

Abstract
Introduction
We have previously shown that the Beta Protein 1 (BP1) homeodomain protein is expressed in 81% of invasive ductal breast carcinomas, and that increased BP1 expression correlates with tumor progression. The purpose of our current investigation was to determine whether elevated levels of BP1 in breast cancer cells are associated with increased cell survival.
Methods
Effects on cell viability and apoptosis of MCF7 cells stably overexpressing BP1 were determined using MTT and Annexin V assays, and through examination of caspase activation. TNFα was used to induce apoptosis. The potential regulation of apoptosis-associated genes by BP1 was studied using real-time PCR and western blot analyses. Electrophoretic mobility shift assays, site-directed mutagenesis, and transient assays were performed to specifically characterize the interaction of BP1 with the promoter of the bcl-2 gene.
Results
Stable overexpression of BP1 led to inhibition of apoptosis in MCF7 breast cancer cells challenged with TNFα. Increased BP1 resulted in reduced processing and activation of caspase-7, caspase-8, and caspase-9, and inactivation of the caspase substrate Poly(ADP-Ribose) Polymerase (PARP). Increased levels of full-length PARP and a decrease in procaspase-8 were also associated with BP1 overexpression. The bcl-2 gene is a direct target of BP1 since: (i) BP1 protein bound to a consensus binding sequence upstream of the bcl-2 P1 promoter in vitro. (ii) MCF7 cells overexpressing BP1 showed increased levels of bcl-2 mRNA and protein. (iii) Transient assays indicated that increased bcl-2 promoter activity is due to direct binding and modulation by BP1 protein. BP1 expression also prevented TNFα-mediated downregulation of bcl-2 mRNA and protein.
Conclusion
These findings suggest mechanisms by which increased BP1 may impart a survival advantage to breast cancer cells, which could lead to increased resistance to therapeutic agents in patients.
Introduction
Homeobox genes are an important class of master regulatory genes that encode transcription factors responsible for orchestrating developmental processes in many species of animals, as well as in plants and fungi. These genes are characterized by a conserved 180-nucleotide sequence coding for a 60-amino-acid homeodomain that directs binding to downstream target genes that may be activated or repressed. An increasing number of investigations support the involvement of homeobox genes in tumorigenesis of prostate, lung, renal, ovarian, colorectal, and breast tissues [1,2]. Specifically in breast cancer, altered levels of various homeobox genes are directly associated with cellular transformation, disruption of the cell cycle, apoptosis, and progression to a metastatic phenotype [3-7].
Beta Protein 1 (BP1) belongs to the Distal-less subfamily of the homeobox gene family [8]. BP1 maps to chromosome 17q21-22, a region of DNA that is often amplified in breast cancer and that contains the tumor suppressor gene BRCA1 and the oncogene ErbB2 [9]. We have found that BP1 is expressed in 81% of invasive ductal breast tumors [10,11]. Notably, BP1 expression correlates with breast cancer progression [11], suggesting BP1 may be important in breast tumorigenesis. We have yet to fully understand, however, the functional consequences of its increased expression. Our earlier studies demonstrated that BP1 is expressed in 63% of acute myeloid leukemias but is not detectable in normal lymphoid cells or in normal bone marrow [12]. In clonogenic assays, K562 erythroleukemia cell lines stably overexpressing BP1 showed a 45-fold increase in the number of cells able to grow in soft agar compared with control cells, but we did not observe differences in cell number per colony [12]. These results indicate that BP1 may play an oncogenic role by increasing cell survival.
Tumor cells are notorious for escaping cell death and often develop resistance to therapeutic agents through activation of antiapoptotic mechanisms. Apoptosis is coordinated by cascades of caspases, a family of cysteine proteases that cleave various substrates, ultimately leading to the destruction of the cell. Two primary pathways of apoptosis have been established. The death-receptor pathway, or extrinsic pathway, is triggered through binding of cytokines (TNFα, TRAIL, Fas ligand) to their respective receptors that belong to the TNF receptor family [13]. The mitochondrial pathway, or intrinsic pathway, is regulated by proapoptotic and antiapoptotic members of the Bcl-2 family, which collectively govern the permeability of the mitochondrial membrane [13,14]. Crosstalk between these two pathways can occur, whereby the mitochondrial pathway is triggered following death receptor activation [15,16].
Our objective in the present investigation was to determine whether BP1 impacts antiapoptotic pathways in breast cancer cells. Specifically, we demonstrate that increased BP1 expression protects MCF7 cells challenged with TNFα, resulting in inhibition of apoptosis. We also show that BP1 protein binds to and directly activates expression of bcl-2, an antiapoptotic gene. These findings provide evidence of a role for BP1 in cell survival and define mechanisms by which BP1 expression may be tumorigenic.
