

Abstract
Hydrophobins are small fungal proteins that are highly surface active and possess a unique ability to form amphiphilic membranes through spontaneous self-assembly. The first crystal structure of a hydrophobin, Trichoderma reesei HFBII, revealed the structural basis for the function of this amphiphilic protein—a patch consisting of hydrophobic side chains on the protein surface. Here, the crystal structures of a native and a variant T. reesei hydrophobin HFBI are presented, revealing the same overall structure and functional hydrophobic patch as in the HFBII structure. However, some structural flexibility was found in the native HFBI structure: The asymmetric unit contained four molecules, and, in two of these, an area of seven residues was displaced as compared to the two other HFBI molecules and the previously determined HFBII structure. This structural change is most probably induced by multimer formation. Both the native and the N-Cys-variant of HFBI were crystallized in the presence of detergents, but an association between the protein and a detergent was only detected in the variant structure. There, the molecules were arranged into an extraordinary detergent-associated octamer and the solvent content of the crystals was 75%. This study highlights the conservation of the fold of class II hydrophobins in spite of the low sequence identity and supports our previous suggestion that concealment of the hydrophobic surface areas of the protein is the driving force in the formation of multimers and monolayers in the self-assembly process.
Keywords: hydrophobin, amphiphile, surfactant, class II, pseudomerohedral twinning, high solvent content
Hydrophobins are a group of proteins with a unique property to spontaneously self-assemble into amphiphilic layers and thus invert the hydropathy of a surface. Hydrophobins are found in filamentous fungi only, and they play important roles in fungal growth, e.g., in lowering the surface tension of water to enable the growth of the hyphae into the air and the coating of the surfaces of aerial hyphae to conceal the hydrophilic cell wall in the air environment (Wösten et al. 1999; Linder et al. 2005). A fungal species may carry several hydrophobin genes, expressed at different times during growth, located in different parts of fungi, and targeted for a specific function. Hydrophobins are nontoxic but may act in pathogenic infections by mediating the attachment to the host organism (Ebbole 1997).
The unique properties of hydrophobins make them potential candidates for various medical and technical applications (Wessels 1997; Scholtmeijer et al. 2001; Wösten 2001), e.g., to be used as coatings to increase biocompatibility of medical implants, in drug delivery using oil vesicles stabilized by hydrophobins, to immobilize enzymes on surfaces, to be used as an anti-fouling agent in surgical tools and window panes, or in the cosmetic industry for hair-care products (Scholtmeijer et al. 2002; Janssen et al. 2002, 2004). Hydrophobins can also cause harmful effects, such as the gushing of beer (Sarlin et al. 2005).
The application potential of hydrophobins has recently been carefully reviewed (Hektor and Scholtmeijer 2005) and some of the potential applications have already proved to be possible, in practice. Hydrophobins can be used in an efficient separation technique (Linder et al. 2004) where a hydrophobin tag is used in the purification of a recombinant protein in a surfactant-based two-phase system. Also, an endoglucanase fused to a Trichoderma reesei hydrophobin HFBI has been successfully immobilized to a hydrophobic surface (Linder et al. 2002). This particular hydrophobin can also be produced on a g scale (Askolin et al. 2001), which makes it a tempting candidate for industrial applications. Kisko and coworkers (Kisko et al. 2005) have shown that the preparation of organized polycrystalline hydrophobin multilayers is possible for T. reesei hydrophobins HFBI and HFBII.
Hydrophobins have been divided into two classes according to the hydropathy patterns of their sequence and the solubility of the assembled layers (Wessels 1994). The assemblages of class I hydrophobins are more resistant to dissociation as compared to assemblies formed by class II hydrophobins. In general, the assemblages formed by hydrophobins require quite harsh conditions before they break down, trifluoroacetic acid for class I hydrophobins and typically 60% ethanol for class II hydrophobins. Hydrophobins are also stable at high temperatures. The formation of class I hydrophobin layers has been found to lead to a rodlet-like mosaic pattern that resembles amyloid fibrils, whereas the aggregates of class II hydrophobins are nonamyloidal and needle-like.
Hydrophobin molecules are typically 70–130 amino acid residues in size and include a signal sequence for secretion. By primary structure, hydrophobins may show a remarkable diversity but are characterized by eight conserved cysteine residues that appear in the sequence in a characteristic pattern. Class I hydrophobins have a somewhat longer stretch in sequence between the third and the fourth conserved cysteines than the class II hydrophobins, and the spacing of the conserved cysteine residues is more invariant for class II. Hydrophobins may also be glycosylated.
The high-resolution structural studies of the class II hydrophobin HFBII from T. reesei (Hakanpää et al. 2004a,b) revealed the molecular basis of the amphiphilicity of hydrophobins, namely, a patch on the protein surface that consists of side chains of hydrophobic residues. Disulfide bridges strongly hold together the HFBII structure, consisting of four antiparallel β-strands and an α-helix. The disulfide bridges are formed nonconsecutively, and two of the bridges are located inside a small barrel that the β-strands form.
Crystallographic studies of hydrophobins have so far been limited to the HFBII structure; the structure of HFBII has been deposited with the Protein Data Bank (PDB; Berman et al. 2000) with the code 1R2M and has been recently updated to an ultrahigh resolution, code 2B97 (Hakanpää et al. 2006). In addition, crystallization conditions and preliminary X-ray characterization have been reported for another class II hydrophobin, HFBI from T. reesei (Askolin et al. 2004), and an NMR study of a class I hydrophobin EAS from Neurospora crassa has been recently redescribed (Kwan et al. 2006). EAS was formerly stated to be largely unstructured in solution (Mackay et al. 2001) but has now been re-examined and found to share a similar fold as that of HFBII. However, in the NMR structure of EAS, the α-helix is missing and instead two short β-strands occupy this region. The disulfide bridges are formed by the eight conserved cysteine residues in a manner similar to that of HFBII. The areas that correspond to the hydrophobic patch in the HFBII structure are disordered or not defined in the NMR structure of EAS.
Determining the protein–protein interactions between hydrophobin molecules is a key factor for understanding how hydrophobins function because their most important functions, such as surface activity, formation of coatings, and adhesion to surfaces, seem to be dependent on self-assembly. In the crystal structure, HFBII forms a dimer with the hydrophobic patches of the two molecules packed against each other, thereby concealing a large portion of the hydrophobic patch from solvent contacts. This type of protein–protein interaction is also most likely important in determining how hydrophobins function in solution. For example, HFBII is highly soluble in water (to concentrations as high as 100 mg/mL), and our previous data show that a micelle-like association of amphiphilic hydrophobin molecules occurs in an aqueous environment. However, instead of micelles, most likely the protein forms dimers and tetramers in solution (Torkkeli et al. 2002; Szilvay et al. 2006). Because data on the structural features of the multimers have been very difficult to obtain using other methods such as NMR, the packing of molecules in crystals is very useful in suggesting functional protein–protein interactions.
In this study we present new crystallization conditions for HFBI and its variant, dubbed N-Cys HFBI, and the solved crystal structures for both. The work revealed several new protein packing geometries that can suggest functional interactions. The N-Cys HFBI exists in solution as a covalently linked dimer and was chosen for the study because the dimeric form of the protein leads to increased protein–protein interactions. Therefore, new structural data on these interactions could be obtained from this variant.
