

Abstract
Efficient in vitro systems to study the life cycle of hepatitis C virus (HCV) were recently developed for JFH1 (genotype 2a), which has unique replication capacity in Huh7 cells. We developed 4a/JFH1 intergenotypic recombinants containing the structural genes (Core, E1, and E2), p7, and all or part of NS2 of the 4a prototype strain ED43 that, after transfection of Huh7.5 cells with RNA transcripts, produced infectious viruses. Compared with the J6/JFH control virus, production of viruses was delayed. However, efficient spread of infection and high HCV RNA and infectivity titers were obtained in serial passages. Sequence analysis of recovered viruses and subsequent reverse genetic studies revealed a vital dependence on one or two NS2 mutations, depending on the 4a/2a junction. Infectivity of ED43/JFH1 viruses was CD81 dependent. The genotype 4 cell culture systems permit functional analyses as well as drug and vaccine research on an increasingly important genotype in the Middle East, Africa, and Europe. We also developed genotype 1a intergenotypic recombinants from H77C with vital mutations in NS3. Using H77C/JFH1 and ED43/JFH1 viruses, we demonstrated high homologous neutralizing antibody titers in 1a and 4a patient sera, respectively. Furthermore, availability of JFH1 viruses with envelope proteins of the six major HCV genotypes permitted cross-neutralization studies; 1a and 4a serum cross-neutralized 1a, 4a, 5a, and 6a but not 2a and 3a viruses. Thus, the JFH1 intergenotypic recombinants will be of importance for future studies of HCV neutralization and accelerate the development of passive and active immunoprophylaxis.
Keywords: Egypt, hepatitis C virus strain ED43, in vitro infection, intergenotypic recombinant, vaccine
Approximately 180 million people are infected with hepatitis C virus (HCV) and are at increased risk of developing severe liver disease. HCV isolates from around the world cluster into six major genotypes, which differ by ≈30% at the nucleotide (nt) and deduced amino acid level (1). Genotype 4 is primarily found in the Middle East and Africa (2, 3). In Egypt, ≈15% of the population is HCV-infected, with genotype 4a comprising ≈90% of cases (2, 3). This particularly high prevalence, presumably caused by an unintended transmission through parenteral treatment for schistosomiasis (3, 4), is at least partly responsible for a still rather high incidence (5), making Egypt a potential region for vaccine trials. Additionally, genotype 4 has been spreading in Europe, resulting in a prevalence of 10% in certain regions (2). Although the only approved treatment for chronic HCV infection, combination therapy with IFN-α and ribavirin, leads to a sustained virologic response in most of genotype 2 or 3 patients, viral clearance is obtained for only approximately half of patients with genotype 1 or 4. There is no vaccine against HCV.
Research on specific antiviral drugs and vaccines has been hampered by the absence of a full viral life cycle cell culture system. However, development of such a system came with cDNA clones of the genotype 2a isolate JFH1 (6, 7) and the intragenotypic 2a/2a recombinant J6/JFH (8), in which the structural genes (Core, E1, and E2), p7, and NS2 of JFH1 were replaced by the corresponding sequence of pJ6CF. For the development of efficient means of HCV prevention and control, it is important to establish culture systems for all major genotypes. Although JFH1-based systems of genotype 1 and 3 were developed (9–11), there are no culture systems producing infectious viruses of genotype 4, 5, and 6.
To establish genotype 4a/JFH1 culture systems, we replaced the JFH1 structural genes, p7 and parts or all of NS2 by the consensus sequence of the genotype 4 prototype strain ED43 (12), and confirmed efficient virus production from two ED43/JFH1 recombinants in the human hepatoma cell line Huh7.5. Similarly, we developed a virus system for reference strain H77C. The biological relevance of these systems was demonstrated in studies on CD81-dependent entry and on virus neutralization. Finally, we analyzed cross-genotype neutralization against our panel of genotype 1-6 JFH1-based recombinant viruses.
Results
Infectivity of ED43/JFH1 Recombinants Depends on the NS2 Junction Site.