Materials and methods
Cell culture and generation of stable cell lines
MCF7 cells were transfected with either the empty vector pcDNA3.2 (Invitrogen, Carlsbad, CA, USA) or a plasmid containing the BP1 open reading frame under control of the cytomegalovirus promoter. Plasmid-containing cell lines were selected in 800 μg/ml G418. Cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum, penicillin/streptomycin, 500 μg/ml G418, and 2 mM glutamine. MTT assays were performed to measure cell viability. Cells were seeded in triplicate in 96-well plates, and were cultured in normal growth media containing 20 ng/ml human TNFα (Sigma-Aldrich, St Louis, MO, USA) or were left untreated. After 72 hours, samples were incubated with 5 mg/ml MTT at 37°C for 4 hours. Formazan crystals were dissolved in dimethylsulfoxide (Sigma-Aldrich). Samples were read at 570 nm with a Versamax microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Annexin V assay
Cell lines were cultured at 3 × 105 cells/well in six-well plates, and were cultured in normal growth media containing 20 ng/ml TNFα for 18 hours or were left untreated. Cells were labeled with a 1:100 dilution of Annexin V–FITC conjugate and 5 μg/ml propidium iodide according to the manufacturer's instructions (Trevigen, Gaithersburg, MD, USA). Each sample was analyzed using a Nikon Eclipse TE300 inverted epifluorescence microscope (Nikon Instruments Inc, Melville, NY, USA) with filter sets for FITC and TRITC. Early apoptotic cells were distinguished by the presence of green staining in the plasma membrane and the absence of red nuclear staining.
Electrophoretic mobility shift assays
Complementary sequences spanning 2,555 to 2,513 nucleotides upstream of the bcl-2 ATG start site were annealed and 5'-end-labeled with γ-32P-ATP using T4 kinase (Invitrogen). The Wheat Germ Coupled Transcription/Translation kit (Promega, Madison, WI, USA) was used to generate BP1 protein from the plasmid pGEM7 containing the BP1 open reading frame. Unlabeled competitor oligonucleotides were added at 500× or 1,000× molar excess to binding reactions. For supershift analyses, binding reactions included BP1 antibody [8]. The following sequences were used as probes and competitors: bcl-2, 5'-ACGGTGGGCCTGAAAGTTACTATATGGAAGTCCTCATCGTGTA-3'; mutant bcl-2, 5'-ACGGTGGGCCTGAAAGTTAGCTCGACGAAGTCCTCATCGTGTA-3'; negative control, 5'-TCTTAGAGGGAGGGCTGAGGGTTTGAAGTCCAACTCCTAAGCC-3'.
Luciferase reporter assays
A construct containing the bcl-2 P1 promoter region linked to a luciferase reporter gene (LB170) was a kind gift from Dr Linda Boxer (Stanford University, Stanford, CA, USA). Cells were transfected with 2.5 μg LB170 and 1 μg plasmid encoding β-galactosidase, using Fugene 6 Transfection Reagent (Roche, Indianapolis, IN, USA) at a 3:2 ratio of Fugene:DNA according to the manufacturer's instructions. Forty-eight hours post transfection, β-galactosidase activity was measured using the Beta-Galactosidase Enzyme Assay System (Promega), and the luciferase reporter activity was assayed using the Luciferase Assay System (Promega). Luciferase activity output was given in relative light units. The relative light unit value for each sample was divided by the β-galactosidase activity to normalize differences in transfection efficiencies. Each transfection was performed three times in duplicate.
Site-directed mutagenesis
Using LB170 as a template, mutation of the BP1 binding site was performed using the Quik Change II XL Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA, USA). HPLC-purified complementary primers (Invitrogen) were designed to delete a seven-nucleotide region of the BP1 consensus binding site (underlined): 5'-'GGTGGGCCTGAAAGT TACTATATGGAAGTCCTCATCGTGTA-3'. Plasmids containing the deletion were designated delLB170. Subsequently, using delLB170 as the template, plasmids were generated to contain the mutant BP1 binding site (GCTCGAC), and were designated mutLB170.
Reverse transcription and quantitative PCR
Total RNA was extracted using Trizol Reagent (Invitrogen) according to the manufacturer's instructions. Reverse transcription of mRNA was performed using the iScript cDNA Synthesis Kit (Biorad, Hercules, CA, USA). TaqMan analyses of BP1 and 18S were performed using QPCR Master Mix Plus reagent (Eurogentec, San Diego, CA, USA). For SYBR Green analyses of bcl-2, the reactions were performed using iTaq SYBR Green Supermix with ROX (Biorad). The cycling conditions were as follows using the ABI Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA): 50°C for 2 minutes, then 95°C for 10 minutes, followed by 40 cycles at 95°C for 15 seconds and 60°C for 1 minute. SYBR Green analyses also included a dissociation protocol.
The ABI Prism software was used to perform an automatic cycle threshold analysis and to generate a standard curve for extrapolation of the sample data. Mean values of each gene were normalized to the corresponding mean value for 18S. The following sequences were used for primers and probes: 18S primers, 5'-GCCGCTAGAGGTGAAATTCTTG-3' and 5'-CATT CTTGGCAAATGCTTTCG-3'; 18S probe, 5'-ACCGGCGCAAGACGGACCAG-3'; BP1 primers, 5'-CCTCCCCCAATTTGTCCTACTC-3' and 5'-GGTTGCTGGCAGGACAGGTA-3'; BP1 probe, 5'-AGCCAGCGAACCCCGGAGACTC-3'; bcl-2 primers, 5'-TGGGATGCCTTTGTGGAACT-3' and 5'-GAGACAGCCAGGAGAAATCAAAC-3'.