Results
The structure of the native HFBI
The overall crystal structure of the native HFBI consisted of four HFBI molecules (designated A–D) in the asymmetric unit, four zinc ions, and 108 water molecules. Hydrophobins are not metalloproteins, and the zinc ions originate from the crystallization solution. No 1-S-octyl-β-D-thioglucoside (OSG) detergent molecules could be found in the asymmetric unit, even though addition of the detergent to the crystallization solution was necessary in order to produce crystals that diffracted X rays well enough for structure determination. The overall structure of HFBI was very similar to HFBII, consisting of four β-sheets and one α-helix (Fig. 1A,C). The same arrangement of disulfide bridges as in HFBII was confirmed for HFBI: The conserved cysteines formed disulfide bridges nonconsecutively. The bridges formed between residues Cys8 and Cys57, Cys18 and Cys48, Cys19 and Cys31, and Cys58 and Cys69. The first bridge connected the N-terminal loop to the small barrel formed by the β-sheets, the second bridge connected the α-helix to the β-barrel, and the last two bridges were located inside the β-barrel.
Figure 1.

(A) The structure of HFBI, molecule A, in stereo. The α-helix is in cyan; β-strands, in purple. The region of the structural change in the HFBI structure is highlighted in red. (B) HFBI, molecule B, showing different conformation compared to molecule A. (C) HFBII, molecule A. (D) Superimposition of molecules A and B of the native HFBI on the area of the structural change. Molecule B is in green.
The B-factors remained rather high throughout the refinement; the average B-factor calculated at the end of the refinement by the Moleman program (Kleywegt et al. 2001) was 44.7 Å2. The final R factors were also relatively high (Table 1) in comparison to the resolution of the data, probably due to the pseudomerohedral twinning present in the crystals. Handling twinned data is problematic, especially when twinning is perfect, as is the case here. However, during validation, the structure was found to be plausible. In the Ramachandran plot, 76% of the residues were found in the most favored regions, 23% in the additionally allowed regions, and 1% in the generously allowed regions.
Table 1.
The statistics of the data collection, processing, and refinement of the HFBI and the N-Cys HFBI-variant data

The electron density maps were mostly of very good quality. However, some of the residues in both the N and the C termini were not clearly visible in the electron density maps, probably due to disorder, and a different number of amino acid residues could be fitted into the density in the termini of each protein molecule. Therefore, molecule A included residues 2–73; molecule B, residues 7–75; molecule C, residues 5–75; and molecule D, residues 6–74. Excluding the termini, the electron density was clear and unambiguous for molecules A and C. Some problems were encountered in the helical part of molecules B and D. In molecule B, residues Lys32, Pro34, Asn46, Val46, and Cys48 showed weak density, and in molecule D, residues Lys32, Ser35, Asp40, Phe44, and Arg45 showed weak density. This lack of clear density was most likely caused by twinning; without the use of the twin operator in the refinement, the electron density in these helical regions was significantly weaker and more ambiguous.
One significant structural difference was found in molecules B and D of the HFBI structure, in comparison to molecules A and C in the same structure, as well as molecules in the HFBII structure: An area of seven residues, from Ala60 to Ala66, had moved to a more extended conformation (Fig. 1D). The direction of the movement was away from the β-barrel, and the greatest distance between the positions of the Cα-atoms of Ala63 in molecule B, and molecule A superimposed onto molecule B, was 8.6 Å. The corresponding distance for molecules D and C was 10.3 Å. The flexible area was located in the second β-hairpin motif, consisting of residues in the β-hairpin loop and in the last β-strand. Some of the residues of the hydrophobic patch were located in this flexible area: Ala60, Val62, Ala63, and Ala66. The sixth and the seventh cysteine residues, Cys57 and Cys58, preceded the flexible area. The position of Val59 was still approximately the same as of that of the corresponding residue in molecule A or C, but the side chain had turned somewhat and there was a flip in the peptide bond between residues Val59 and Ala60. The flexible region ended with Leu67, the position of which was close to that of the corresponding residue in molecule A or C, but the side chain is in a slightly different orientation. Leu67 was followed by Leu68 and Cys69, the eighth conserved cysteine residue, and thus the conserved cysteines delimited the area of the structural changes (Fig. 1D). Movement in the aforementioned area had some effect on the elements of the secondary structure of molecules B and D. The fourth β-strand ran from Gly64 to Thr71 in the HFBI molecules A and C, and also correspondingly in the HFBII structure. In molecules B and D of the HFBI structure, this β-strand was shortened from eight residues to three residues and consisted of residues from Cys69 through Thr71. In molecules A and C of the HFBI structure, the β-strand content in the molecule was 37%, while in molecules B and D it was 31%.
The four molecules in the asymmetric unit of the HFBI structure form a curved structure (Fig. 2B). The curving provided a slight groove in the middle of the HFBI tetramer, where most of the hydrophobic surface areas were buried. Movement in the area of the last β-hairpin motif in molecules B and D appeared to facilitate the formation of this tetramer.
Figure 2.
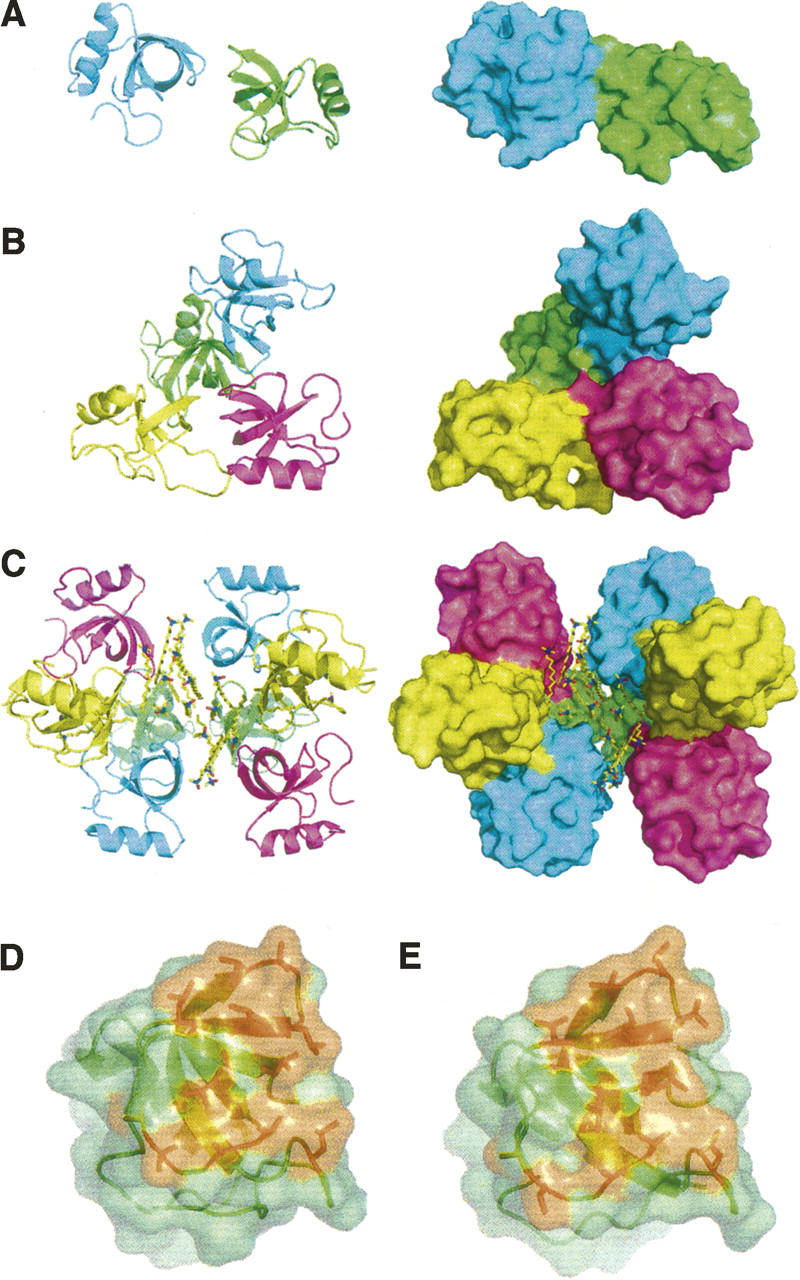
Ribbon (left) and molecular surface (right) representation of (A) HFBII dimer, (B) HFBI tetramer, and (C) N-Cys HFBI detergent-mediated octamer. The molecular surfaces of HFBI (D) and HFBII (E). The hydrophobic patch is in orange.