Three 4a/2a intergenotypic recombinants were constructed. To retain the unique replication capacity of the genotype 2a isolate JFH1 (6), the JFH1 genes Core, E1, E2, p7, and partial or complete NS2 were replaced by the genotype 4a prototype strain ED43 (Fig. 1A). pED43/JFH1-α was constructed with the 3′-terminal intergenotypic junction between the first and second putative NS2 transmembrane domain in analogy to 1a, 1b, and 2a JFH1-based recombinants with enhanced viability (9). In pED43/JFH1-β this junction was located within the NS2 protease domain in analogy to a naturally occurring HCV recombinant (13). pED43/JFH1-γ and a replication deficient GND control contained the entire ED43 NS2 gene.
Fig. 1.

ED43/JFH1 recombinants and their viability in Huh7.5 cells. (A) Genome map of cDNA clones (ED43, purple; JFH1, black). ED43/JFH1-α had its 3′-terminal intergenotypic junction at nt 2,866/2,867 (H77 reference (AF009606) nt 2,867/2,868), whereas the junction of ED43/JFH1-β was at nt 3,185/3,186 (H77 reference nt 3,186/3,187). The complete ED43 NS2 gene was included in ED43/JFH1-γ and the replication-deficient control ED43/JFH1-GND. (B) Huh7.5 cells were transfected in parallel with RNA transcripts from pJ6/JFH, pED43/JFH1-α, -β, -γ, and -GND. After immunostaining, the percentage of HCV Core-positive cells was scored by using confocal fluorescence microscopy. One milliliter of supernatant from day 6 (J6/JFH; ≈104.4 TCID50), day 19 (ED43/JFH1-β; ≈102.9 TCID50 and ED43/JFH1-GND), or day 45 (ED43/JFH1-γ; ≈102.9 TCID50) was used for first passage (arrows) (SI Fig. 6). ED43/JFH1-α Core-positive cells were detected until day 19 but not thereafter.
RNA transcripts from pED43/JFH1-α, -β, -γ and -GND as well as pFL-J6/JFH were transfected into Huh7.5 cells. ED43/JFH1-α, -β and -γ all replicated as evidenced by anti-Core immunostaining on day 1 [Fig. 1B and supporting information (SI) Fig. 6]. However, whereas J6/JFH spread to most cells on day 3, ED43/JFH1-β and -γ spread occurred after an eclipse phase of 16 and 43 days, respectively. ED43/JFH1-α did not spread, and positive cells could not be detected after day 19 (Fig. 1B and SI Fig. 6). Infectivity titration using the 50% tissue culture infectious dose (TCID50) method confirmed the delay in production of infectious particles for ED43/JFH1-β and -γ, whereas ED43/JFH1-α was not infectious (data not shown). After passage of supernatant to naïve Huh7.5 cells, both viable ED43/JFH1 recombinants spread rapidly (SI Fig. 6). As in earlier studies (11, 14), cell death, followed by proliferation of HCV antigen-negative Huh7.5 cells, occurred after infection had spread to most cells.
ED43/JFH1 Viral Spread Kinetics Compare with J6/JFH, but Peak Infectivity Titers Are Lower.
To follow viral titers, infection kinetics and genome adaptation in Huh7.5 cells, we serially passed J6/JFH, ED43/JFH1-β and -γ in four cell-free passages. Included in these passages was a kinetic experiment, in which Huh7.5 cells were inoculated with equal doses of the respective virus. In all cultures, infection spread kinetics were comparable (Fig. 2A) and correlated with an increase in genome titers, which peaked at ≈107 units/ml (Fig. 2B). Similarly, the initial rise in infectivity titer was comparable for J6/JFH and ED43/JFH1 recombinants. However, as observed in all four passages, ED43/JFH1 infectivity titers did not rise above ≈103.5 TCID50 per milliliter, whereas J6/JFH titers continued to rise to ≈104.5 TCID50 per milliliter (Fig. 2C).
Fig. 2.

Comparison of infection kinetics of serially passaged J6/JFH and ED43/JFH1 viruses. Huh7.5 cells were incubated for 6 h with ≈103 TCID50 (MOI = 0.003) first-passage J6/JFH# or third-passage J6/JFH§, ED43/JFH1-β or -γ. (A) Percentage of Core-positive cells. (B) HCV RNA titers in culture supernatants monitored by HCV TaqMan. nd, not detected. (C) Supernatant infectivity titers determined by TCID50 assay (dark colored columns). RNA titers are shown for comparison (light colored columns).