Western blot analysis
Cell lysates were prepared in ice-cold RIPA lysis buffer (50 mM Tris, pH 7.5, 2 mM ethylenediamine teraacetic acid, 100 mM NaCl, 1% NP-40) containing 1× Complete Mini protease inhibitor cocktail (Roche). Proteins were separated by SDS-PAGE and were transferred to a polyvinylidene difluoride membrane. Blots were probed overnight at 4°C with rabbit anti-BP1 (Novus Biologicals, Littleton, CO, USA) at a 1:5,000 dilution, or with a 1:1,000 dilution of mouse anticaspase-7 and anticaspase-8 antibody, rabbit anticaspase-9 and anti-PARP (anti-Poly(ADP-Ribose) Polymerase) antibody (Cell Signaling, Danvers, MA, USA) or mouse anti-Bcl-2 antibody (Santa Cruz, Santa Cruz, CA, USA). After washing, blots were incubated with either horseradish peroxidase-linked goat anti-mouse (1:500 dilution) or donkey anti-rabbit secondary antibodies (1:15,000 dilution). Signals were detected using SuperSignal West Dura Extended Duration Substrate (Pierce, Rockford, IL, USA). Relative band intensities were quantitated using the Kodak Image Station 2000 MM and the Kodak ID software (version 3.6.4; Scientific Imaging System, Eastman Kodak Co., Rochester, NY, USA) and by standardizing protein levels against β-actin.
Statistical methods
Statistical tests comparing mean levels were performed with SAS software based on a priori analysis of variance contrasts. Each replicate was treated as an independent observation. Except where noted, contrasts involving MCF7/EV cells were based on averaging across EV1 and EV2. Luciferase values were log-transformed and the percentage of positive cells stained with Annexin V was arcsine-transformed for significance testing. Results are declared significant at α = 0.02, two-sided.
Results
BP1 inhibits TNFα-mediated cell death through a caspase-dependent mechanism
Three MCF7 cell lines were generated that stably express increased levels of BP1 protein (MCF7/BP1-1, MCF7/BP1-2, and MCF7/BP1-4), as well as two control cell lines containing the empty vector (MCF7/EV1 and MCF7/EV2) (Figure 1a). We first compared the viability of MCF7/EV and MCF7/BP1 cell lines that were grown in the presence or absence of TNFα. As shown in Figure 1b, an average of 43% of MCF7/EV cells survived 3 days post TNFα treatment, whereas all three BP1-overexpressing cell lines displayed an approximately twofold increase in viability (74%, 90%, and 80% for BP1-1, BP1-2, and BP1-4, respectively; P < 0.0001 for each comparison). Furthermore, MCF7/BP1 cells exposed to TNFα showed a twofold to threefold decrease in Annexin V binding compared with MCF7/EV cell lines (Figure 1c, P < 0.0001 for all three overexpressing cell lines), indicating that increased BP1 expression decreases the ability of MCF7 cells to undergo apoptosis.
Figure 1.
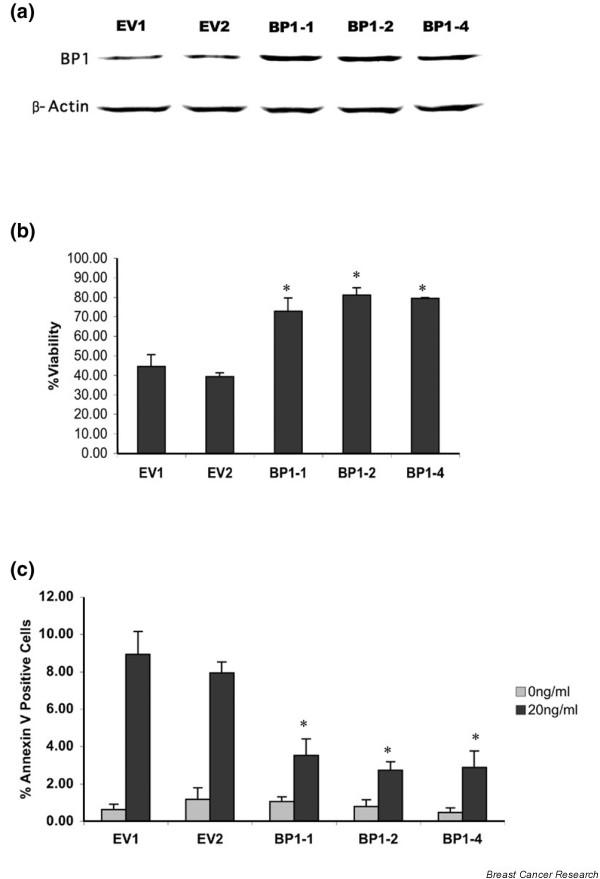
Effect of BP1 on TNFα-induced cell death. (a) Western blot analysis of Beta Protein 1 (BP1) protein expression in MCF7/EV cell lines and MCF7/BP1 cell lines. (b) MCF7/EV and MCF7/BP1 cell lines were treated with 20 ng/ml TNFα for 72 hours. MTT assays were performed to assess cell viability. To calculate the percentage viability, absorbance values were compared in treated cells versus untreated cells. *Statistically significant differences (P < 0.0001). (c) MCF7/EV and MCF7/BP1 cell lines were treated with 20 ng/ml TNFα for 18 hours. Cells were labeled with both an Annexin V–FITC conjugate and propidium iodide to distinguish early apoptotic cells. Five fields of cells were photographed and counted for each sample in three independent experiments. The percentage of cells in each field with Annexin V staining in the plasma membrane, but showing exclusion of propidium iodide, is reported. *P < 0.0001.