Four zinc ions were located in the asymmetric unit of the HFBI structure, coordinated with aspartic acid residues on the protein surface. These zinc ions mediated symmetry contacts between adjacent molecules and were, therefore, crucial for the crystal packing. One coordination site was between molecules A and B, and an equivalent site was also found between molecules C and D. The residue interacting with the zinc ion was Asp30 in each molecule. The crystal structure of HFBII contains a manganese ion in a location identical to that of the aforementioned zinc ion in the HFBI structure, but the coordination is different. The corresponding aspartic acid residues (Asp25) are involved in the HFBII structure, but, in addition, an aspartic acid residue (Asp34) and a glutamine residue (Gln60) from adjacent dimers coordinate with the manganese ion. Two additional metal coordination sites in the HFBI structure were located between molecule A and a symmetry molecule C and molecule B and a symmetry molecule D. Here, residues Asp40 in molecules A, C, and D were involved, as was Asp43 in molecules A, B, and C. In the HFBII structure, this type of coordination is not possible, since the corresponding residues are Thr35 and Ile38.
The structure of the N-Cys HFBI variant
The overall crystal structure of the N-Cys HFBI variant consisted of four HFBI molecules (designated A–D), two zinc ions, and 10 lauryldimethylamine oxide (LDAO) detergent molecules in the asymmetric unit (Fig. 3). One hundred eighteen water molecules were included in the refinement. Most of the LDAO molecules were well ordered in the corresponding electron density, but density for some of the polar head groups was not visible (Fig. 4). The extended, hydrophobic carbon chain was, however, always clearly observable. It is possible that some of the detergent molecules had coordinated to the same place, but the chains run in opposite directions, which explains the weak density for the head group.
Figure 3.
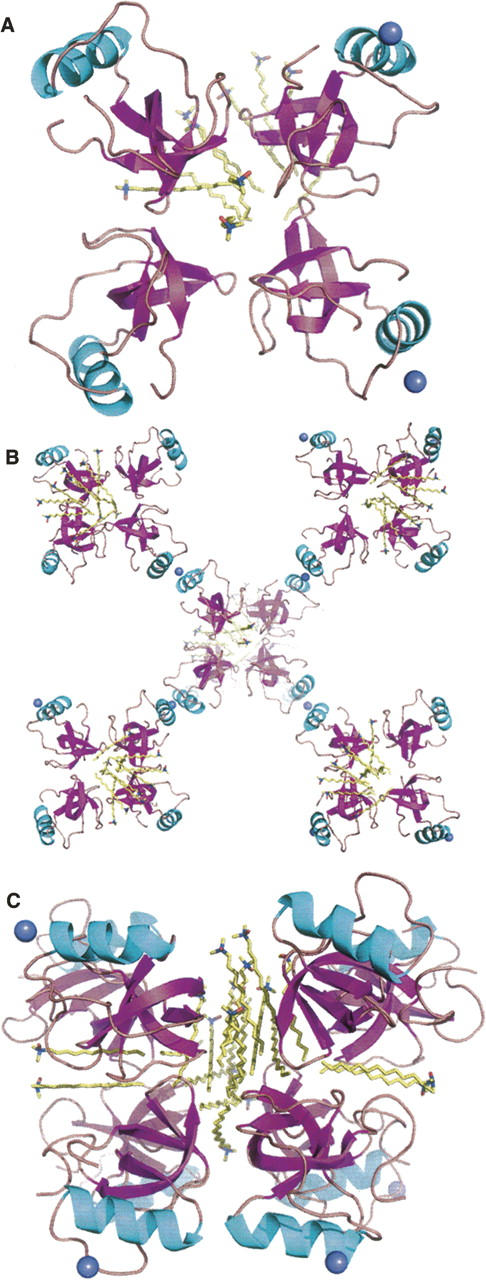
The N-Cys HFBI-variant structure: (A) Contents of the asymmetric unit, (B) packing through the zinc ions (clover), and (C) detergent interactions (detergent-associated octamer).
Figure 4.

The electron density map of the N-Cys HFBI structure, showing the density around the LDAO detergent molecules as 2 |Fo| − |Fc| map at 1.5 σ.
The electron density maps were of very good quality, and the density was clear and unambiguous for the protein main and side chains. In all four N termini of the protein molecules, the amino acid residues could be fitted to the electron density starting only from Asn6. The N-terminal extensions, through which the covalent dimer was formed in the variant structure, could not be identified anywhere in the electron density maps. The lack of electron density for these areas could be due to the long and flexible nature of the extensions, since the N termini of adjacent HFBI molecules were close enough to allow the molecules to be linked through the extensions. However, since the protein material also contained fragmented forms of N-Cys HFBI variant, the absence of the N-terminal extension in the solved structure could indicate that the crystallizing form was not the covalent dimer but fragments of it. Some weak residual density was visible in the N termini, which might indicate that the crystal consists of HFBI molecules with the N termini of different length.
