

Abstract
In this study we evaluate, at full atomic detail, the folding processes of two small helical proteins, the B domain of protein A and the Villin headpiece. Folding kinetics are studied by performing a large number of ab initio Monte Carlo folding simulations using a single transferable all-atom potential. Using these trajectories, we examine the relaxation behavior, secondary structure formation, and transition-state ensembles (TSEs) of the two proteins and compare our results with experimental data and previous computational studies. To obtain a detailed structural information on the folding dynamics viewed as an ensemble process, we perform a clustering analysis procedure based on graph theory. Moreover, rigorous pfold analysis is used to obtain representative samples of the TSEs and a good quantitative agreement between experimental and simulated Φ values is obtained for protein A. Φ values for Villin also are obtained and left as predictions to be tested by future experiments. Our analysis shows that the two-helix hairpin is a common partially stable structural motif that gets formed before entering the TSE in the studied proteins. These results together with our earlier study of Engrailed Homeodomain and recent experimental studies provide a comprehensive, atomic-level picture of folding mechanics of three-helix bundle proteins.
Keywords: transition state ensemble, Villin, protein A
An eventual solution to the protein-folding problem will involve a close calibration of theoretical methods to experimental data (1–4). In the endeavor of obtaining a quantitative agreement between theory and experiments, two small α-helical proteins have played a central role, namely the B domain of protein A from Staphylococcus aurues and the Villin headpiece subdomain from chicken. Although these proteins belong to different SCOP fold classes (5), both have simple three-helix bundle native topologies and fold autonomously on the microsecond time scale (6, 7), which makes them ideal test cases for protein simulations and numerous simulation studies, ranging from simple Cα Go-type to all-atom models with explicit water, have been undertaken for both protein A (8–21) and Villin (16, 17, 22–29).
Important advances have been made toward agreements with experiments for both proteins, but several key issues remain unresolved (6, 30, 31). The need for additional studies also is emphasized by recent experiments. Fersht et al. (31, 32) performed a comprehensive mutational analysis on protein A by obtaining Φ values at <30 aa positions, providing an important benchmark for simulation studies. The obtained Φ values suggest that the transition-state ensemble (TSE) is characterized mainly by a well formed H2 (we denote the three individual helices from N- to C-terminal by H1, H2, and H3, following previous convention) stabilized by hydrophobic interactions with H1. Recent experimental studies of Villin (6, 33) have focused mainly on achieving fast-folding mutants, although new biophysical characterization of wild-type Villin also was obtained. Interestingly, the results indicate that these mutants are approaching the “speed-limit” for folding. Nonetheless, a limited free-energy barrier for folding remains so that the TSE (and by consequence Φ analysis) still is a meaningful concept for Villin.
To obtain a complete picture of the folding kinetics for a protein, the observation of a large number of folding trajectories is crucial. This might be particularly important for protein A, given that the inconsistencies between various computational studies for this protein may lie in the existence of multiple transition states and pathways (34). We recently developed a minimalist transferable all-atom model (35), which was successfully used to fold a diverse set of proteins, including α, β, α + β, and α/β types, to their near-native conformations. Here we apply the same model to protein A and a single-point mutant of Villin but go beyond structure prediction by carefully exploring their folding behavior as an ensemble process. Moreover, we determine the TSEs for two proteins. To achieve this, we make use of pfold analysis, which requires additional simulations but is the most reliable method for identifying the TSE (36, 37). Combining the results obtained here with previous results for the Engrailed Homeodomain (ENH) (38) allows us to formulate a universal framework for the folding of small three-helix bundle proteins within which we find a substantial diversity in the details of the folding mechanism.
Results
We perform 2,000 Monte Carlo (MC) dynamics folding trajectories for protein A and Villin at a single temperature (T ≈ 300 K), starting from random initial conformations [see supporting information (SI) Text]. The large number of folding trajectories and the long total simulation time (≈70 ms) can be achieved because of the relative simplicity of our transferable all-atom protein model (Eq. 1 in Methods). Our objective, based on these simulation results, is to identify and compare robust features of the folding mechanism for the two proteins.
Initial Selection of Trajectories.