We then examined whether constitutive BP1 expression affected TNFα-mediated cell death through modulation of caspase pathways. Upstream initiator caspase-8 and caspase-9, as well as the downstream effector caspase-7 and its substrate PARP, were analyzed by Western blot analysis in MCF7/EV and MCF7/BP1 cell lines treated with TNFα for various times (Figure 2a). MCF7 cells are deficient in caspase-3 due to a genomic deletion in exon 3 [17], so this caspase was not examined. Processed fragments of each caspase are readily apparent after 12 and 24 hours of exposure to TNFα. Across each cell line, processing of PARP is also seen by 12 hours, with the amount of cleaved protein accumulating through 24 hours. In each MCF7/BP1 cell line, however, there is a clear reduction in cleavage of every caspase as well as of PARP. Analyses of band intensities of each fragment revealed a ≥50% decrease in caspase and PARP cleavage in cells overexpressing BP1, relative to the levels of cleaved products in MCF7/EV cells. Strikingly, untreated MCF7/BP1 cells showed a threefold to fourfold increase in levels of full-length PARP relative to MCF7/EV cells (Figure 2b). In addition, MCF7/BP1 cells show a 1.6-fold to 2.0-fold downregulation of procaspase-8 (Figure 2c), indicating that BP1 may affect the early stages of apoptosis. Together, our findings demonstrate a role for BP1 in caspase-dependent pathways of TNFα-mediated cell death.
Figure 2.

BP1 inhibits TNFα-mediated caspase activation. (a) MCF7/EV and MCF7/Beta Protein 1 (BP1) cells were treated with 20 ng/ml TNFα. At the indicated times, protein was analyzed by western blot to examine the expression levels and processing of caspase-8, caspase-9, and caspase-7, as well as the substrate Poly(ADP-Ribose) Polymerase (PARP). In each case, the top band represents the uncleaved, inactive procaspase or full-length active PARP. Arrow, intermediate fragments; arrowheads, position of the expected cleaved product. (b) and (c) Western blot analyses of PARP and procaspase-8 expression in MCF7/EV and MCF7/BP1 cell lines.
BP1 regulates the expression of Bcl-2
We next sought to define transcriptional targets of BP1 that might explain why its overexpression results in increased cell viability in the presence of TNFα. bcl-2, a well-established antiapoptotic oncogene, is often associated with resistance to various cell-death-inducing agents [18]. The bcl-2 gene contains two promoters: P1, located 1,386 to 1,423 bp upstream of the translational start site; and P2, located 1.3 kb downstream of P1 [19]. The sequence 5'-TACTATATG-3' matches a consensus binding site for BP1 protein [8] and is located upstream of the P1 promoter at -2539 bp relative to the ATG translational start site.
An electrophoretic mobility shift assay was used to demonstrate that BP1 protein can specifically bind to a dsDNA oligonucleotide probe containing this site (Figure 3a). A shifted band was observed in the presence of in vitro transcribed and translated BP1 protein (lane 2), while a faint band was observed at this location when wheatgerm extract alone was mixed with the bcl-2 probe (lane 1). Specificity of the interaction was evidenced by the loss of the shifted band upon addition of 500× or 1,000× molar excess of competitor DNA of the same sequence as the bcl-2 probe (lanes 3 and 4). Addition of excess negative control DNA that lacks a BP1 binding site did not reduce the intensity of the band (lanes 5 and 6). In the presence of anti-BP1 antibody we observed both a decrease in the shifted band as well as the appearance of a supershifted band (arrow, lanes 7 and 8), verifying that BP1 protein bound to the bcl-2 probe DNA. These data indicate that the bcl-2 gene is a potential target for regulation by BP1.
Figure 3.

Identification of bcl-2 as a putative target gene of BP1. (a) Electrophoretic mobility shift assays were performed to detect potential binding of in vitro transcribed and translated Beta Protein 1 (BP1) to a consensus binding sequence located in bcl-2. Binding of BP1 to a 32P end-labeled dsDNA probe containing the putative BP1 binding site and surrounding sequence is observed as a shifted band (arrow). 500× and 1,000× molar excess unlabeled probes or a negative control (NC) sequence lacking a BP1 binding site were added as a cold competitor for BP1 binding. Addition of 1 or 2 μl BP1 antibody (Ab) resulted in a supershift of the original band (arrowhead). Wheat germ extract alone (WG) served as a control. (b) Western blot analysis of Bcl-2 protein expression in MCF7/EV and MCF7/BP1 cell lines. (c) bcl-2 mRNA from each cell line was analyzed by real-time PCR. MCF7/EV and MCF7/BP1 cells were cultured in the presence or absence of TNFα for 72 hours. Data shown represent bcl-2 levels normalized to 18S. *P < 0.0001. (d) Western blot analysis after culture of cell lines in the presence or absence of TNFα for 72 hours.
In support of this finding, a comparison of bcl-2 expression levels in MCF7/EV and MCF7/BP1 cells by western blot analysis and by real-time PCR revealed a twofold increase in both bcl-2 protein (Figure 3b) and mRNA (Figure 3c, black bars; P < 0.0001 comparing the average of untreated EV1 and EV2 with BP1-1, BP1-2, and BP1-4).
