at hand. This is similar to the investigation of Xiang and Honig (2001) that assessed the applicability of the united-atom CHARMM and AMBER force fields for the side-chain packing problem. To account for the electrostatics, we have used a DDD of 2.0rij where rij is the distance between the atoms i and j.
Protein test set
The test set used in this study comprises 76 high-resolution crystal structures obtained from the PDB (Berman et al. 2000). These proteins were selected based on the test sets used in other investigations (Xiang and Honig 2001; Liang and Grishin 2002; Peterson et al. 2004). This set includes the 65 proteins used by Peterson et al. (2004), 31 out of 33 structures used by Xiang and Honig (2001), and 15 structures used by Liang and Grishin (2002) that were not used to train their potential functions. The PDB codes for all the proteins used in this study are listed in Table 1. All prosthetic groups and nonprotein inclusions were stripped from the PDB files. The structures were optimized using REDUCE (Word et al. 1999). REDUCE optimizes the χ3 dihedral for glutamine and the χ2 dihedral for asparagine and histidine, in cases where the current dihedral χ + 180° leads to clearly better hydrogen bonding. Subsequent to this optimization, the protonation states for the histidines were decided using the GROMACS pdb2gmx module (Berendsen et al. 1995). Two cysteine residues were considered to form a disulfide bridge if the distance between their sulfur atoms was <2.5 Å. The resulting PDB files protonated and converted to the AMBER naming conventions, using AMBER (Pearlman et al. 1995). Generation of the topology files was done using an in-house program, GRAPPLE. No preminimization of the native structure was carried out. It has been shown that preminimization of the native structures can lead to improvements in the accuracy of the prediction (Xiang and Honig 2001).
Rotamer library
The backbone-independent version of the Dunbrack rotamer library was used in this study. This library was derived from a statistical analysis of 850 proteins. The total number of rotamers in the original library is 341 (Dunbrack and Cohen 1997). Using only the dihedral angles from the above rotamer library, the rotamers were generated using bond lengths and angles from the AMBER library files. The bond lengths and angles used to generate the rotamer configurations were not changed during the course of the SPRUCE iterations. However, all degrees of freedom associated with the flexible side-chains were allowed to change during the final minimization. In order to overlay the rotameric coordinates onto the fixed backbone of the proteins in the test set, a quaternion alignment was performed using the backbone N − Cα − C as the basis. The position of Cβ can change from the one in the crystal structure, though the change is usually very small. The rotamer library was enhanced to include flexibility in the χ5 dihedral for arginine. The dihedrals involving the polar hydrogens on serine, threonine, and tyrosine were also made flexible by adding four rotamer populations at 0°, 90°, 180°, and −90°. Three additional rotamers were added for histidine at χ2 = 0, for each of the three χ1 populations. The rotamers were read into the program along with the indices that indicate the population number for the dihedrals. For each population of a dihedral, a list was constructed that consisted of the rotamers that belong to that population. Finally, the bounds [l h]_{p}^{\chi}, on the dihedral values of the rotamers that comprise part of the same population were determined. For example, the rotamer library for asparagine consists of three populations for χ1 and six populations for χ2. The bounds on the dihedral angles are [50.3 69.3]11, [−176.0 −160.3]12, [−64.7 −79.5]13 for χ1. The bounds for χ2 are [−137.6 −114.6]21, [−66.3 −53.9]22, [−23.2 1.5]23, [24.3 57.0]24, [60.6 101.9]25 and [121.5 175.1]26.
Algorithm
The SPRUCE grows the side-chains for the flexible residues one unit at a time. The protocol employed for SPRUCE is shown below in the form of a pseudocode.