The structure of the HFBI molecules greatly resembled that of hydrophobin HFBII (Fig. 1A,C). The secondary structure elements and the arrangement of the disulfide bridges were identical to the HFBII structure and molecules A and C of the native HFBI structure. Movement in the second β-hairpin region, as seen in molecules B and D of the native HFBI structure, was not observed. As with the native HFBI, the N-Cys HFBI-variant structure also contained four HFBI molecules in the asymmetric unit (Fig. 3), but the packing of monomers was different. Also, the interactions within the asymmetric units were different, since in the native HFBI structure the molecules were in contact through a rather large area in the region of the hydrophobic patch, whereas in the N-Cys HFBI variant structure the hydrophobic areas mainly interacted with the detergents. The four HFBI molecules in the N-Cys HFBI-variant structure were positioned in a manner similar to a four-leaf clover, all in the same plane. At the tip of each leaf, a zinc ion was found, connecting each clover to the adjacent one through coordination to aspartic acid residues (Asp40 in molecules B and D and Asp43 in all of the molecules) on the protein surface. The location of the zinc ion was identical to the latter one described for the HFBI structure. Ten LDAO detergent molecules were located in the asymmetric unit, and two of these were coordinated in the middle of the HFBI clover (Figs. 3, 4). A part of the hydrophobic patch of each HFBI molecule in the clover faced these detergents. Eight more LDAO molecules lay on one side of the clover, concealing the rest of the hydrophobic patches from the solvent. Another clover, produced by crystal symmetry from (x,y,z,) with a symmetry operation (−1-x, y, −1/2-z), was located on the other side of the detergents, so that a sandwich-like structure of two HFBI clovers with detergent molecules in between was formed. Therefore, the quaternary structure was a detergent-associated octamer, composed of eight HFBI molecules and 20 LDAO molecules (Fig. 3C).
Discussion
Structural changes
The overall structures of the hydrophobin molecules in the HFBI, N-Cys HFBI, and HFBII structures were very similar with molecules B and D in the HFBI structure being the only exception, with a flexible area in the second β-hairpin motif. This type of movement was not possible in the crystal structures of the N-Cys HFBI variant, HFBII, or the A and C molecules of the HFBI structure, because the extended region would have collided with another molecule in the asymmetric unit or with a symmetry molecule; i.e., the crystal contacts prevented the area from moving. It is probable that the movement in the HFBI structure was not forced by the crystal contacts, since the flexible region faced a solvent channel in the crystal structure. Rather, the movement in the β-hairpin area was most likely driven by the formation of the HFBI tetramer. Also, if molecules B and D did retain the conformation present in molecules A and C, Gln65 of molecule B or D would have collided with Ala20 of an adjacent molecule. Although the movement in the second β-hairpin area is not present in the dimeric crystal structure of HFBII, one might speculate that HFBII could form a tetrameric structure similar to that of HFBI, the formation of which would require similar conformational changes. In any case, it seems that conformational changes are mostly limited to the loop areas of class II hydrophobins, whereas the central β-barrel structure, reinforced by the disulfide bridges, seems to remain unchanged.
For comparison, all the molecules in each structure were superimposed pairwise by least-square fitting by Cα in the Shelxpro program (Sheldrick and Schneider 1997). The residues used in the least-square fitting were Val7–Val73 in the HFBI molecules and Val2–Gly68 in the HFBII molecules to insure that the length of the match is the same for each comparison. In the HFBI structure, there were two types of molecules with respect to the conformation of the second β-hairpin motif. Molecules A and C had this area in a similar conformation and the molecules superimposed with an root-mean-square distance (RMSD) of 0.45 Å. Molecules B and D both possessed extended conformation, and there were other types of subtle differences between these two molecules, since they superimposed with an RMSD of 1.00 Å. Superimposing molecule A or C with B or D produced an average RMSD value of 2.53 Å. The four HFBI molecules in the N-Cys HFBI-variant structure were all in almost identical conformations; superimposing any two of these molecules produced an average RMSD of 0.29 Å. When molecules A or B of the HFBII structure were superimposed with molecules A or C of the HFBI structure, the average RMSD was 0.59 Å, and with molecules B or D or the HFBI structure, the average RMSD was 2.67 Å. When molecules A or B of the HFBII structure were superimposed with any of the molecules in the N-Cys HFBI-variant structure, the RMSD was 0.59 Å.
Some of the RMSD values appeared to be relatively high, and to some extent this may be affected by the crystallographic resolution of the models (Carugo 2003). However, high RMSD values also indicated that plasticity is an intrinsic property of hydrophobin structures. When the residues in the β-strands only were least-square-fitted by Cα, the RMSD values were somewhat lower, indicating that the β-barrel structure is more stable than the entire molecule; e.g., the RMSD for superimposing the β-strands of any two molecules from the N-Cys HFBI structure was on average 0.16 Å, and superimposing the β-strands of molecules B and D in the HFBI structure produced an RMSD value of 0.54 Å. The plasticity may be a property of hydrophobins that is necessary in order to be able to bind to surfaces (which are often coarse) and to form monolayers.
Hydrophobic patch
The hydrophobic patch, the functional site of hydrophobins, could be distinctly identified on the surface of HFBI molecules, both in the HFBI and the N-Cys HFBI-variant structures. This area correlated very well with the hydrophobic patch of HFBII. In an HFBI molecule, the hydrophobic side chains forming the hydrophobic patch consisted of residues Leu12, Val23, Leu24, Leu26, Ile27, Leu29, Val59, Ala60, Val62, Ala63, Ala66, Leu67, and Leu68. In an HFBII molecule, the corresponding residues were Leu7, Val18, Leu19, Leu21, Ile22, Val24, Val54, Ala55, Val57, Ala58, Ala61, Leu62, and Leu63. When the sequences of HFBI and HFBII were aligned according to the conserved cysteines (Fig. 5), the residues in the hydrophobic patch were exactly the same, except for the residue corresponding to Leu29 in the HFBI structure, which was a valine (Val24) in the HFBII structure. In the vicinity of Leu29 lay Gln65 and Gly64, the corresponding residues in the HFBII sequence being Gln60 and Asp59. It is noteworthy that close to the hydrophobic patch of HFBII lies a charged residue (Asp59) in a location that is typically a glysine (as for HFBI, Gly64) in the sequence of class II hydrophobins. In the HFBI structure, the side chain of Gln65 stuck out from the surface as compared to Gln60 of HFBII, but this may have been due to the crystal packing. In addition, residues Phe13, Ala20, and Val33 were located on the protein surface in the HFBI structure and on the edges of the hydrophobic patch and could have been considered a part of this functional site. These residues are the same also in the HFBII structure, except for Thr28, which corresponds to Val33 in the HFBI structure.
Figure 5.

Sequence comparison of class II hydrophobins. Residues, corresponding to those of hydrophobic patch of HFBI and HFBII, are highlighted with a gray background. The conserved cysteine residues are indicated in bold.
The shape of the hydrophobic patch was relatively flat in the HFBI structure, for molecules A and C, and in the N-Cys HFBI-variant structure. The hydrophobic patch areas of hydrophobins HFBI and HFBII appeared very similar (Fig. 2D,E). In molecules B and D of the HFBI structure, the shape of the hydrophobic surface area was altered due to movement in the second β-hairpin motif, and a slightly curved hydrophobic patch had been produced.