Constitutive expression of bcl-2 abrogates cell death in MCF7 cells exposed to TNFα [20,21]. To examine whether regulation of bcl-2 by BP1 is associated with the observed increase in MCF7/BP1 cell viability, bcl-2 mRNA expression was analyzed in TNFα-treated cells. Although bcl-2 mRNA was downregulated by TNFα in MCF7/EV cells (Figure 3c, gray bars; P = 0.004 and P < 0.0001 for EV1 and EV2, respectively), BP1-overexpressing cells showed no significant change in bcl-2 mRNA after treatment. Consistent with these data, Bcl-2 protein levels are not reduced by TNFα treatment, in contrast to the empty vector control (Figure 3d).
BP1 directly targets the bcl-2 promoter
We next determined whether increased levels of bcl-2 expression in MCF7/BP1 cells could be attributed to direct regulation of the bcl-2 gene by BP1 protein. A schematic diagram of the promoter region of bcl-2 is shown in Figure 4a. MCF7/EV and MCF7/BP1 cell lines were transfected with LB170 (a gift from Dr Linda Boxer, Stanford University), a construct containing the bcl-2 P1 promoter region and the 5'-flanking sequence [22], including the BP1 binding site, linked to the luciferase reporter gene. MCF7/BP1-1 and MCF7/BP1-4 consistently showed a fivefold activation of the P1 promoter, whereas MCF7/BP1-2 showed up to an 11-fold increase, compared with levels seen in MCF7/EV control cells (Figure 4b, black bars; P < 0.0001 for all three overexpressing cell lines). These results show that BP1 overexpression increased transcriptional activation through the bcl-2 promoter. The results do not, however, distinguish between a direct effect, caused by binding of BP1 protein to the promoter, and an indirect effect by BP1, due to regulation of other factors that bind and activate transcription of bcl-2. Site-directed mutagenesis and deletion of the BP1 consensus binding site were carried out to differentiate these possibilities.
Figure 4.

bcl-2 is a direct transcriptional target of BP1. (a) Schematic diagram of bcl-2 P1 promoter constructs. LB170 contains the Beta Protein 1 (BP1) binding site (underlined). delLB170 contains a deletion of seven of the nine nucleotides of the binding site (indicated by X), and mutLB170 contains the mutated BP1 binding site (lowercase, underlined). LUC, luciferase. (b) MCF7/EV and MCF7/BP1 cells were transiently transfected with LB170, delLB170, or mutLB170, as well as a plasmid encoding β-galactosidase. Forty-eight hours post transfection, protein was extracted and assayed for luciferase activity. Relative light units were normalized to β-galactosidase expression units to signify levels of bcl-2 P1 promoter activity (RLU/Bgal). *P < 0.0001, • P < 0.05. (c) Electrophoretic mobility shift assay: in vitro transcribed and translated BP1 protein (BP1) was incubated with a 32P end-labeled DNA oligonucleotide probe containing the sequence from the bcl-2 promoter including the BP1 binding site (arrow). Cold competitor DNAs including a bcl-2 sequence identical to the probe (bcl-2), mutated bcl-2 (mbcl-2), and a negative control (NC) lacking the BP1 binding site, were added at 500× or 1,000× molar excess. Wheat germ extract (WG) incubated with the bcl-2 probe served as a control.
Using the LB170 construct as a template, a two-step site-directed mutagenesis procedure was performed. First, seven nucleotides of the nine-nucleotide sequence in the BP1 binding site were deleted to generate delLB170, followed by insertion of the mutated sequence, described in [23], to create mutLB170 (Figure 4a). An electrophoretic mobility shift assay was performed to determine whether this mutation could inhibit binding of BP1 to bcl-2 (Figure 4c). As before, BP1 protein (WG/BP1) bound to the bcl-2 probe, as indicated by the shifted band (lane 2, arrow). No protein binding to the bcl-2 probe was observed at this location using the wheatgerm extract (lane 1). Competition with 500× and 1,000× molar excess of unlabeled probe DNA (bcl-2) resulted in the loss of the shifted band signal (lanes 3 and 4). If excess competitor DNA containing a seven-nucleotide mutation of the BP1 binding site was added (mbcl-2), however, little competition for binding was observed (lanes 5 and 6). A negative control DNA also did not compete for binding (lanes 7 and 8). This mutation is thus sufficient to disrupt binding of BP1 protein to DNA.
MCF7/EV and MCF7/BP1 cell lines were then transiently transfected with the wild-type LB170, delLB170, or mutLB170. Notably, deletion of the BP1 binding site resulted in an average 45% to 51% decrease in bcl-2 promoter activation across all cell lines (Figure 4b, grey bars; P < 0.05). Mutation of this site caused an average 37% to 49% reduction in activation of the bcl-2 promoter, which was statistically significant for BP1-1 (white bars, P = 0.02) but not for BP1-2 or BP1-4, perhaps due to residual BP1 binding to the mutant site (Figure 4c). We thus conclude that BP1 protein can bind to the bcl-2 promoter and directly contribute to activation of its expression in MCF7 cells.