%————————– Begin pseudocode —————–———%
% Initialization
for each flexible residue r
persist[r] = 0
Select and substitute a random rotamer for r
end
% Begin iterations for side-chain packing
for each iteration i,
% Determine the patch to be regrown
R = random from list of flexible residues
if persist[R] > = lp
patch = {R}
else
patch = {R + N−1 flexible residue neighbors of R}
end
Energy_old = Energy of residues in patch
% Determine whether growth is from χ1 or χ2
for each residue r in patch,
current_χ [r] = 1 or 2
end
% Start the growth process
while any residue in patch incomplete
for each residue r in patch,
trial_count = 0
if residue r incomplete
for each population p of current_χ[r]
% Trials over range [l h]pχ for population p
for trials over range of p in increments of d
trial_dihedral = l + trial_count*dχ + rand(−0.5,0.5)*dχ
crds[trial_count] = generate coordinates for unit at trial_dihedral
energy[trial_count] = energy for unit
increment trial_count by 1
end
% Trials over the flexibility range
for Df random dihedral trials in the range [l-f l]
crds[trial_count] = generate coordinates for unit
energy[trial_count] = energy for unit
increment trial_count by 1
end
for Df random dihedral trials in the range [h h+f]
crds[trial_count] = generate coordinates for unit
energy[trial_count] = energy for unit
increment trial_count by 1
end
end
pick one trial i, based on Equation 1
substitute crds[i] into system
end
% Goto next dihedral in subsequent iteration
increment current_chi[r] by 1
end
end
Compare old patch with new patch
if patch is the same
for each residue r, in patch
increment persist[r] by 1
end
end
Energy_new = (Energy of residues in patch)
Accept or Reject based on Tacc, ΔUfull
if Accept and patch change
for each residue r, in patch
persist[r] = 0
end
end
end
%————————— End pseudocode—————————%
After the completion of the above iterations, a conjugate-gradient (Press et al. 1988) energy minimization was performed using the full Hamiltonian, keeping the backbone rigid. Tprop and Tacc were set to 1000 K and 20 K, respectively. The extent of variability surrounding the primary rotameric angles was ±45° for all residues. No attempt was made to optimize this value for specific dihedrals or residue types. The number of iterations used was 200 per flexible residue. N, lp, Df, and dχ were set to 6, 4, 3.0, and 5.0°, respectively. The extent of variability, limit for the persistence counter, patch size, and number of trial moves can be changed to alter the execution time for this algorithm. For the above choice of parameters, ∼22 h are required for a single run over the entire test set using one processor. The calculations were performed on a 3.2-GHz Intel Xeon processor. A unit in the above algorithm refers to a group of atoms that are used during the selection of a trial for a side-chain dihedral. For example, for the selection of χ1 for aspartate, the unit comprises the following atoms: hydrogens on Cβ and Cγ. For the selection of χ2, the corresponding unit consists of Oδ1 and Oδ2. At the stage where the final dihedral for a side-chain is being selected, all the side-chain atoms have formed part of one or more unit. Detailed topology files for each residue type that incorporate information regarding these units were compiled and used for this algorithm. The SPRUCE algorithm was implemented in an in-house program. In the current implementation, we intend to use SPRUCE as a sampling technique for generating side-chain configurations, hence, the detailed balance condition is not satisfied for the moves. However, the method can be extended to perform rigorous Monte Carlo sampling in the canonical ensemble, by including the total probabilities of the forward and reverse moves in the acceptance criterion. Tests were conducted to ensure that the energy calculations were in agreement with the AMBER program (Pearlman et al. 1995). We carried out five independent runs on the test set. The results from these runs were combined to give a final predicted structure that was used to calculate the statistics regarding the accuracy of placement of χ1 and χ1+2, and the RMSD. The procedure used for generating the final conformation is similar to the one used in Peterson et al. (2004). For each residue in a protein, the value of the dihedral is compared with the dihedral angle at the corresponding position in the rotamer library. Since the amount of flexibility is ±45°, the current conformation could only have resulted from rotamers that have the values of their dihedrals within ±45° of the angle in the conformation. These rotamers then have their counter incremented by one. This procedure is repeated for all the conformations in the different runs. The rotamer with the highest value of its counter is then selected as the rotamer in the final conformation. The precise value of the dihedral is obtained by averaging over the runs that led to an increment of the selected rotamer. Furthermore, these runs are labeled as active. For the subsequent dihedral along the side-chain, the procedure is repeated using only the active runs. In cases where a simple majority is absent within the active runs, an average is performed over the dihedrals in the runs that contribute to the highest valued rotamer counters. The prediction accuracies resulting from generating the combined structure are approximately better by [1.0 2.0]40 than those resulting from a simple average over the five independent runs.
Performance measures
Two widely implemented performance measures were used to judge the accuracy of the results from our method. They are the percentage of correctly predicted χ1 and χ1+2, and mass-weighted side-chain heavy-atom overall RMSD deviation. Backbone coordinates were not allowed to change during the SPRUCE protocol. A side-chain dihedral angle was said to be correctly predicted if it was within a certain absolute deviation from the angle in the native structure. Deviations of 40° and 20° were considered, since these are the tolerances that have been used in the literature. Due consideration was given to the symmetry of aspartate, glutamate, tyrosine, phenylalanine, and arginine, while calculating the dihedral angle values and the overall RMSD. In the cases where alternate positions were specified in the PDB files, the predicted configuration was considered correct if it satisfied the criteria for either of the alternate structures. Alternate structures were also used by Peterson et al. (2004) for calculating their prediction accuracies. However, Liang and Grishin (2002) excluded residues with alternate conformations during the calculation of prediction accuracies with their method.