Not all of the 2,000 trajectories contain native-like low-energy structures. Therefore, before turning to the folding kinetics, we make an initial objective selection of a set of “representative” trajectories that fold into native-like conformations. This selection of trajectories is based on a simple clustering procedure of the lowest-energy structures obtained for each trajectory (this procedure is different from the structural kinetic cluster analysis performed on the full trajectories below). Hence, we first collect for each trajectory the lowest-energy structure observed in that trajectory. This set of 2,000 conformations (each one representing a trajectory) is then clustered by using their pairwise root-mean-square deviation (rmsd) into a simple single-link graph. In this graph, each node represents a conformation, and edges are drawn between any two conformations (nodes) whose rmsd is below a threshold value dc. Finally, we select the cluster with the highest average connectivity, <k>, where k is the number of edges of a node and < > is the cluster average. With our choice of dc (1.1 and 1.5 Å for protein A and Villin, respectively), the two clusters selected for protein A and Villin contain roughly the same number of structures (147 and 149 for protein A and Villin, respectively). All structures within these clusters are highly similar to each other, and, more importantly, they are structurally highly similar to the respective experimental structures (<rmsd> = 2.7 and 2.8 Å for protein A and Villin, respectively).
Having made this selection of lowest-energy structures based on their structural connectivity, we will in what follows focus on the corresponding 147 and 149 trajectories, respectively, that are now guaranteed to proceed to low-energy, highly native-like states. A selection criterion based on structural connectivity among minimum-energy structures is objective because it does not require the knowledge of native conformation. It also is robust with respect to the choice of the cutoff value dc, although non-native-like clusters can sometimes have comparable connectivities <k> (see SI Fig. 5). In particular, we find that clusters representing the “mirror image” topologies of the helix bundles are highly connected. A similar-in-spirit connectivity criterion has been used previously to identify high-quality candidates in protein structure prediction contexts (39). We find that the center structures within each of the two selected clusters for protein A and Villin, i.e., the top-k structures, are indeed at least as native-like as the cluster average (see Fig. 1).
Fig. 1.

Comparison between the native structures (in blue) and superimposed minimum-energy top-k structures (in red) obtained through a clustering procedure for protein A (Left) and Villin (Right), as described in the text. The rmsd values between top-k and experimental structures are 2.7 and 2.1 Å for protein A and Villin, respectively. Structures were created by using PyMOL (50).
Chain Collapse Versus Secondary Structure Formation.
We begin by examining the relaxation behavior of the two proteins. Fig. 2 compares the chain collapse and helix formation as obtained from the selected trajectories. A common property for the two proteins is a relatively rapid initial collapse of the chain, although it is slightly faster for Villin. Because of this fast “burst” phase, we find that the Rg relaxation is well described by a double-exponential function for both protein A and Villin (see Fig. 2 Upper). Similar fits are obtained for the total energy E and the rmsd with similar fit parameters (see SI Fig. 6). We find that the two time constants are separated roughly by an order of magnitude, and we associate the slowest relaxation phase (time constant τslow) with the overall folding process. Averaging over the three observables (Rg, E, and, rmsd), we find <τslow> = 43.0 × 106 MC steps and <τslow> = 42.8 × 106 MC steps for protein A and Villin, respectively, so that protein A and Villin fold approximately at the same rate in our model. This result is in rough agreement with the corresponding experimental time constants, which are 8.6 μs at 310 K (extrapolated) for protein A (7) and 5 μs at 300 K for Villin (6).
Fig. 2.

Relaxation behavior of the average radius of gyration, Rg (Upper) and average fraction helicity (Lower) of each individual helix, obtained at T ≈ 300 K. Helicity is determined with the criterion of Kabsch and Sander (51). The Rg relaxation data are fitted (red curves, Upper) to a double-exponential function, f(t) = a1 exp(−t/τfast) + a2 exp(−t/τslow) + b, using a Levenberg–Marquardt fit procedure with a1, a2, τfast, τslow, and b as free parameters.
Although the collapse behavior is similar for the two proteins, we observe differences in the way secondary structure is formed, as can be seen from Fig. 2 Lower. For protein A, the initial collapse phase coincides with the formation of H3. H1 and H2 also are formed during the collapse, albeit only partially. Hence, there is a substantial overall coil-to-helix transition in the initial phase of folding. In contrast to protein A, Villin exhibits relatively fast formation of both H2 and H3 during the initial chain collapse, whereas H1 forms at a slower rate. Although helix formation can be fast in our model, as exemplified by H3 in protein A, we find that it is not unrealistically fast however. Laser-induced T-jump experiments have been obtained for protein A by using IR spectroscopy (7) and for the Villin headpiece by using tryptophan fluorescence (6). In both studies, a fast phase (≈τslow/100) was detected and interpreted to be related to fast helix melting and formation. It is important to note that these studies are T-jump unfolding experiments and, as such, cannot be directly related to our folding kinetics results. However, they show that it is possible for helix formation to occur on very fast time scales relative to the overall folding transition even for extremely fast-folding proteins like protein A and Villin.
Structural Kinetic Cluster Analysis.