The solvent accessible areas (SAA) for the HFBI tetramer and the HFBII dimer were 14,100 Å2 and 7300 Å2, respectively. For the N-Cys HFBI structure, the SAA of the four HFBI molecules was 14,200 Å2, when the interactions with the detergent molecules were excluded. The SAA of an HFBI molecule was, on average, ∼3500 Å2 in the presence of the other molecules in the tetramer and 4100 Å2 in the absence of the others. The corresponding figures for a N-Cys HFBI molecule were 3600 Å2 and 4100 Å2 and, for a HFBII molecule, 3700 Å2 and 4000 Å2. We calculated the SAA of the hydrophobic patch by using the side-chain atoms of the aforementioned 13 aliphatic residues that constitute the hydrophobic patch. This SAA in the absence of the adjacent molecules was ∼19% of the total SAA of one molecule for all the structures. For HFBII, the size of the hydrophobic patch was previously estimated to be ∼12% of the total surface area. Calculated with the side-chain atoms, the patch size for HFBII is also ∼19%. The SAA values for individual molecules are presented in Table 2.
Table 2.
The solvent accessible areas (SAA) in HFBI, N-Cys HFBI, and HFBII structures

The area buried by the other molecules in the asymmetric unit was ∼14% and 19% of the total SAA of one HFBI molecule in HFBI and N-Cys HFBI structures, respectively. In the N-Cys HFBI structure, the detergent molecules took part in concealing the protein surface from the solvent; in fact, the area buried by the protein molecules in the N-Cys HFBI structure was only 13%. For HFBII, the area buried between the dimer interface was 8% of the total SAA of one HFBII molecule. Thus, the area buried between the dimer interface in the HFBII structure is about half of what is buried between the tetramer interface in the HFBI structure. Consequently, the tetrameric structure is more efficient in concealing the hydrophobic surface areas in comparison to the dimeric form, suggesting that the formation of a larger multimer is an energetically favorable process. Also, the SAA of the hydrophobic patch for the monomeric, detergent-interacting form of HFBI in the N-Cys HFBI structure was found to be considerably smaller than those of the tetrameric and dimeric forms, indicating that interaction with a hydrophobic counterpart is favored over the multimer formation.
When considering the area of the hydrophobic patch, ∼34%, 45%, and 68% of the patch was buried in the HFBII, HFBI, and N-Cys HFBI structures, respectively. However, in the HFBI structure, concealment of the hydrophobic surface areas was more efficient for molecules A and C. For these molecules, ∼51% of the patch areas were buried, whereas for molecules B and D ∼40% of the patch areas were buried. The buried patch area in the N-Cys HFBI structure varied from molecule to molecule and ranged from 55% to 82%, with the average value being 68%. Also, the amount of areas that were buried by the adjacent protein molecules, and those buried by detergent molecules varied. On average, the adjacent protein molecules buried 38% and the detergent molecules buried 30% of the patch area.
We have argued before that multimers of hydrophobin are formed in solution, driven by the formation of an energetically more favorable state, where parts of the hydrophobic patch are concealed from the solvent by multimerization, in comparison to a monomeric hydrophobin where the entire hydrophobic surface area would be exposed to the solvent (Fig. 6). We suppose that upon monolayer formation multimers dissociate first to less soluble monomers. These monomers are able to form a monolayer in which packing of monomers is considerably different than in multimers, allowing the formation of an exposed hydrophobic surface on one side. Self-assembly onto a surface of a hydrophobic substrate is probably even more energetically favorable than formation of multimers. The structure of the N-Cys HFBI variant, where the hydrophobin monomers are gathered around the detergent molecules, conceals more of the hydrophobic surface areas of the protein than do the dimeric or tetrameric forms and thus provides further evidence that supports the above-mentioned scheme.
Figure 6.

A proposal for a model of multimerization in solution and the formation of monolayer.
Multimerization
The multimerization of HFBI and HFBII has been shown to be very concentration-dependent. It has been found that HFBI and HFBII form tetramers at a high protein concentration (10 mg/mL), while at a lower concentration (0.5 mg/mL), dimers and monomers are formed (Torkkeli et al. 2002). It was recently found that monomers of HFBI and HFBII are formed at low concentrations, and a maximum level of multimerization is attained at ∼150 μg/mL, where the tetrameric state is found to be predominant (Szilvay et al. 2006). Also in the crystal structures, the multimerization states correlated with the protein concentrations in the crystallization drops: The concentrations of the proteins in the crystallization drops were 8 mg/mL, 1 mg/mL, and 4 mg/mL for HFBI, N-Cys HFBI, and HFBII, respectively. Thus, the highest protein concentration produced the highest multimer, i.e., the HFBI tetramer, while the lower protein concentrations produced the HFBII dimer and the monomeric structure of the N-Cys HFBI. Tetramer and dimer formation has also been observed for a class I hydrophobin SC3 (Wang et al. 2004).
The multimerization states of all current hydrophobin structures differ from each other. Hydrophobin HFBII is in a dimeric state, with parts of the hydrophobic patches of the molecules buried in the dimer interface. From the neighboring dimer, there is partial contact with the unburied part of the hydrophobic patch, but the interaction is much weaker than the interaction within the dimer, and, therefore, the multimerization state was determined as a dimer rather than a loose tetramer.
In the N-Cys HFBI-variant structure, the hydrophobin molecules were in a monomeric state, if we only consider the contacts between the adjacent protein molecules. The hydrophobic patch of each molecule was partially packed against the detergent molecules and, thus, concealed. Most of the contacts with the side-chain atoms of the residues in the hydrophobic patch were made with the detergent molecules; there were 356 protein–detergent interactions, with a distance <5.0 Å. The interchain protein–protein contacts on the hydrophobic surface, with a distance of <5.0 Å, were limited to 82 interactions. Thus, the quaternary structure was clearly a detergent-associated octamer, formed by the eight hydrophobin HFBI molecules and the 20 detergent molecules (Fig. 3). On the basis of the current three-dimensional structures of hydrophobins, it seems that hydrophobins require additional hydrophobic moiety, such as detergents, in order to form octameric structures.
In the native HFBI structure, the molecules in the asymmetric unit were in contact via hydrophobic interaction through the hydrophobic patch, coordinated through two of the zinc ions, hydrogen bonds, and van der Waals interactions. Between adjacent molecules in the tetramer, there were 83 protein–protein interactions with a distance of <4.0 Å. In between the side-chain atoms of the residues in the hydrophobic patch of adjacent molecules, there were 114 interactions with a distance of <5.0 Å. The closest contact between hydrophobic patches of adjacent molecules was 3.3 Å. Therefore, the HFBI structure was considered a tetramer. There were more contacts between the molecule pairs of A–B and C–D than between the pairs A–C and B–D. Therefore, it is possible that the dimers A–B and C–D are first formed and the final tetramer is formed from two dimers. Overall, this study shows that hydrophobins are able to form different kinds of multimers. It seems that in solution there is a concentration-dependent equilibrium of monomers, dimers, and tetramers (Fig. 6). Multimerization is an efficient method for protecting the hydrophobic patch or “the active surface” prior to formation of monolayers.