Definition of core residues
There have been several different conventions for designating residues as core versus surface. In this study, the surface area was calculated for each residue in its native conformation both in the absence (i.e., exposed) and presence of the rest of the native protein. The surface area was calculated using a solvent probe radius of 1.4 Å. If the surface area in the presence of the protein was less than a certain percentage PB of the exposed surface area, the residue was designated as buried or core. For a value of PB = 12.5%, we find that ∼44% of the residues are buried. This number is close to the range of 40%–45% buried residues from other studies (Holm and Sander 1992; Xiang and Honig 2001; Liang and Grishin 2002). Table 3 lists the fraction of core residues as a function of PB and the corresponding prediction accuracies.
Program availability
The executable, topology, and input files required for SPRUCE can be obtained from the authors. SPRUCE has been programmed in C.
Acknowledgments
This research was supported in part by grants from the National Science Foundation, the National Institutes of Health, the Howard Hughes Medical Institute, the National Biomedical Computation Resource, the San Diego Supercomputer Institute, and the UCSD Achievement Rewards for Collegiate Scholars Program.
Footnotes
Reprint requests to: Tushar Jain, Howard Hughes Medical Institute, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0365, USA; e-mail: tjain@mccammon.ucsd.edu; fax: (858) 534-4974.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062165906.
References
- Abagyan R. and Totrov M. 1994. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235: 983–1002. [DOI] [PubMed] [Google Scholar]
- Berendsen H.J.C., van der Spoel D., van Drunen R. 1995. GROMACS—A message-passing parallel molecular-dynamics implementation. Comput. Phys. Commun. 91: 43–56. [Google Scholar]
- Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower M.J., Cohen F.E., Dunbrack R.L. 1997. Prediction of protein side-chain rotamers from a backbone-dependent rotamer library: A new homology modeling tool. J. Mol. Biol. 267: 1268–1282. [DOI] [PubMed] [Google Scholar]
- Canutescu A.A., Shelenkov A.A., Dunbrack R.L. 2003. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 12: 2001–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claussen H., Buning C., Rarey M., Lengauer T. 2001. Flexe: Efficient molecular docking considering protein structure variations. J. Mol. Biol. 308: 377–395. [DOI] [PubMed] [Google Scholar]
- Cornell W.D., Cieplak P., Bayly C.I., Gould I.R., Merz K.M., Ferguson D.M., Spellmeyer D.C., Fox T., Caldwell J.W., Kollman P.A. 1995. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117: 5179–5197. [Google Scholar]
- Cracknell R., Nicholson D., Parsonage N., Evans H. 1990. Rotational insertion bias: A novel method for simulating dense phases of structured particles, with particular application to water. Mol. Phys. 71: 931–943. [Google Scholar]
- Dahiyat B.I. and Mayo S.L. 1997. De novo protein design: Fully automated sequence selection. Science 278: 82–87. [DOI] [PubMed] [Google Scholar]
- De Maeyer M., Desmet J., Lasters I. 1997. All in one: A highly detailed rotamer library improves both accuracy and speed in the modelling of sidechains by dead-end elimination. Fold. Des. 2: 53–66. [DOI] [PubMed] [Google Scholar]
- Desjarlais J.R. and Handel T.M. 1995. De novo design of the hydrophobic cores of proteins. Protein Sci. 4: 2006–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet J., De Maeyer M., Hazes B., Lasters I. 1992. The dead-end elimination theorem and its use in protein side-chain positioning. Nature 356: 539–542. [DOI] [PubMed] [Google Scholar]
- Dunbrack R.L. and Cohen F.E. 1997. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 6: 1661–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbrack R.L. and Karplus M. 1993. Backbone-dependent rotamer library for proteins—Application to side-chain prediction. J. Mol. Biol. 230: 543–574. [DOI] [PubMed] [Google Scholar]
- Escobedo F. and Chen Z. 2000. A configurational-bias approach for the simulation of inner sections of linear and cyclic molecules. J. Chem. Phys. 113: 11382–11392. [Google Scholar]
- Escobedo F. and de Pablo J.J. 1994. Extended continuum configurational bias Monte Carlo methods for simulation of flexible molecules. J. Chem. Phys. 102: 2636–2652. [Google Scholar]
- Fernandez-Recio J., Totrov M., Abagyan R. 2002. Soft protein–protein docking in internal coordinates. Protein Sci. 11: 280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel D. and Smit B. 1996. Complex fluids. In Understanding molecular simulation pp. 271–313. 1st ed., Academic Press, New York.