Although the time-dependence of secondary structure formation and chain collapse in Fig. 2 give useful information, this type of analysis does not provide details about structural states during the folding process. We therefore turn to a structural cluster procedure developed by our group (38). The basic idea is centered around the concept of a “structural graph” (for an extensive discussion see ref. 38 and SI Fig. 7), which aims to provide structural and kinetic information about coarse-grained features of the folding process. The structural graph is created in two steps. In the first step, all snapshots from all trajectories (147 and 149 for protein A and Villin, respectively) are treated on an equal footing and clustered together into a single-link graph; this aspect sets it apart from some other cluster procedures used to analyze folding trajectories (40, 41). Each conformation is represented by a node, and two nodes are linked by an edge if their structural similarity d is less than a cutoff, dc. We determine dc based on the total number of conformations in the Giant Component (GC), i.e., the largest cluster, which represents the native basin of attraction N (for suitable d measures). In the second step, information about the trajectories is reintroduced to kinetically characterize the clusters. A key quantity is the flux, F, defined as the fraction of all trajectories passing through the cluster. Hence, clusters through which all trajectories pass have F = 1. This is the situation for the GC, for example, as every trajectory eventually reaches N. Other clusters with F = 1 can be interpreted as obligatory intermediate states (38). In addition to F, we calculate for each cluster the mean first-passage time (MFPT) and the mean least-exit time (MLET). The F, MFPT, and MLET quantities along with the structural characteristics of the obtained clusters provide a powerful yet simple way of understanding details about the folding process from an ensemble perspective.
The clustering of the snapshots in principle can be performed by using any structural similarly measure d. Here we follow Hubner et al. (38) and construct structural graphs using the three order parameters rmsd, distance rmsd (drms), and ΔRg. Because of the different characteristics of the parameters, each one provides a different perspective on the folding process. Note that the cluster properties we focus on here are “coarse-grained” in nature, which is necessary in a MC study where the dynamics at very short time scales may depend on chain update properties. At longer time scales, however, the detailed balance criterion guarantees that averaged properties will become increasingly accurate.
Fig. 3 shows the results of our structural graph analysis for the two proteins. Clusters are represented as horizontal lines color-coded according to their flux F and drawn from t = MFPT to MLET. Starting with protein A, we find the rmsd and drms structural graphs to be dominated by a single high-flux cluster (i.e., the GC), which we associate with the native state. The absence of clusters at early times t during the folding process (t < τslow) means that, during pretransition state folding times, no accumulation of structurally similar conformations occurs—no structurally defined intermediate is observed. The ΔRg-structural graph provides a somewhat different perspective by reporting only on chain size and, by contrast, exhibits several early high-flux clusters, which in fact have F ≈ 1. This finding means that during early folding times, although the rmsd and drms graphs exclude the possibility of significant populations of structurally coherent states, all trajectories fluctuate widely in size. In fact, transitions between the early clusters in the ΔRg structural graph are numerous, ≈5–10 per trajectory (see Fig. 3). In the ΔRg structural graph, we see that the GC is a low-Rg cluster with MFPT ≈ 4 × 106 MC steps ≪ τslow. As opposed to the rmsd and drms structural graphs, where the GC represents the native state, the GC in the Rg graph therefore must contain not only conformations that are part of the native basin of attraction but also pre-TSE compact conformations. A key question then is whether the native state is reached through a path within this low-Rg GC cluster or through another path involving more extended conformations. To answer this question, it is useful to consider the location of the TSE, which is shown as a shaded area in Fig. 3. The determination of the TSE is discussed in detail below. From the somewhat extended nature of the TSE, we see that it is highly unlikely that N is reached by remaining in the low-Rg GC cluster, i.e., through a series of compact states. Instead, the TSE is located during fluctuations to more extended conformations after which the chain collapses into the native state. In this sense, the early low-Rg states are “off-pathway” [a similar behavior was found for another three-helix bundle protein, ENH (38)]. For the Villin headpiece we find, as for protein A, that the largest fluctuations in chain size occur during early times in the folding process. However, after the initial collapse phase, the Villin chain remains fairly compact throughout the rest of the folding process, which is clear from the ΔRg structural graph in Fig. 3. This also is consistent with the TSE obtained for Villin, which is relatively compact, as shown below.
Fig. 3.