Identification of Putative Adaptive Mutations in Recovered ED43/JFH1 Viruses.
The eclipse phase preceding spread of ED43/JFH1 recombinants in transfection cultures was in contrast to the immediate viral spread during subsequent passages, indicating a requirement for adaptation. Thus, we analyzed the ORF sequence of viruses recovered from culture supernatant in first, second, and third passage. In addition, the complete 5′UTR of first-passage virus was sequenced. In first passage, ED43/JFH1-β acquired amino acid change T827A in the 4a part of NS2 (Table 1). Interestingly, the same change occurred in ED43/JFH1-γ first-passage virus, which had two additional dominant amino acid changes; T329S in E1 and T977S in the γ-specific 4a part of NS2 (Table 2). All dominant mutations observed in first passage were consistently observed in second- and third-passage viruses, except for the γ-specific T329S. Further dominant changes were not observed until third passage (Tables 1 and 2). In contrast, J6/JFH did not acquire mutations in the 5′UTR or the ORF during three passages.
Table 1.
Mutations of ED43/JFH1-β during serial passages in Huh7.5 cells
| HCV gene | ED43 |
JFH1 |
|||||
|---|---|---|---|---|---|---|---|
| Core | E2 | NS2 | NS2 | NS3 | NS5A | ||
| Nucleotide number* | |||||||
| ED43/JFH1-β | 787 | 2206 | 2819 | 3305 | 4222 | 6306 | 7646 |
| H77 Abs ref. | 788 | 2207 | 2820 | 3306 | 4223 | 6307 | 7593 |
| pED43/JFH1-β | G | C | A | G | C | C | G |
| Direct sequencing† | |||||||
| First passage | • | • | G | • | • | • | • |
| Second passage | G/a | • | G | G/a | C/t | • | G/t |
| Third passage | A/g | C/T | G | G/A | C/T | C/T | T/g |
| Third-passage clonal distribution‡ | |||||||
| 3/10 | A | T | G | A | T | T | T |
| 2/10 | A | T | G | A | T | • | T |
| 2/10 | • | • | G | • | • | T | T |
| 1/10 | A | • | G | • | • | T | T |
| 1/10 | • | • | G | • | • | • | T |
| 1/10 | • | • | G | • | • | • | • |
| Amino acid number* | |||||||
| ED43/JFH1-β | 827 | 989 | 1989 | 2436 | |||
| H77 Abs ref. | 827 | 989 | 1989 | 2418 | |||
| Change | T → A | E → K | T → I | V → L | |||
*Positions are numbered according to the HCV sequence of pED43/JFH1-β. Corresponding H77 reference positions (AF009606) are given.
†Mutations representing ≥50% of the sequence read in at least one passage. Positions with mixtures are written with the dominant sequence in upper case and the minor sequence in lower case letters, or with both upper case wherever a dominant nucleotide was not determinable. Dots indicate identity with the original plasmid sequence.
‡In addition to indicated mutations, G7147C (E2269D) and A7640G (T2434A) were present in three clones and C1944G (T535S), T2021C (F561L), A2772G (D811G), T3392C (Y1018H), and T5836C [non coding (nc)] were present in two clones. In each clone, a number of single mutations were found, yielding an average of 22 mutations in total per clone. Thus, all clones had unique sequences.
Table 2.
Mutations of ED43/JFH1-γ during serial passages in Huh7.5 cells
| HCV gene | ED43 |
JFH1 |
||||||
|---|---|---|---|---|---|---|---|---|
| E1 | NS2 | NS3 | NS5A | |||||
| Nucleotide number* | ||||||||
| ED43/JFH1-γ | 986 | 1325 | 1336 | 2819 | 3269 | 4459 | 4918 | 7148 |
| H77 Abs ref. | 987 | 1326 | 1337 | 2820 | 3270 | 4460 | 4919 | 7161 |
| pED43/JFH-γ | G | A | A | A | A | C | G | T |
| Direct sequencing | ||||||||
| First passage | • | T/a | G | G | T | • | • | • |
| Second passage | • | A/T | G/a | G | T | C/T | G/A | • |
| Third passage | A/g | A/t | G/a | G | T | T | A | C |
| Third-passage clonal distribution† | ||||||||
| 7/10 | A | • | G | G | T | T | A | C |
| 2/10 | • | • | G | G | T | • | • | • |
| 1/10 | • | • | • | G | T | • | • | • |
| Amino acid number* | ||||||||
| ED43/JFH1-γ | 216 | 329 | 827 | 977 | 2270 | |||
| H77 Abs ref. | 216 | 329 | 827 | 977 | 2274 | |||
| Change | A → T | T → S | T → A | T → S | C → R | |||
See Table 1 legend for details.