- Gallicchio E., Zhang L.Y., Levy R.M. 2002. The sgb/np hydration free energy model based on the surface generalized born solvent reaction field and novel nonpolar hydration free energy estimators. J. Comput. Chem. 23: 517–529. [DOI] [PubMed] [Google Scholar]
- Gelin B.R. and Karplus M. 1979. Side-chain torsional potentials: Effect of dipeptide, protein, and solvent environment. Biochemistry 18: 1256–1268. [DOI] [PubMed] [Google Scholar]
- Gordon D.B. and Mayo S.L. 1998. Radical performance enhancements for combinatorial optimization algorithms based on the dead-end elimination theorem. J. Comput. Chem. 19: 1505–1514. [Google Scholar]
- Gordon D.B. and Mayo S.L. 1999. Branch-and terminate: A combinatorial optimization algorithm for protein design. Structure 7: 1089–1098. [DOI] [PubMed] [Google Scholar]
- Gordon D.B., Hom G.K., Mayo S.L., Pierce N.A. 2003. Exact rotamer optimization for protein design. J. Comput. Chem. 24: 232–243. [DOI] [PubMed] [Google Scholar]
- Gray J.J., Moughon S., Wang C., Schueler-Furman O., Kuhlman B., Rohl C.A., Baker D. 2003. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 331: 281–299. [DOI] [PubMed] [Google Scholar]
- Havranek J.J. and Harbury P.B. 2003. Automated design of specificity in molecular recognition. Nat. Struct. Biol. 10: 45–52. [DOI] [PubMed] [Google Scholar]
- Hawkins G.D., Cramer C.J., Truhlar D.G. 1996. Parameterized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem. 100: 19824–19839. [Google Scholar]
- Holm L. and Sander C. 1992. Fast and simple monte carlo algorithm for side-chain optimization in proteins: Application to model-building by homology. Proteins 14: 213–223. [DOI] [PubMed] [Google Scholar]
- Huang E.S., Koehl P., Levitt M., Pappu R.V., Ponder J.W. 1998. Accuracy of side-chain prediction upon near-native protein backbones generated by ab initio folding methods. Proteins 33: 204–217. [DOI] [PubMed] [Google Scholar]
- Hwang J.K. and Liao W.F. 1995. Side-chain prediction by neural networks and simulated annealing optimization. Protein Eng. 8: 363–370. [DOI] [PubMed] [Google Scholar]
- Jacobson M.P., Friesner R.A., Xiang Z.X., Honig B. 2002a. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 320: 597–608. [DOI] [PubMed] [Google Scholar]
- Jacobson M.P., Kaminski G.A., Friesner R.A., Rapp C.S. 2002b. Force field validation using protein side chain prediction. J. Phys. Chem. B 106: 11673–11680. [Google Scholar]
- Jain T. and de Pablo J. 2002a. A biased Monte Carlo technique for calculation of the density of states of polymer films. J. Chem. Phys. 116: 7238–7243. [Google Scholar]
- Jain T. and de Pablo J. 2002b. Monte Carlo simulation of free-standing polymer films near the glass transition temperature. Macromolecules 35: 2167–2176. [Google Scholar]
- Kossiakoff A.A., Randal M., Guenot J., Eigenbrot C. 1992. Variability of conformations at crystal contacts in BPTI represent true low-energy structures: Correspondence among lattice packing and molecular dynamics structures. Proteins 14: 65–74. [DOI] [PubMed] [Google Scholar]
- Kraemer-Pecore C.M., Lecomte J.T.J., Desjarlais J.R. 2003. A de novo redesign of the WW domain. Protein Sci. 12: 2194–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman B., Dantas G., Ireton G.C., Varani G., Stoddard B.L., Baker D. 2003. Design of a novel globular protein fold with atomic-level accuracy. Science 302: 1364–1368. [DOI] [PubMed] [Google Scholar]
- Kumar S., Szleifer I., Panagiotopoulos A. 1991. Determination of the chemical potentials of polymeric systems from Monte Carlo simulations. Phys. Rev. Lett. 66: 2395–2398. [DOI] [PubMed] [Google Scholar]
- Lasters I., De Maeyer M., Desmet J. 1995. Enhanced dead-end elimination in the search for the global minimum energy conformation of a collection of protein side-chains. Protein Eng. 8: 815–822. [DOI] [PubMed] [Google Scholar]