Results of the structural kinetic cluster analysis for protein A and Villin. Each cluster is represented by a line, from t = MFPT to t = MLET, and color-coded by its flux, F, as indicated by the color scale. Only clusters with F > 0.1 are shown. The vertical location of each cluster is determined by the average radius of gyration, <Rg>, where < > is the cluster average. The location of the two TSEs for protein A and Villin are indicated by shaded areas centered (+) around the average Rg and time t, with averages taken over the two ensembles; the sizes of the two shaded areas reflect 1σ deviations in both the Rg and t directions. The rmsd structural graphs are obtained by using the cutoffs dc = 1.1 Å and 1.5 Å for protein A and Villin, respectively, whereas for drms we use dc = 0.9 Å and 1.2 Å, respectively. With these choices of dc, the rmsd and drms GCs contain ≈35% of all conformations, a reasonable number given that the trajectory time is ≈2τslow. For ΔRg, all reasonable choices of dc give larger GCs than for rmsd and drms, indicating that it contains not only native-like structures (see text). The results for ΔRg are not very sensitive to the specific choice of dc. Results are shown obtained for dc = 0.0080 Å and 0.0040 Å, respectively.
Finally, we note that both protein A and Villin exhibit a semihigh flux cluster (F ≈ 0.6–0.7) in the later stages of the folding process (see Fig. 3), and their structural properties and role in the folding process appear to be remarkably similar. Both clusters are overlapping in time with the native basin of attraction. A closer analysis of the drms structural graph for Villin reveals that, for 61% of the conformations in this late semihigh F cluster, the corresponding trajectories have passed previously into the GC. For protein A, the corresponding fraction is 71%. From this perspective, these two clusters can be characterized as nonobligatory “post-TSE” intermediate states [similar to “hidden intermediates” found recently (42)]. On the other hand, we also find that there is a small but significant overlap between the TSEs and these late intermediates: 9 of 46 and 16 of 57 TSE structures, respectively, are present in the protein A and Villin intermediates. Structurally, too, it is evident that the two intermediates resemble a small part of the “conformational space” of the TSE. Both intermediates are characterized by a well formed H3 detached from a native-like H1–H2 segment, which fits into the overall pattern of the TSEs (see below). It should be pointed out, however, that the two intermediate states are much more structurally coherent than the TSEs. Clearly then, this means that the stability of conformations within our transition states is not uniform, making the distinction between transition state and intermediate a nontrivial issue. Transition states with some degree of polarization have been noted previously in the Src homology 3 (SH3) domain (43). Delineating this intriguing issue is important in particular in the context of simple protein models, but it is beyond the scope of the present work. However, a kinetic observable such as pfold (see below) is clearly necessary to examine the issue. For now, we simply note that the two intermediates detected here are nonobligatory states that partially overlap with the respective TSEs.
Transition State Ensembles.
The transition state is key to understanding the folding process as it defines the rate-limiting step for folding. We construct the TSEs directly through the pfold analysis, which is a natural and highly reliable way of determining the TSE. This analysis is based on the notion that each conformation in the TSE has a unique property, namely, that trajectories starting from such a conformation have an equal chance of first reaching the native state and the unfolded state, given random initial conditions. We make use of this definition in finding the “true” TSE for our two proteins by first identifying a set of putative transition-state structures and then confirming or rejecting them based on the probability of folding, pfold, obtained by additional simulations. Because the rmsd GC cluster corresponds to the native state, we hypothesize that most viable putative transition-state structures can be found by selecting structures that immediately precede entry into the GC in the structural graph, which gives us a set of 783 and 798 putative transition-state structures for protein A and Villin, respectively. For each conformation in these putative sets, 100 independent trajectories are initiated randomly, and conformations with 0.4 < pfold < 0.6 are taken to be part of the TSE (see SI Text). This procedure generates a set of 46 and 57 “true” transition-state structures for protein A and Villin, respectively. The structures are illustrated in SI Fig. 8, and the coordinates are published as SI Data Sets 1 and 2.
Having obtained a representative sample of the transition state by using the stringent pfold criterion, we use this set of structures to calculate theoretical Φ values for our two proteins. We follow previous convention and interpret Φi for a residue i as the number of contacts present in the TSE for residue i divided by the number of native contacts (in i) (Eq. S3). We included all Φsim values with standard deviation, calculated over all TSE conformations, σ < 0.5 and the result of our Φ value calculations is given in Fig. 4. Experimental Φ values have been obtained previously for protein A (31, 32), and the agreement between theory and experiment is excellent, with an average absolute deviation | Φexp − Φsim | of 0.16 taken over all data points. We find that the most structured regions in the TSE are H1 and H2, as indicated by high Φ values, which form a native-like helical hairpin, whereas H3 is only weakly interacting with H1 and H2. This picture is in good agreement with two other detailed all-atom studies (12, 21) but not with results from a simpler Go-type study (9) that indicated initial H2–H3 formation instead. Of the obtained Φsim values, only one differs significantly from the experimental value, namely, ΦL23sim > ΦL23exp, located in the H1–H2 turn region. Our model thus predicts a highly ordered H1–H2 turn region in the TSE that may be related to the use of implicit water in our model. With implicit water, unsatisfied intramolecular hydrogen bonds cannot be compensated for by hydrogen bonding to water molecules, which is likely to happen in a poorly ordered H1–H2 helix loop. We also underestimate somewhat Φ values in H3, which may indicate that H3 participates less in the TSE than suggested by experiments. For all other positions, a good quantitative agreement between Φsim and Φexp exists, which has been challenging to obtain in previous simulation studies (31).