*Positions are numbered according to the HCV sequence of pED43/JFH1-γ. In addition, A2785G (passage 1-3) and G7022A and A7128G coding for D2228N and E2263G (passage 1-2) were observed as quasispecies.
†In addition to indicated mutations, G1026A (C229Y), T1150C (nc), C2480T (L714F), A2995G (nc), C3001G (D887E), G7291A (nc), and T7985C (nc) were present in three clones; G723A (C128Y), T1211G (F291V), T1369C (nc), A2114G (T592A), G2251A (nc), T2916C (V859A), T2937C (V866A), G3208A (M956I), T4540C (nc), A5668G (nc), A6248G (I1970V), A7103G (M2255V), G7534A (nc), and G7584A (G2415E) were present in two clones.
To investigate the potential for coexistence of different ED43/JFH1 quasispecies, nt 86-8335 of recovered third-passage viruses were amplified in a single long RT-PCR and 10 clones of each recombinant were sequenced (Tables 1 and 2). The analysis confirmed the presence of the T827A amino acid change, which was found in all clones from both constructs. Furthermore, all 10 ED43/JFH1-γ clones acquired T977S. Interestingly, T329S, which dominated in first passage, was not observed in any third-passage γ-clones.
Identification of Vital Adaptive Mutations in ED43/JFH1 by Reverse Genetic Analyses.
To identify mutations necessary for production of infectious ED43/JFH1 recombinant viruses, we initially introduced mutations found to be dominant in first passage into the original plasmids, and tested their viability in Huh7.5 cells. Although the percentage of Core positive cells declined for the original constructs as earlier observed, ED43/JFH1-βT827A, ED43/JFH1-γT827A,T977S and ED43/JFH1-γT329S,T827A,T977S immediately spread in culture (Fig. 3A), yielding relatively high infectivity titers (Fig. 3 B and C). ED43/JFH1-γT827A did not spread in culture until day 42, where it had acquired the T977S amino acid change and six other mutations (A387C, C436A, G3154A, A4153G, T6184G, and T7879C) coding for one amino acid change (N16T). Likewise, the ED43/JFH1-γT977S transfection culture produced low or undeterminable infectivity titers (Fig. 3C), until the virus finally infected most cells on day 30 where it had acquired the T827A amino acid change as well as C4944T and T7125C coding for T1535I and L2262P. This emphasizes the prerequisite of the two NS2 mutations for production of infectious ED43/JFH1-γ viruses. Day 5 transfection supernatants were passed to naïve Huh7.5 cells and the complete ORF of viruses recovered after 17 days of culture was analyzed. Neither ED43/JFH1-βT827A nor ED43/JFH1-γT827A,T977S acquired additional mutations. However, ED43/JFH1-γT329S,T827A,T977S acquired three nucleotide changes (C781A, A5592G, and T7809C) resulting in amino acid changes Q1751R and V2490A. To test whether the additional coding mutations observed in third-passage clonal analysis of ED43/JFH1-β and ED43/JFH1-γ (Tables 1 and 2) could further improve viral infectivity, these mutations were introduced into pED43/JFH1-βT827A or pED43/JFH1-γT827A,T977S respectively, alone or in combinations. However, no significant increase of peak titers was observed after transfection of Huh7.5 cells (Fig. 3C).
Fig. 3.

Viability of ED43/JFH1 recombinants with putative adaptive mutations. (A and B) Transfection of RNA transcripts from pJ6/JFH, pED43/JFH1-GND, and pED43/JFH1-β and -γ constructs with or without mutations observed in first passage. (A) Percentage of Core-positive cells. Day 5 supernatants of relevant cultures were used for inoculation of first viral passages (arrow). (B) TCID50 determinations on transfection supernatants. (C) Transfection of RNA transcripts from pJ6/JFH, pED43/JFH1-GND, and pED43/JFH1-γT977S as well as pED43/JFH1-βT827A and pED43/JFH1-γT827A,T977S constructs with or without mutations observed in third passage. TCID50 determinations on transfection supernatants are shown. †, none (TCID50 = 0); *, one (TCID50 < 0.7); ‡, two (TCID50 < 0.7) of six replicates infected by undiluted supernatant. ED43/JFH1-GND was confirmed negative.