In this work we have been able to obtain a large amount of significant information by comparing the crystal structures of HFBI and its variant and the previously determined HFBII structure. On the basis of the structural change in the second β-hairpin motif and especially the relatively high RMSD values, we propose that plasticity plays an important role in the function of HFBI. Yet, the central β-barrel seems to be more rigid than other parts of the structure. It has been suggested that significant structural changes are essential for the function of class I hydrophobins (Wang et al. 2002; Zangi et al. 2002). Even so, the detailed description of structural changes opens the door toward designing new experiments so that we may understand the functional role of such changes. In addition, the finding that surfactants interact in a defined way with hydrophobins in the HFBI-variant structure may lead to exploration of new functions for hydrophobins. The various arrangements in the crystal packing observed for hydrophobins also show that the molecular architecture of these proteins truly is in favor of the formation of a variety of multimeric structures. We still know very little about the role of the self-assembly of hydrophobins in fungal biology, but the current results indicate that a magnitude of molecular interactions and self-assembled structures can be expected. We also note that the versatility of interactions and arrangements seen for hydrophobins can make these molecules very useful in the developing field of nanotechnology.
Materials and methods
Protein production
The native HFBI was produced using an overexpressing strain of T. reesei as previously described (VTT D-98692) (Linder et al. 2001). The sequence of the native HFBI is SNGNGNVCPPGLFSNPQCCATQVLGLIGLDCKVPSQNVYDGTDFRNVCAKTGAQPLCCVAPVAGQALLCQTAVGA. The N-Cys HFBI variant (T. reesei strain: Rut-C30/F18A/pGZ12) had an additional segment of 13 amino acids engineered to its N terminus. The sequence of this extension was SCPATTTGSSPGP. The second cysteine residue in the extension was included in order to enable a site-specific bioconjugation with synthetic functional groups. Details of the genetic methods used are described elsewhere (Kostiainen et al. 2006; G.R. Szilvay, unpubl.). The name N-Cys HFBI was chosen for the variant because the presence of Cys2 in the N terminus affected some of its properties; e.g., it caused covalent dimerization of the molecule.
Purification
Both the native protein and the N-Cys HFBI variant were purified in essentially the same way. Biomass from bioreactor cultivations was subjected to treatment with 4 M hydrochloride in 200 mM Tris-buffer at pH 7.5 for 2 h. The mixture was centrifuged for 15 min at 4000g. The supernatant was recovered, diluted with water, and subjected to extraction with a surfactant, as previously described (Linder et al. 2001). The protein was then purified by reverse-phase high-performance liquid chromatography (RP-HPLC). The fractions, containing native and variant HFBI, were identified by mass spectroscopy. The pooled peak fractions were then lyophilized.
Characterization by mass spectrometry
The exact molecular weights of the proteins were verified using a high resolution ESI-FTICR mass spectrometer (data not shown). The average molecular weight of the native HFBI was determined to be 7532.6 Da and the material was homogenous; it had no deaminations, nor was proteolytic cleavage detected. Hydrophobin HFBI had previously been reported to be deaminated and/or to be cleaved from the N terminus (Askolin et al. 2001). The covalent dimer of the N-Cys HFBI-variant was determined to have a mass of 17,351.05 Da, which is equal to the mass of two native protein molecules and linkers. Fragments of the N-Cys HFBI variant were also present in the protein material. These fragments were heterogenic, often consisting of one N-Cys HFBI molecule with the N-terminal extension cleaved in various positions. Analysis of the fragments in the mass spectra suggested that some of the material already bound together as a covalent dimer had been cleaved after binding, since fragments existed that corresponded to a mass of one HFBI molecule, its N-terminal extension, and fragments of another extension.
Crystallization
Lyophilized HFBI material was dissolved in pure water at a concentration of 16 mg/mL for crystallization. The N-Cys HFBI variant was found less water soluble and was dissolved in pure water at a concentration of 6 mg/mL. The hanging-drop vapor-diffusion method was applied at 293 K.
The crystals of HFBI grew from a mother liquor containing 0.1 M zinc sulfate as a precipitant and 0.1 M sodium cacodylate as a buffer at pH 6.5. Crystals formed very rapidly; when inspected under the microscope immediately after pipetting, small crystals could already be seen. However, these crystals diffracted X rays extremely poorly. When the detergent OSG was added to the crystallization drop, the growth rate of the crystals slowed down and the diffraction quality improved. Still, the crystals grew from the precipitant that formed immediately after pipeting. Drop size was 5 μL, and the concentrations of the protein and the detergent in the drop were 8 mg/mL and 9 mM, respectively. The size of the crystal used for data collection was 0.35 × 0.35 × 0.02 mm.
The crystals of the N-Cys HFBI variant grew from a mother liquor of 0.1 M zinc acetate as the precipitant, 0.1 M HEPES as a buffer at pH 7, and the detergent LDAO) as an additive. The drop size used was 5 μL, and the concentrations of the protein and the detergent in the drop were 1 mg/mL and 2 mM, respectively. The size of the crystal used for data collection was 0.2 × 0.2 × 0.05 mm.
Data collection
The data were collected using synchrotron radiation from the EMBL beamlines BW7B, located at the DORIS storage ring, DESY, Hamburg. The data were collected on a Mar345 Image Plate detector at 100 K. The crystals were shock-frozen in liquid nitrogen after a cryo-protectant soak. The cryo-protectant solution was formulated by replacing 30% and 35% of the water in the mother liquor of crystallization with 2-methyl-2,4-pentanediol and glycerol for the crystals of the native HFBI and the N-Cys HFBI variant, respectively. The cryo-treated crystals were stored in liquid nitrogen, and the data were collected with the crystal placed under a nitrogen stream. The data collection times were ∼6 h and 4 h for the native and the variant data sets, respectively. An oscillation angle of 1.0° was used, and the total angular range covered was 180° for the HFBI-data and 120° for the N-Cys HFBI-variant data.
The data were processed and scaled with the XDS program (Kabsch 1993). The crystals of the native HFBI diffracted to 2.1 Å and appeared to belong to space group F222 with unit cell dimensions a = 49.60 Å, b = 108.94 Å, c = 132.55 Å. The data processed well in this space group (Rmeas and I/σ(I) in the last shell were 28.7% and 8.22, respectively), but after the molecular replacement and the refinement the R factors remained high, and part of the electron density for the model was ambiguous. This led us to believe that the crystals might be twinned in a way that is undetectable from the diffraction images, i.e., by pseudomerohedral twinning, and the twin operator mimics a symmetry operator of a higher crystal system. This type of twinning has been previously observed for acetyl coenzyme A synthetase with true space group C2 and apparent space group F222 (Lehtiö et al. 2005), and γδ T-cell ligand T10 with true space group P21 and apparent space group C222 (Rudolph et al. 2004). Pseudomerohedral twinning occurs in a monoclinic space group when the condition c cosβ = −a / 2 is fulfilled. This rule is true for HFBI data; thus the twin law was assumed to be −h, −k, h + l, as for acetyl coenzyme A synthetase (Lehtiö et al. 2005). HFBI data were then processed in the space group C2. The statistics are presented in Table 1.
Crystals of the N-Cys HFBI variant diffracted to a 2.3 Å resolution and were of the orthorhombic crystal system, space group C2221 or C222, of which C2221 produced much lower crystallographic R factors when the structure was refined. These statistics are presented in Table 1.