Discussion
Inhibition of apoptosis is a key step in tumor development and growth, promoting the selection and propagation of cells that can resist destruction by various cellular stresses. Evasion of apoptosis by tumor cells has been attributed to downregulation or inactivation of tumor suppressor genes, and to increased activation or expression of oncogenic factors [24]. The studies presented here reveal that high-level BP1 expression is associated with enhanced survival of breast cancer cells challenged with TNFα. Potential mechanisms by which BP1 promotes continued cell viability were identified, involving genes in both extrinsic and intrinsic apoptotic pathways. Specifically, we demonstrated that BP1 can activate bcl-2 and PARP, and can repress procaspase-8. BP1 transcriptionally activates bcl-2 through direct binding upstream of the P1 promoter region, resulting in a twofold increase in Bcl-2 protein.
Upon either deletion or mutation of the BP1 binding site, we observed an approximately 40% to 50% decrease in bcl-2 promoter activity. One possible reason for the remaining activity is that the mutation did not completely prevent BP1 binding. Another possibility is that there may be other factors present that promote bcl-2 expression independent of BP1 binding. The plasmid LB170, used in our studies of the bcl-2 promoter, contains several binding sites for known transcriptional regulators of bcl-2, including Wilms' Tumor 1, SP1, and cAMP response element binding proteins. Wilms' Tumor 1 protein has been associated with aggressive phenotypes of breast cancer and was recently shown to upregulate bcl-2 expression in BT-474 breast cancer cells [25]. Additionally, SP1 sites and a cAMP response element are necessary for estradiol-induced bcl-2 gene expression in MCF7 and T47D cells [26].
Furthermore, high BP1 expression prevents TNFα-induced downregulation of bcl-2 mRNA and protein. This is consistent with data from other laboratories demonstrating that high expression of Bcl-2 promotes cell survival in the presence of TNFα [20,21]. These results not only support our observation that bcl-2 is a transcriptional target of BP1, but identify the upregulation of bcl-2 as a probable mechanism by which BP1 inhibits cell death.
As mentioned, our previous findings demonstrate BP1 expression in 100% of estrogen-receptor-alpha-negative breast cancers studied, compared with 73% of estrogen-receptor-alpha-positive tumors [11]. This raises the intriguing possibility that BP1 protein and estrogen receptor alpha protein may interact and modulate bcl-2 gene expression and action. There is consequently a possibility that a more robust interaction occurs between BP1 protein and bcl-2 in the absence of estrogen receptor alpha; hence, this would provide an interesting area for future study.
Our data further point to a role for BP1 in modulation of caspase-dependent pathways in apoptosis. Increased expression of BP1 reduced TNFα-induced processing of caspase-7, caspase-8, caspase-9, and the caspase substrate PARP by approximately 50%, consistent with the ability of BP1 to enhance cell viability by twofold. These findings suggest a model by which BP1 may modulate TNFα-induced cell death at several points (Figure 5).
Figure 5.
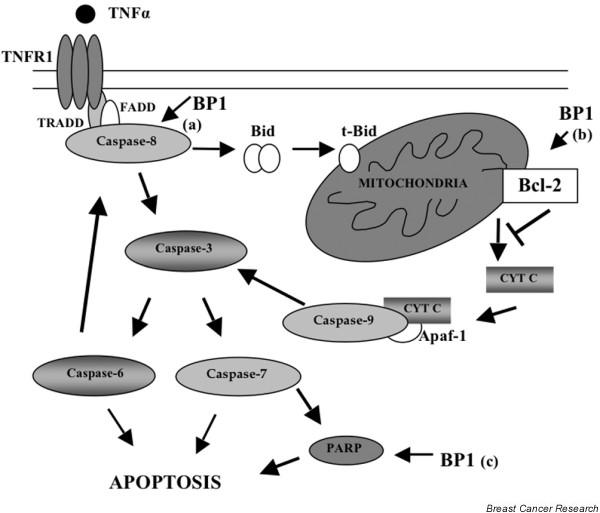
Model for BP1-mediated disruption of caspase activation after TNFα treatment. Arrows indicate the three points (a, b and c) at which Beta Protein 1 (BP1) may impact apoptosis. Bid, BH3 interacting domain death agonist; CYT C, cytochrome c; FADD, Fas (TNFRSF6)-associated via death domain; PARP, Poly(ADP-Ribose) Polymerase; t-Bid, truncated Bid; TNFR1, TNF receptor 1; TRADD, TNFRSF1A-associated via death domain.
First, full-length procaspase-8 expression is decreased in MCF7/BP1 cell lines; lower levels of procaspase-8 may result in less available activated caspase-8, which would lead to decreased activation of downstream caspases and PARP, as we observed. Scanning of the caspase-8 DNA sequence has revealed possible binding sites for BP1 protein, indicating that caspase-8 is a potential transcriptional target of BP1.
Second, Bcl-2 controls the release of cytochrome c from the mitochondria. Following cytochrome c release, crosstalk between the death-receptor and mitochondrial pathways of apoptosis can lead to additional processing of caspase-8 mediated by effector caspases-3 and -6 [27,28]. Owing to its regulation of bcl-2, BP1 may reduce activation of those caspases downstream of the mitochondria [29].
A third point at which BP1 may affect apoptosis is through regulation of PARP. We discovered increased levels of full-length PARP in MCF7/BP1 cells. PARP has been shown to be overexpressed in 57% of breast tumors [30]. PARP has multiple roles in cell death, and in regulation of gene expression, proliferation, and differentiation, and is well known for its ability to mediate DNA repair in response to DNA damage [31]. Of relevance here, PARP inhibitors, when used in conjunction with chemotherapeutic drugs or radiotherapy, are known to increase the cytotoxic effects of these agents in tumor cells [32]. We have located potential binding sites for BP1 protein in the PARP genomic sequence, suggesting that PARP is also a possible target gene for regulation by BP1.