Fig. 4.

Comparison between simulated and experimental (32) Φ values. At positions where more than one experimental Φ value is reported, the average value is used, and their individual experimental Φ values on different types of mutations are shown in SI Fig. 9. Error bars denote the standard deviation, σ.
For Villin, only one Φ value has been published so far, ΦK65 ≈ 1.3 in H3, which was obtained by a lysine to norleucine mutation designed to speed up folding (33). It is unclear to what extent this somewhat unconventional mutation can be interpreted in standard Φ value language but it appears to suggest that the N-terminal side of H3 is highly involved in the TSE. Although we were unable to obtain a Φ value at position K65, our results indicate by contrast that the most organized region of the TSE is centered around the H1–H2 segment. Overall, the situation is therefore similar to that of protein A. However, we note that the TSE is overall less organized for Villin (SI Fig. 8). Also, we find quite large variations in the Φ values within both H1 and H2. Low Φ values are observed in the N-terminal ends of both H1 and H2.
Discussion
Using a relatively simple transferable sequence-based all-atom model, we performed a large number of ab initio protein folding runs for protein A and Villin headpiece that provided us with necessary data to study the folding kinetics as an ensemble process. By combining our results, we obtain a coarse-grained picture of the folding processes of two three-helix bundle proteins. Qualitatively the folding scenarios are similar for both proteins and for another three-helix bundle protein, ENH (38). For protein A, the initial collapse phase coincides with the formation of H3 and a partial formation of H1 and H2. In the subsequent slower phase, H1 and H2 continue to form while the chain visits both compact and noncompact states. Although there is significant secondary structure, these states lack specific global structural characteristics as we see from the absence of high-flux rmsd and drms early structural clusters (Fig. 3). Only when H1 and H2 are sufficiently structured can the transition state be reached in which the H1–H2 segment is native-like, forming a relatively ordered helical hairpin. H3, by contrast, has only limited interaction with this H1–H2 “nucleus” of the transition state. The folding process for Villin differs from that of protein A in some aspects. The initial collapse in Villin is accompanied by the formation of both H2 and H3, whereas H1 forms at a slower rate. We also find that the Villin chain remains mostly compact during the remaining part of the folding process, as shown by the cluster analysis that exhibits only low-Rg clusters. The TSE of Villin is characterized by relatively well formed secondary elements and a native-like H1–H2 segment, similar to the situation in protein A but the TSE is structurally less coherent. The Villin TSE also is quite compact although it is slightly more extended than both the native structure and the early disordered compact states that follow the initial collapse.
The chain collapse behavior in the initial phase of folding that we find in our simulations has been observed, although with some variations, in several other simulation studies of both protein A (11, 12, 14, 16, 17, 38) and Villin (16, 17, 25–27). For protein A, this type of behavior may at first glance seem inconsistent with experiments that generally indicate that protein A folds in apparent two-state kinetics (31, 32, 44). Two observations most often used to support two-state kinetics are single-exponential relaxation and a linear dependence of ln kf on denaturant concentration [D], where kf is the folding rate, i.e., V-shaped Chevron plots. However, the folding relaxation kinetics for protein A have only been resolved for times t > 150 μs (31), whereas our collapse phase occurs on a much faster time scale. Hence, the fast collapse phase we observe would only be detected if much higher time-resolution folding experiments can be obtained for protein A. Highly time-resolved experiments have been performed on apomyoglobin, where folding was initiated from a cold-denatured state, and a fast initial collapse phase was detected and found to be completed within 7 μs (45). Linear refolding arms in the Chevron plots of protein A and various mutants have been observed in several studies (31, 32, 44, 46), but the folding rate kf usually can not be determined at very low [D], which might be important. A unique insight into the folding of protein A was obtained in a recent single-molecule study of protein A using FRET (46). Although two distinct populations were observed in the FRET efficiency (showing that U and N are distinct populations), a shift of the peak corresponding to U was observed with varying [D], indicating a chain compaction of U as [D] decreases. Whether this compaction of U would result in Chevron rollovers for [D] approaching 0, as observed when a collapsed state is artificially stabilized (47), remains to be seen. Regardless, we find that our results are in good agreement with the bimodal distribution of the FRET signal (see figure 6 in ref. 47), which can be seen from a clear separation in the probability distributions of 1/r6ee (mimicking FRET efficiency) for conformations in the U and N states, respectively, where ree is the chain end-to-end distance (SI Fig. 10). Finally, we note that direct evidence for a compact unfolded state with high degree of nonnative hydrophobic interactions has been observed recently in the Trp-cage miniprotein TC5b by using a novel type of NMR pulse-labeling experiment (48).