Structure solution
The HFBI data processed in space group C2 were used for molecular replacement with the T. reesei hydrophobin HFBII as a search model in the Molrep program (Vagin and Teplyakov 1997) in the CCP4 suite (Collaborative Computational Project, Number 4 1994). The amino acid identity of these two proteins is 69%. A calculation of the Matthews coefficient (Matthews 1968) suggested four (VM = 2.98 Å3/Da) to six (VM = 1.99 Å3/Da) molecules are present in the asymmetric unit. Four molecules were located in the asymmetric unit, but no hints of a presence, i.e., corresponding electron density for additional molecules, could be found. With four molecules in the asymmetric unit, the solvent content of the crystal was 58.8%.
The structure solution of the N-Cys HFBI variant was also carried out in Molrep using one of the molecules in the newly solved HFBI structure as a search model. Molrep located four HFBI molecules in the asymmetric unit, but no extra density, due to protein, could be detected when the electron density map was inspected. This corresponds to a Matthews coefficient of 4.9 Å3/Da and a solvent content of 74.6%, which is exceptionally high in comparison to usual values for the Matthews coefficient, which lie between 1.62 and 3.53 Å3/Da. However, structures with a solvent content of ∼70% can be found in the PDB, e.g., transcriptional regulatory protein with a 71% solvent content (PDB code 1QKK) and tropomyosin with a 70% solvent content (PDB code 1KQL).
Refinement
The structure of the native HFBI was refined with the Shelxl program (Sheldrick and Schneider 1997) due to the convenience of this refinement program in handling twinned data. When refined without the twin instructions, the R factors remained high, with R ∼ 30% and Rfree ∼ 40%. With the twin law (−h, −k, h + l) included (Shelx command TWIN −1 0 0 0–1 0 1 0 1), the first round of refinement in the space group C2 yielded R factors R = 26.4% and Rfree = 30.9% for all data. A BASF instruction was included in the refinement in order to estimate the twin fraction. The twin fraction was also estimated with DETWIN in the CCP4 suite and the twin test indicated a twin fraction close to 0.5. The BASF value refined to ∼0.49 and the twinning in the HFBI data could be thought of as perfect. This was expected, since the data processed so well in the higher symmetry space group. Also, detwinning of the data was not possible due to the perfect pseudo-merohedral twinning, and the structure was refined until convergence, using the untreated twinned data with the twin instructions. The refinement statistics are presented in Table 1.
The variant HFBI structure was refined with the program CNS (Brünger et al. 1998). The refinement statistics are presented in Table 1. Program O (Jones et al. 1991) was used to inspect the electron density maps. For the HFBI and the N-Cys HFBI-variant structures, the solvent accessible areas were calculated with the Areaimol program (Lee and Richards 1971) in the CCP4 suite, using a solvent molecule probe with a radius of 1.4 Å. The geometries of the structures were checked with Procheck (Laskowski et al. 1993) and Whatif (Vriend 1990). The figures were generated with the PyMOL program (DeLano Scientific). The structures have been deposited with the PDB under codes 2FZ6 (native HFBI) and 2GVM (N-Cys HFBI variant).
Acknowledgments
The ISB National Graduate School has supported J.H. and G.Sz. European Community—Research Infrastructure Action under the FP6 “Structuring the European Research Area Programme” contract no. RII3/CT/2004/5060008 is acknowledged for access to EMBL-DESY Hamburg beamlines. We would like to thank the beamline staff of the EMBL-Hamburg/DESY beamline BW7B for their skilled help. We would also like to thank Dr. Janne Jänis and Ms. Hanne Kinnunen for analysis of the protein material by ESI-FTICR, Prof. Markku Ahlgren for consultation in determining the correct twin operator, and Mrs. Reetta Kallio-Ratilainen for skilled technical assistance.
Footnotes
Reprint requests to: Juha Rouvinen, Department of Chemistry, University of Joensuu, P.O. Box 111, 80101 Joensuu, Finland; e-mail: juha.rouvinen@joensuu.fi; fax: +358-13-251-3390.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062326706.
Abbreviations: LDAO, lauryldimethylamine oxide; OSG, 1-S-octyl-β-D-thioglucoside; PDB, the Protein Data Bank; RMSD, root-mean-square distance; RP-HPLC, reversed-phase high-performance liquid chromatography; SAA, solvent accessible area.
References
- Askolin S., Nakari-Setälä T., Tenkanen M. 2001. Overproduction, purification, and characterization of the Trichoderma reesei hydrophobin HFBI. Appl. Microbiol. Biotechnol. 57: 124–130. [DOI] [PubMed] [Google Scholar]
- Askolin S., Turkenburg J.P., Tenkanen M., Uotila S., Wilson K.S., Penttilä M., Visuri K. 2004. Purification, crystallization and preliminary X-ray diffraction analysis of the Trichoderma reesei hydrophobin HFBI. Acta Crystallogr. D Biol. Crystallogr. 60: 1903–1905. [DOI] [PubMed] [Google Scholar]
- Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissing I.N., Bourne P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S. et al. 1998. Crystallography & NMR System: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. [DOI] [PubMed] [Google Scholar]
- Carugo O. 2003. How root-mean-square distance (r.m.s.d.) values depend on the resolution of protein structures that are compared. J. Appl. Crystallogr. 36: 125–128. [Google Scholar]
- Collaborative Computational Project Number 4. 1994. The CCP4 Suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50: 760–763. [DOI] [PubMed] [Google Scholar]
- Ebbole D.J. 1997. Hydrophobins and fungal infection of plants and animals. Trends Microbiol. 5: 405–408. [DOI] [PubMed] [Google Scholar]
- Hakanpää J., Paananen A., Askolin S., Nakari-Setälä T., Parkkinen T., Penttilä M., Linder M.B., Rouvinen J. 2004a. Atomic resolution structure of the HFBII hydrophobin, a self-assembling amphiphile. J. Biol. Chem. 279: 534–539. [DOI] [PubMed] [Google Scholar]
- Hakanpää J., Parkkinen T., Hakulinen N., Linder M., Rouvinen J. 2004b. Crystallization and preliminary X-ray characterization of Trichoderma reesei hydrophobin HFBII. Acta Crystallogr. D Biol. Crystallogr. 60: 163–165. [DOI] [PubMed] [Google Scholar]
- Hakanpää J., Linder M., Popov A., Schmidt A., Rouvinen J. 2006. Hydrophobin HFBII in detail—Ultra-high resolution structure of 0.75 Å. Acta Crystallogr. D Biol. Crystallogr. 62: 356–367. [DOI] [PubMed] [Google Scholar]
- Hektor H.J. and Scholtmeijer K. 2005. Hydrophobins: Proteins with potential. Curr. Opin. Biotechnol. 16: 1–6. [DOI] [PubMed] [Google Scholar]
- Janssen M.I., van Leeuwen M.B., Scholtmeijer K., van Kooten T.G., Dijkhuizen L., Wösten H.A. 2002. Coating with genetic engineered hydrophobin promotes growth of fibroblasts on a hydrophobic solid. Biomaterials 23: 4847–4854. [DOI] [PubMed] [Google Scholar]
- Janssen M.I., van Leeuwen M.B., van Kooten T.G., de Vries J., Dijkhuizen L., Wösten H.A. 2004. Promotion of fibroblast activity by coating with hydrophobins in the β-sheet end state. Biomaterials 25: 2731–2739. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zhou J.Y., Cowan S., Kjeldgaard M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47: 110–119. [DOI] [PubMed] [Google Scholar]
- Kabsch W. 1993. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26: 795–800. [Google Scholar]
- Kisko K., Torkkeli M., Vuorimaa E., Lemmetyinen H., Seeck O.H., Linder M., Serimaa R. 2005. Langmuir-Blodgett films of hydrophobins HFBI and HFBII. Surf. Sci. 584: 34–40. [DOI] [PubMed] [Google Scholar]
- Kleywegt G.J., Zou J.Y., Kjelgaard M., Jones T.A. 2001. Around O. In International tables for crystallography, Vol. F. Crystallography of biological macromolecules. Kluwer Academic Publishers(eds. Rossmann M.G. and Arnold E.) . , Dordrecht, The Netherlands 366–367.