Conclusion
Our findings reveal details of a role for BP1 in caspase-dependent and bcl-2-linked mechanisms of tumor cell survival, and suggest BP1 could serve as a marker for drug resistance and a therapeutic target. This is the first study to define a function for increased BP1 expression in breast cancer and to highlight pathways important for further exploration.
Abbreviations
bp = base pairs; BP1 = Beta Protein 1; FITC = Fluorescein Isothiocyanate; HPLC = high-performance liquid chromatography; MTT = thiazolyl blue tetrazolium bromide; PARP = Poly(ADP-Ribose) Polymerase; PCR = polymerase chain reaction; TNF = tumor necrosis factor.
Competing interests
George Washington University holds the patent for BP1 antibody, and Novus licenses the technology from George Washington University. The Novus BP1 antibody was used in this research. PEB has previously acted as a consultant for Novus, but is no longer doing so.
Authors' contributions
HSS carried out the experimental procedures, participated in the design of the study, and drafted the manuscript as part of her PhD requirements at the George Washington University Medical Center. SWF generated the stable cell lines used in the study. JJP assisted in the design of the study and in performing the viability assays. JR carried out western blot analyses of BP1 and SJS performed the statistical analyses. PEB conceived of the study, directed its design and coordination, and helped to draft the manuscript. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
This research was supported by the Susan G. Komen Foundation (PEB) and National Institutes of Health grants CA91149 (PEB), CA102928 (SWF) and CA101875 (JJP).
Contributor Information
Holly S Stevenson, Email: stevensonhol@mail.nih.gov.
Sidney W Fu, Email: sfu@gwu.edu.
Joseph J Pinzone, Email: Joseph.Pinzone@osumc.edu.
Jinguen Rheey, Email: jgrheey@gwu.edu.
Samuel J Simmens, Email: simmens@gwu.edu.
Patricia E Berg, Email: bcmpeb@gwumc.edu.
References
- Cillo C, Cantile M, Faiella A, Boncinelli E. Homeobox genes in normal and malignant cells. J Cell Physiol. 2001;188:161–169. doi: 10.1002/jcp.1115. [DOI] [PubMed] [Google Scholar]
- Grier DG, Thompson A, Kwasniewska A, McGonigle GJ, Halliday HL, Lappin TR. The pathophysiology of HOX genes and their role in cancer. J Pathol. 2005;205:154–171. doi: 10.1002/path.1710. [DOI] [PubMed] [Google Scholar]
- Care A, Felicetti F, Meccia E, Bottero L, Parenza M, Stoppacciaro A, Peschle C, Colombo MP. HOXB7: a key factor for tumor-associated angiogenic switch. Cancer Res. 2001;61:6532–6539. [PubMed] [Google Scholar]
- Chu MC, Selam FB, Taylor HS. HOXA10 regulates p53 expression and matrigel invasion in human breast cancer cells. Cancer Biol Ther. 2004;3:568–572. doi: 10.4161/cbt.3.6.848. [DOI] [PubMed] [Google Scholar]
- Coletta RD, Christensen K, Reichenberger KJ, Lamb J, Micomonaco D, Huang L, Wolf DM, Muller-Tidow C, Golub TR, Kawakami K, et al. The Six1 homeoprotein stimulates tumorigenesis by reactivation of cyclin A1. Proc Natl Acad Sci USA. 2004;101:6478–6483. doi: 10.1073/pnas.0401139101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman V, Martensen SA, Reisman D, Evron E, Odenwald WF, Jaffee E, Marks J, Sukumar S. Compromised HOXA5 function can limit p53 expression in human breast tumors. Nature. 2000;405:974–978. doi: 10.1038/35016125. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhu T, Chen Y, Mertani HC, Lee KO, Lobie PE. Human growth hormone-regulated HOXA1 is a human mammary epithelial oncogene. J Biol Chem. 2003;278:7580–7590. doi: 10.1074/jbc.M212050200. [DOI] [PubMed] [Google Scholar]
- Chase MB, Fu S, Haga SB, Davenport G, Stevenson H, Do K, Morgan D, Mah AL, Berg PE. BP1, a homeodomain-containing isoform of DLX4, represses the beta-globin gene. Mol Cell Biol. 2002;22:2505–2514. doi: 10.1128/MCB.22.8.2505-2514.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Stevenson H, Strovel JW, Haga SB, Stamberg J, Do K, Berg PE. Distinct functions of two isoforms of a homeobox gene, BP1, and DLX7, in the regulation of the beta-globin gene. Gene. 2001;278:131–139. doi: 10.1016/S0378-1119(01)00716-8. [DOI] [PubMed] [Google Scholar]
- Fu SW, Schwartz A, Stevenson H, Pinzone JJ, Davenport GJ, Orenstein JM, Gutierrez P, Simmens SJ, Abraham J, Poola I, et al. Correlation of expression of BP1, a homeobox gene, with estrogen receptor status in breast cancer. Breast Cancer Res. 2003;5:R82–R87. doi: 10.1186/bcr602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man YG, Fu SW, Schwartz A, Pinzone JJ, Simmens SJ, Berg PE. Expression of BP1, a novel homeobox gene, correlates with breast cancer progression and invasion. Breast Cancer Res Treat. 2005;90:241–247. doi: 10.1007/s10549-004-4492-9. [DOI] [PubMed] [Google Scholar]