In terms of achieving a good agreement between theory and experiment, the excellent correspondence we find between Φexp and Φsim obtained for protein A is encouraging and means that, overall, the characteristics of the TSE is in very good agreement with the Φ analysis performed by Fersht et al. (31, 32) across the entire chain. We note that this agreement is quite robust and is independent on computational details such as the definition of the folded state (SI Fig. 11). A remaining issue is the extent to which secondary structure is present in the TSE. Our results indicate the presence of significant amounts of helicity in all three helices, including H3, which scores low Φsim values mainly because of its weak interaction with H1 and H2 in the TSE. Our finding that H3 is structured in the TSE appears to be at odds with the conclusion made by Fersht et al. from their Ala → Gly scanning study (31, 32). Φ values from such mutations, when performed at protein surface positions, were interpreted as mainly probing the secondary structure content of the TSE because no tertiary contacts are deleted upon mutation (49). In light of these experiments, it therefore is possible that the stability of secondary structure elements, in particular H3, are somewhat overestimated in our model. However, we note that low Ala → Gly Φexp values in H3 also could be explained by residual H3 secondary structure in the denatured state D under folding conditions. This situation would produce small ΔΔGD-‡ values upon mutation and consequently small Φ values. There are two factors that indicate that this might indeed be the case. First, all Ala → Gly Φexp values in H3 are markedly larger at 2 M GdmCl than at 0 M GdmCl (see table 1 in ref. 32), which is consistent with the melting of residual secondary structure in the denatured state at 2 M GdmCl compared with 0 M GdmCl. Importantly, this trend does not exist for H1 or H2. Second, H3 is the only one of the helices that exhibits some stability on its own, i.e., as an individual fragment (42). Hence, it appears likely that some residual secondary structure exists in H3 in the denatured state D under folding conditions, which may be an alternative explanation for the low Ala → Gly Φexp values in H3.
Conclusions and Outlook
We have demonstrated that a simple and computationally tractable transferable all-atom model can capture details of the folding behavior of two small helical proteins at a quantitative level. In particular, we find that the obtained Φ values for protein A fit experimental data to a degree that has not been achieved by previous simulation studies, whereas future experiments will have to be conducted to test the validity of the obtained Φ values for the Villin headpiece.
This study along with a previous investigation of the ENH (38) provide a comprehensive analysis of folding processes for three-helix bundle proteins at an atomistically detailed level. When we combine the results from these studies, a universal picture of the folding of three-helix bundle proteins emerges. The first step is an initial collapse of the chain accompanied by partial formation of the α-helices (to a greater or lesser extent). On average, the chain remains relatively compact, but frequent visits to more extended structures occur. During such fluctuations, the TSE can be located after which the chain collapses to N. The TSE consists of relatively well formed helices organized into a two-helix hairpin and a third helix, which is partially detached. Within this general framework, there can be significant differences in the details, however. For example, the initial collapse phase can be accompanied by the formation of a single helix (such as H3 for protein A) or two helices (H2 and H3 for Villin). Moreover, there are two possibilities for the helical hairpin in the TSE, which is dominated by a H1–H2 in both protein A and Villin. Our simulations (38) and recent experimental work of Fersht and coworkers found that H2–H3 hairpin in ENH forms an independently stable domain (32). Our analysis suggests that formation of a helix–turn–helix motif before entering the TSE is perhaps a universal mechanism observed in folding of three-helix bundle proteins, although details of which hairpin is formed may vary.
Methods
Energy Function.
The all-atom energy function E in our previous study (35) has been further developed and now takes the form:
where Econ is the pairwise atom–atom contact potential, Ehb is the hydrogen-bonding potential, Etrp is the sequence-dependent local torsional potential based on the statistics of sequential amino acid triplets, and Esct is the side-chain torsional angle potential (see SI Text). Detailed information on the first three energy terms can be found in our previous publication (35). It should be noted that secondary structure information from PSIPRED is not used in this study, which enables us to observe true ab initio folding of proteins.