ED43/JFH1 Infection Depends on CD81 and Can Be Neutralized by Homologous Antiserum.
Blockage of the tetraspanin cell surface molecule CD81 was shown to inhibit genotype 1a, 1b, 2a and 3a infection of Huh7 cells (6, 9–11). To investigate the CD81 dependence of genotype 4a entry, we incubated Huh7.5 cells with anti-CD81 antibody before infection with ≈100 TCID50 ED43/JFH1-β or -γ. CD81 antibody but not an isotype-matched control reduced the number of focus forming units (FFUs) in a concentration dependent manner; ≥90% inhibition was achieved with 0.5 μg/ml CD81 antibody (Fig. 4).
Fig. 4.

Blocking of CD81 inhibits ED43/JFH1 infection. Huh7.5 cells were preincubated with anti-CD81 antibody or anti-HIV-p24 isotype-matched control antibody before addition of ≈100 TCID50 ED43/JFH1-γ third-passage virus. The count of FFUs per well after an incubation period of 2 days is indicated. Each data point represents triplicate experiments. Error bars indicate standard errors of the mean. nd, not determined.
To further investigate the biological relevance of ED43/JFH1 viruses, ≈100 TCID50 ED43/JFH1-γ were incubated with serial 2-fold dilutions of chronic phase serum from a genotype 4a infected patient (AA) before infection of Huh7.5 cells. In contrast to incubation with HCV negative control serum, incubation with AA serum reduced the number of FFUs in a concentration dependent manner, yielding a 50% neutralization titer of 1:6,400 (Fig. 5 and Table 3). Thus, the 4a/JFH1 viruses could be efficiently neutralized with homologous patient serum.
Fig. 5.

Neutralization of ED43/JFH1 virus. Approximately 100 TCID50 of ED43/JFH1-γ first-passage virus was incubated with serial 2-fold dilutions of genotype 4a (AA, purple) or genotype 1a (H06, orange) chronic-phase patient samples or a mixture of sera from four HCV-negative controls (black) in final dilutions as indicated, before incubation with Huh7.5 cells. The count of FFUs per well after an incubation period of 2 days is indicated. Each data point represents triplicate experiments. Error bars indicate standard errors of the mean.
Table 3.
Reciprocal titers of neutralizing antibodies in chronic-phase serum from patients infected with HCV genotype 1a (H06) and 4a (AA) against JFH1-based viruses representing the six HCV genotypes
| Envelope genotype | Reciprocal serum neutralizing antibody titer |
|||
|---|---|---|---|---|
| 90% |
50% |
|||
| 1a (H06) | 4a (AA) | 1a (H06) | 4a (AA) | |
| 1a | 50 | <50 | 1,600 | 50 |
| 2a | <50 | <50 | <50 | <50 |
| 3a | <50 | <50 | <50 | <50 |
| 4a | 800 | 400 | 12,800 | 6,400 |
| 5a | 3,200 | 800 | 25,600 | 3,200 |
| 6a | 25,600 | 3,200 | 204,800 | 25,600 |
Approximately 100 TCID50 of JFH1-based recombinant virus containing Core-NS2 of each of the six major genotypes was incubated in triplicates with a 2-fold dilution series of genotype 1a or 4a chronic-phase patient serum or a mixture of sera from four HCV negative controls and tested in Huh7.5 cells. Reciprocal neutralization titers are indicated as the highest dilution showing a reduction in FFUs of at least 90% or 50% compared to HCV-negative controls.
Development of Viable H77C/JFH1 Recombinants and Homologous Neutralization with Chronic Phase Patient H Serum.