- Liang S.D. and Grishin N.V. 2002. Side-chain modeling with an optimized scoring function. Protein Sci. 11: 322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looger L.L. and Hellinga H.W. 2001. Generalized dead-end elimination algorithms make large-scale protein side-chain structure prediction tractable: Implications for protein design and structural genomics. J. Mol. Biol. 307: 429–445. [DOI] [PubMed] [Google Scholar]
- Looger L.L., Dwyer M.A., Smith J.J., Hellinga H.W. 2003. Computational design of receptor and sensor proteins with novel functions. Nature 423: 185–190. [DOI] [PubMed] [Google Scholar]
- Mendes J., Baptista A.M., Carrondo M.A., Soares C.M. 1999. Improved modeling of side-chains in proteins with rotamer-based methods: A flexible rotamer model. Proteins 37: 530–543. [DOI] [PubMed] [Google Scholar]
- Metropolis N., Rosenbluth A.W., Rosenbluth M.N., Teller A.H., Teller E. 1953. Equation of state calculations by fast computing machines. J. Chem. Phys. 21: 1087–1092. [Google Scholar]
- Okur A., Strockbine B., Hornak V., Simmerling C. 2003. Using PC clusters to evaluate the transferability of molecular mechanics force fields for proteins. J. Comput. Chem. 24: 21–31. [DOI] [PubMed] [Google Scholar]
- Onufriev A., Bashford D., Case D.A. 2004. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 55: 383–394. [DOI] [PubMed] [Google Scholar]
- Pearlman D.A., Case D.A., Caldwell J.W., Ross W.S., Cheatham T.E., Debolt S., Ferguson D., Seibel G., Kollman P. 1995. Amber, a package of computer-programs for applying molecular mechanics, normal-mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 91: 1–41. [Google Scholar]
- Peterson R.W., Dutton P.L., Wand A.J. 2004. Improved side-chain prediction accuracy using an ab initio potential energy function and a very large rotamer library. Protein Sci. 13: 735–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrella R.J., Lazaridis T., Karplus M. 1998. Protein sidechain conformer prediction: A test of the energy function. Fold. Des. 3: 353–377. [DOI] [PubMed] [Google Scholar]
- Press W.H., Teukolsky S.A., Vetterling W.T., Flannery B.P. 1988. Minimization or maximization of functions. In Numerical recipes in C pp. 420–425. 2d Cambridge University Press, New York.
- Siepmann J. and Frenkel D. 1992. Configurational bias Monte Carlo—A new sampling scheme for flexible chains. Mol. Phys. 75: 59–70. [Google Scholar]
- Siepmann J. and Wick C. 2000. Self-adapting fixed-end-point configurational-bias Monte Carlo method for the regrowth of interior segments of chain molecules with strong intramolecular interactions. Macromolecules 33: 7207–7218. [Google Scholar]
- Tuffery P., Etchebest C., Hazout S., Lavery R. 1991. A new approach to the rapid-determination of protein side-chain conformations. J. Biomol. Struct. Dyn. 8: 1267–1289. [DOI] [PubMed] [Google Scholar]
- van Gunsteren W.F. and Berendsen H.J.C. 1984. Computer simulation as a tool for tracing the conformational differences between proteins in solution and in the crystalline state. J. Mol. Biol. 176: 559–564. [DOI] [PubMed] [Google Scholar]
- Vasquez M. 1996. Modeling side-chain conformation. Curr. Opin. Struct. Biol. 6: 217–221. [DOI] [PubMed] [Google Scholar]
- Wang C., Schueler-Furman O., Baker D. 2005. Improved side-chain modeling for protein–protein docking. Protein Sci. 14: 1328–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Word J.M., Lovell S.C., Richardson J.S., Richardson D.C. 1999. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 285: 1735–1747. [DOI] [PubMed] [Google Scholar]
- Wu M. and Deem M. 1999. Analytical rebridging Monte Carlo: Application to cis/trans isomerization in proline-containing, cyclic peptides. J. Chem. Phys. 111: 6625–6632. [Google Scholar]
- Xiang Z. and Honig B. 2001. Extending the accuracy limits of prediction for side-chain conformations. J. Mol. Biol. 311: 421–430. [DOI] [PubMed] [Google Scholar]
- Zacharias M. 2003. Protein–protein docking with a reduced protein model accounting for side-chain flexibility. Protein Sci. 12: 1271–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]