Supplementary Material
ACKNOWLEDGMENTS.
We thank Dr. Chaok Seok for help with the implementation of the local move set that conserves detailed balance. This work is supported by the National Institutes of Health.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0707284105/DC1.
References
- 1.Alm E, Baker D. Curr Opin Struct Biol. 1999;9:189–196. doi: 10.1016/S0959-440X(99)80027-X. [DOI] [PubMed] [Google Scholar]
- 2.Gianni S, Guydosh NR, Khan F, Caldas TD, Mayor U, White GW, DeMarco ML, Daggett V, Fersht AR. Proc Natl Acad Sci USA. 2003;100:13286–13291. doi: 10.1073/pnas.1835776100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laurents DV, Baldwin RL. Biophys J. 1998;75:428–434. doi: 10.1016/S0006-3495(98)77530-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pande VS. Proc Natl Acad Sci USA. 2003;100:3555–3556. doi: 10.1073/pnas.0830965100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lo Conte L, Ailey B, Hubbard TJ, Brenner SE, Murzin AG, Chothia C. Nucleic Acids Res. 2000;28:257–259. doi: 10.1093/nar/28.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kubelka J, Eaton WA, Hofrichter J. J Mol Biol. 2003;329:625–630. doi: 10.1016/s0022-2836(03)00519-9. [DOI] [PubMed] [Google Scholar]
- 7.Vu DM, Myers JK, Oas TG, Dyer RB. Biochemistry. 2004;43:3582–3589. doi: 10.1021/bi036203s. [DOI] [PubMed] [Google Scholar]
- 8.Mayor U, Guydosh NR, Johnson CM, Grossmann JG, Sato S, Jas GS, Freund SM, Alonso DO, Daggett V, Fersht AR. Nature. 2003;421:863–867. doi: 10.1038/nature01428. [DOI] [PubMed] [Google Scholar]
- 9.Berriz GF, Shakhnovich EI. J Mol Biol. 2001;310:673–685. doi: 10.1006/jmbi.2001.4792. [DOI] [PubMed] [Google Scholar]
- 10.Cheng S, Yang Y, Wang W, Liu H. J Phys Chem B. 2005;109:23645–23654. doi: 10.1021/jp0517798. [DOI] [PubMed] [Google Scholar]
- 11.Favrin G, Irback A, Wallin S. Proteins. 2002;47:99–105. doi: 10.1002/prot.10072. [DOI] [PubMed] [Google Scholar]
- 12.García AE, Onuchic JN. Proc Natl Acad Sci USA. 2003;100:13898–13903. doi: 10.1073/pnas.2335541100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh A, Elber R, Scheraga HA. Proc Natl Acad Sci USA. 2002;99:10394–10398. doi: 10.1073/pnas.142288099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo Z, Brooks CL, III, Boczko EM. Proc Natl Acad Sci USA. 1997;94:10161–10166. doi: 10.1073/pnas.94.19.10161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hubner IA, Deeds EJ, Shakhnovich EI. Proc Natl Acad Sci USA. 2005;102:18914–18919. doi: 10.1073/pnas.0502181102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jang S, Kim E, Shin S, Pak Y. J Am Chem Soc. 2003;125:14841–14846. doi: 10.1021/ja034701i. [DOI] [PubMed] [Google Scholar]
- 17.Kim SY, Lee J, Lee J. J Chem Phys. 2004;120:8271–8276. doi: 10.1063/1.1689643. [DOI] [PubMed] [Google Scholar]
- 18.Kussell E, Shimada J, Shakhnovich EI. Proc Natl Acad Sci USA. 2002;99:5343–5348. doi: 10.1073/pnas.072665799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Linhananta A, Zhou Y. J Chem Phys. 2002;117:8983–8995. [Google Scholar]
- 20.Zhou Y, Karplus M. Nature. 1999;401:400–403. doi: 10.1038/43937. [DOI] [PubMed] [Google Scholar]
- 21.Boczko EM, Brooks CL., III Science. 1995;269:393–396. doi: 10.1126/science.7618103. [DOI] [PubMed] [Google Scholar]
- 22.De Mori GM, Colombo G, Micheletti C. Proteins. 2005;58:459–471. doi: 10.1002/prot.20313. [DOI] [PubMed] [Google Scholar]
- 23.Herges T, Wenzel W. Structure (London) 2005;13:661–668. doi: 10.1016/j.str.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 24.Kleiner A, Shakhnovich E. Biophys J. 2007;92:2054–2061. doi: 10.1529/biophysj.106.081257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zagrovic B, Snow CD, Shirts MR, Pande VS. J Mol Biol. 2002;323:927–937. doi: 10.1016/s0022-2836(02)00997-x. [DOI] [PubMed] [Google Scholar]