- Haga SB, Fu S, Karp JE, Ross DD, Williams DM, Hankins WD, Behm F, Ruscetti FW, Chang M, Smith BD, et al. BP1, a new homeobox gene, is frequently expressed in acute leukemias. Leukemia. 2000;14:1867–1875. doi: 10.1038/sj.leu.2401912. [DOI] [PubMed] [Google Scholar]
- Zimmermann KC, Green DR. How cells die: apoptosis pathways. J Allergy Clin Immunol. 2001;108:S99–S103. doi: 10.1067/mai.2001.117819. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH, Hengartner MO. Programmed cell death: alive and well in the new millennium. Trends Cell Biol. 2001;11:526–534. doi: 10.1016/S0962-8924(01)02173-0. [DOI] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase-8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/S0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid a Bcl-2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/S0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- Janicke RU, Sprengart ML, Wati MR, Porter AG. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem. 1998;273:9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH, Vaux DL. Alterations in the apoptotic machinery and their potential role in anticancer drug resistance. Oncogene. 2003;22:7414–7430. doi: 10.1038/sj.onc.1206945. [DOI] [PubMed] [Google Scholar]
- Seto M, Jaeger U, Hockett RD, Graninger W, Bennett S, Goldman P, Korsemeyer SJ. Alternative promoters and exons, somatic mutation and deregulation of the Bcl-2-Ig fusion gene in lymphoma. EMBO J. 1988;7:123–131. doi: 10.1002/j.1460-2075.1988.tb02791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burow ME, Weldon CB, Tang Y, Navar GL, Krajewski S, Reed JC, Hammond TG, Clejan S, Beckman BS. Differences in susceptibility to tumor necrosis factor alpha-induced apoptosis among MCF-7 breast cancer cell variants. Cancer Res. 1998;58:4940–4946. [PubMed] [Google Scholar]
- Burow ME, Weldon CB, Tang Y, McLachlan JA, Beckman BS. Oestrogen-mediated suppression of tumour necrosis factor alpha-induced apoptosis in MCF-7 cells: subversion of Bcl-2 by anti-oestrogens. J Steroid Biochem Mol Biol. 2001;78:409–418. doi: 10.1016/S0960-0760(01)00117-0. [DOI] [PubMed] [Google Scholar]
- Wilson BE, Mochon E, Boxer LM. Induction of bcl-2 expression by phosphorylated CREB proteins during B-cell activation and rescue from apoptosis. Mol Cell Biol. 1996;16:5546–5556. doi: 10.1128/mcb.16.10.5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebb D, Tang DC, Dre L, Chin K, Berg PE, Rodgers GP. Identification of upstream regulatory elements that repress expression of adult beta-like globin genes in a primitive erythroid environment. Blood Cells Mol Dis. 1998;24:356–369. doi: 10.1006/bcmd.1998.0202. [DOI] [PubMed] [Google Scholar]
- Johnstone RW, Ruefi AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/S0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Tuna M, Chavez-Reyes A, Tari AM. HER2/neu increases the expression of Wilms' Tumor 1 (WT1) protein to stimulate S-phase proliferation and inhibit apoptosis in breast cancer cells. Oncogene. 2005;24:1648–1652. doi: 10.1038/sj.onc.1208345. [DOI] [PubMed] [Google Scholar]
- Dong L, Wang W, Wang F, Stoner M, Reed JC, Harigai M, Samudio I, Kladde MP, Vyhlidal C, Safe S. Mechanisms of transcriptional activation of bcl-2 gene expression by 17β-estradiol in breast cancer cells. J Biol Chem. 1999;274:32099–32107. doi: 10.1074/jbc.274.45.32099. [DOI] [PubMed] [Google Scholar]
- Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, et al. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol. 1999;144:281–292. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Lahti JM, Kidd VJ. Caspase-8 activation and bid cleavage contribute to MCF7 cellular execution in a caspase-3-dependent manner during staurosporine-mediated apoptosis. J Biol Chem. 2000;275:9303–9307. doi: 10.1074/jbc.275.13.9303. [DOI] [PubMed] [Google Scholar]
- Ruiz de Almodovar C, Ruiz-Ruiz C, Munoz-Pinedo C, Robledo G, Lopez-Rivas A. The differential sensitivity of Bcl-2-overexpressing human breast tumor cells to TRAIL or doxorubicin-induced apoptosis is dependent on Bcl-2 protein levels. Oncogene. 2001;20:7128–7133. doi: 10.1038/sj.onc.1204887. [DOI] [PubMed] [Google Scholar]
- Bieche I, de Murcia G, Lidereau R. Poly(ADP-ribose) polymerase gene expression status and genomic instability in human breast cancer. Clin Cancer Res. 1996;2:1163–1167. [PubMed] [Google Scholar]
- Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- Plummer ER. Inhibition of poly(ADP-ribsose) polymerase in cancer. Curr Opin Pharmacol. 2006;6:364–368. doi: 10.1016/j.coph.2006.02.004. [DOI] [PubMed] [Google Scholar]