- Kostiainen M.A., Szilvay G.R., Smith D.K., Linder M.B., Ikkala O. 2006. Multivalent dendrons for high-affinity adhesion of proteins to DNA. Angew. Chem. Int. Ed. Engl. 45: 1–6. [DOI] [PubMed] [Google Scholar]
- Kwan A.H.Y., Winefield R.D., Sunde M., Matthews J.M., Haverkamp R.G., Templeton M.D., Mackay J.P. 2006. Stuctural basis for rodlet assembly in fungal hydrophobins. Proc. Natl. Acad. Sci. 103: 3621–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26: 283–291. [Google Scholar]
- Lee B. and Richards F.M. 1971. The interpretation of protein structures: Estimation of static accessibility. J. Mol. Biol. 55: 379–400. [DOI] [PubMed] [Google Scholar]
- Lehtiö L., Fabrichnig I., Hansen T., Schönheit P., Goldman A. 2005. Unusual twinning in an acetyl coenzyme A synthetase (ADP-forming) from Pyrococcus furiosus. Acta Crystallogr. D Biol. Crystallogr. 61: 350–354. [DOI] [PubMed] [Google Scholar]
- Linder M., Selber K., Nakari-Setälä T., Qiao M., Kula M.R., Penttilä M. 2001. The hydrophobins HFBI and HFBII from Trichoderma reesei showing efficient interactions with nonionic surfactants in aqueous two-phase systems. Biomacromolecules 2: 511–517. [DOI] [PubMed] [Google Scholar]
- Linder M., Szilvay G.R., Nakari-Setälä T., Söderlund H., Penttilä M. 2002. Surface adhesion of fusion proteins containing the hydrophobins HFBI and HFBII from Trichoderma reesei. Protein Sci. 11: 2257–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder M., Qiao M., Laumen F., Selber K., Hyytiä T., Nakari-Setälä T., Penttilä M.E. 2004. Efficient purification of recombinant proteins using hydrophobins as tags in surfactant-based two-phase systems. Biochemistry 43: 11873–11882. [DOI] [PubMed] [Google Scholar]
- Linder M., Szilvay G., Nakari-Setälä T., Penttilä M. 2005. Hydrophobins: The protein amphiphiles of fungi. FEMS Microbiol. Rev. 29: 877–896. [DOI] [PubMed] [Google Scholar]
- Mackay J.P., Matthews J.M., Winefield R.D., Mackay L.G., Haverkamp R.G., Templeton M.D. 2001. The hydrophobin EAS is largely unstructured in solution and functions by forming amyloid-like structures. Structure 9: 83–91. [DOI] [PubMed] [Google Scholar]
- Matthews B.W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33: 491–497. [DOI] [PubMed] [Google Scholar]
- Rudolph M.G., Wingren C., Crowley M.P., Chien Y., Wilson I.A. 2004. Combined pseudo-merohedral twinning, non-crystallographic symmetry and pseudo-translation in a monoclinic crystal form of the γδ T-cell ligand T10. Acta Crystallogr. D Biol. Crystallogr. 60: 656–664. [DOI] [PubMed] [Google Scholar]
- Sarlin T., Nakari-Setälä T., Linder M., Penttilä M., Haikara A. 2005. Fungal hydrophobins as predictors of the gushing activity of malt. J. Inst. Brew. 111: 105–111. [Google Scholar]
- Scholtmeijer K., Wessels J.G., Wösten H.A. 2001. Fungal hydrophobins in medical and technical applications. Appl. Microbiol. Biotechnol. 56: 1–8. [DOI] [PubMed] [Google Scholar]
- Scholtmeijer K., Janssen M.I., Gerssen B., de Vocht M.L., van Leeuwen B.M., van Kooten T.G., Wösten H.A., Wessels J.G. 2002. Surface modifications created by using engineered hydrophobins. Appl. Environ. Microbiol. 68: 1367–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G.M. and Schneider T.R. 1997. SHELXL: High-resolution refinement. Methods Enzymol. 277: 319–343. [PubMed] [Google Scholar]
- Szilvay G.R., Nakari-Setälä T., Linder M. 2006. Behavior of Trichoderma reesei hydrophobins in solution: Interactions, dynamics, and multimer formation. Biochemistry (in press). [DOI] [PubMed]
- Torkkeli M., Serimaa R., Ikkala O., Linder M. 2002. Low-resolution structural models. Biophys. J. Trichoderma reesei 83: 2240–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A. and Teplyakov A. 1997. MOLREP: An automated program for macromolecular replacement. J. Appl. Crystallogr. 30: 1022–1025. [Google Scholar]
- Vriend G. 1990. WHAT IF: A molecular modeling and drug design program. J. Mol. Graph. 8: 52–56. [DOI] [PubMed] [Google Scholar]
- Wang X., de Vocht M.L., de Jonge J., Poolman B., Robillard G.T. 2002. Structural changes and molecular interactions of hydrophobin SC3 in solution and on a hydrophobic surface. Protein Sci. 11: 1172–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Graveland-Bikker J.F., de Kruif C.G., Robillard G.T. 2004. Oligomerization of hydrophobin SC3 in solution: From soluble state to self-assembly. Protein Sci. 13: 810–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels J.G.H. 1994. Developmental regulation of fungal cell wall formation. Annu. Rev. Phytopathol. 32: 413–437. [Google Scholar]
- Wessels J.G. 1997. Hydrophobins: Proteins that change the nature of the fungal surface. Adv. Microb. Physiol. 38: 1–45. [DOI] [PubMed] [Google Scholar]
- Wösten H.A.B. 2001. Hydrophobins: Multipurpose proteins. Annu. Rev. Microbiol. 55: 625–646. [DOI] [PubMed] [Google Scholar]
- Wösten H.A.B., van Wetter M.A., Lugones L.G., van der Mei H.C., Busscher H.J., Wessels J.G.H. 1999. How a fungus escapes the water to grow into the air. Curr. Biol. 9: 85–88. [DOI] [PubMed] [Google Scholar]
- Zangi R., de Vocht M.L., Robillard G.T., Mark A.E. 2002. Molecular dynamics study of the folding of hydrophobin SC3 at a hydrophilic/hydrophobic interface. Biophys. J. 83: 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]