- 26.Zagrovic B, Snow CD, Shirts MR, Pande VS. J Mol Biol. 2002;323:927–937. doi: 10.1016/s0022-2836(02)00997-x. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez A, Shen MY, Colubri A, Sosnick TR, Berry RS, Freed KF. Biochemistry. 2003;42:664–671. doi: 10.1021/bi026510i. [DOI] [PubMed] [Google Scholar]
- 28.Duan Y, Kollman PA. Science. 1998;282:740–744. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 29.Kinnear BS, Jarrold MF, Hansmann UH. J Mol Graphics Model. 2004;22:397–403. doi: 10.1016/j.jmgm.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 30.Wolynes PG. Proc Natl Acad Sci USA. 2004;101:6837–6838. doi: 10.1073/pnas.0402034101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato S, Religa TL, Fersht AR. J Mol Biol. 2006;360:850–864. doi: 10.1016/j.jmb.2006.05.051. [DOI] [PubMed] [Google Scholar]
- 32.Sato S, Religa TL, Daggett V, Fersht AR. Proc Natl Acad Sci USA. 2004;101:6952–6956. doi: 10.1073/pnas.0401396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiu TK, Kubelka J, Herbst-Irmer R, Eaton WA, Hofrichter J, Davies DR. Proc Natl Acad Sci USA. 2005;102:7517–7522. doi: 10.1073/pnas.0502495102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Itoh K, Sasai M. Proc Natl Acad Sci USA. 2006;103:7298–7303. doi: 10.1073/pnas.0510324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang JS, Chen WW, Skolnick J, Shakhnovich EI. Structure (London) 2007;15:53–63. doi: 10.1016/j.str.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 36.Snow CD, Rhee YM, Pande VS. Biophys J. 2006;91:14–24. doi: 10.1529/biophysj.105.075689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du R, Pande VS, Grosberg AY, Tanaka T, Shakhnovich EI. J Chem Phys. 1998;108:334–350. [Google Scholar]
- 38.Hubner IA, Deeds EJ, Shakhnovich EI. Proc Natl Acad Sci USA. 2006;103:17747–17752. doi: 10.1073/pnas.0605580103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shortle D, Simons KT, Baker D. Proc Natl Acad Sci USA. 1998;95:11158–11162. doi: 10.1073/pnas.95.19.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karpen ME, Tobias DJ, Brooks CL., III Biochemistry. 1993;32:412–420. doi: 10.1021/bi00053a005. [DOI] [PubMed] [Google Scholar]
- 41.Rao F, Caflisch A. J Mol Biol. 2004;342:299–306. doi: 10.1016/j.jmb.2004.06.063. [DOI] [PubMed] [Google Scholar]
- 42.Bai Y, Karimi A, Dyson HJ, Wright PE. Protein Sci. 1997;6:1449–1457. doi: 10.1002/pro.5560060709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Riddle DS, Grantcharova VP, Santiago JV, Alm E, Ruczinski I, Baker D. Nat Struct Biol. 1999;6:1016–1024. doi: 10.1038/14901. [DOI] [PubMed] [Google Scholar]
- 44.Myers JK, Oas TG. Nat Struct Biol. 2001;8:552–558. doi: 10.1038/88626. [DOI] [PubMed] [Google Scholar]
- 45.Ballew RM, Sabelko J, Gruebele M. Proc Natl Acad Sci USA. 1996;93:5759–5764. doi: 10.1073/pnas.93.12.5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang F, Sato S, Sharpe TD, Ying L, Fersht AR. Proc Natl Acad Sci USA. 2007;104:123–127. doi: 10.1073/pnas.0609717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Otzen DE, Oliveberg M. Proc Natl Acad Sci USA. 1999;96:11746–11751. doi: 10.1073/pnas.96.21.11746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mok KH, Kuhn LT, Goez M, Day IJ, Lin JC, Andersen NH, Hore PJ. Nature. 2007;447:106–109. doi: 10.1038/nature05728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scott KA, Alonso DOV, Sato S, Fersht AR, Daggett V. Proc Natl Acad Sci USA. 2007;104:2661–2666. doi: 10.1073/pnas.0611182104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeLano WL. The PYMOL Molecular Graphics System. San Carlos, CA: DeLano; 2002. [Google Scholar]
- 51.Kabsch W. Acta Crystallogr A. 1978;34:827–828. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
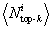{kind=link}
{kind=link}