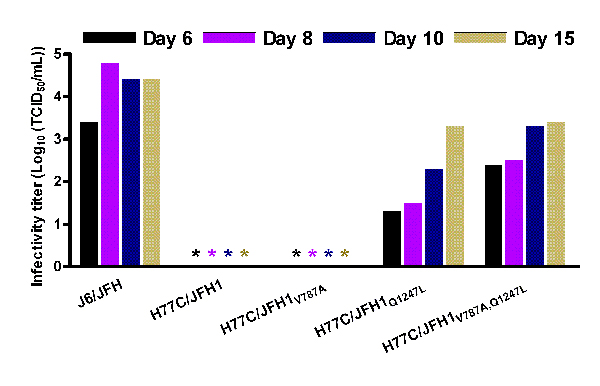
Meunier et al. (15) showed that chronic phase serum from Patient H with persistent genotype 1a infection efficiently neutralized HCV pseudoparticles (HCVpp) of different genotypes, including 1a and 4a. To study homologous neutralization with Patient H sera, we constructed pH77C/JFH1 with Core-NS2 from H77C (16). After two independent transfections H77C/JFH1 spread to most cells after 41 or 19 days of culture, respectively. Infectious virus could be passed to naïve cells yielding peak infectivity titers of ≈103.5 TCID50 per milliliter and HCV RNA titers of ≈107 units/ml, comparable with the values observed for ED43/JFH1 (data not shown). Sequencing of the ORF of first-passage virus from the two transfections identified dominant amino acid changes in p7 and NS3 (V787A and Q1247L) or in NS3 (R1408W), respectively (SI Table 4). Reverse genetic studies showed that introduction of either Q1247L or R1408W in NS3 allowed production of infectious viral particles with relatively high infectivity titers immediately after transfection (SI Fig. 7 A and B). V787A alone did not confer viability (SI Fig. 7A), but continuous propagation of H77C/JFH1V787A led to viral spread after acquisition of I1312V (NS3) and D2169A (NS5A). None of three recombinants with mutations introduced in NS3 acquired additional changes after passage to naïve cells (SI Table 4).
Homologous neutralization of recombinant H77C/JFH1 virus was demonstrated with serum from Patient H, taken 29 years after acute infection (H06). Serial 2-fold dilutions of H06 serum were used to neutralize ≈100 TCID50 of H77C/JFH1, yielding a 50% neutralization titer of 1:1,600 (SI Fig. 8 and Table 3).
Testing of Cross-Genotype Neutralization of Genotype 1-6 Recombinant Viruses with 1a and 4a Antisera.
The H06 1a serum efficiently neutralized ED43/JFH1-γ with a 50% titer of 1:12,800, whereas the AA 4a serum showed low-level neutralization of H77C/JFH1 with a 50% titer of 1:50 (Table 3). To further broaden the investigation of cross-genotype neutralization, serial 2-fold dilutions of 1a and 4a sera were tested against ≈100 TCID50 of JFH1-based recombinant viruses expressing the envelope proteins of genotype 2a (8), 3a (11), 5a (T.B.J., unpublished work), and 6a (J.M.G., unpublished work). Genotype 2a and 3a viruses could not be neutralized at a 1:50 dilution of either serum. However, genotype 5a and 6a viruses were efficiently neutralized by both sera with 50% neutralization titers of at least 1:3,200 (Table 3).
Discussion
We have established robust cell culture systems for studying HCV genotype 4; resulting viruses encode the Core and envelope proteins, p7 and NS2 from the 4a prototype strain ED43. Except for HCVpp systems expressing 4a envelope proteins (15, 17, 18), no other systems for in vitro studies of genotype 4 are currently available. The developed systems represent a significant step in the extension of the currently available JFH1-based cell culture systems of genotypes 1, 2, and 3 (6, 8–11).
Three ED43/JFH1 recombinants with different intergenotypic junctions replicated in Huh7.5 cells. The ED43/JFH1-β and -γ recombinants with junctions inside NS2 and at the NS2/NS3 cleavage site, respectively, were viable. Interestingly, naturally occurring intergenotypic recombinant isolates with the same 3′-terminal junctions as used in the β- and γ- but not in the α-construct have been described (13, 19). A study on JFH1-based inter- and intragenotypic recombinants suggested that the α-junction (termed C3), located in the N terminus of NS2, in general was favorable in obtaining the most efficient phenotype (9). However, we found that no infectious ED43/JFH1-α viruses were produced, suggesting that the optimal recombination site depends on the respective isolate. The fact that infectious viruses were produced from ED43/JFH1-β but not -α suggested that ED43 sequences between the two junction sites are needed for interaction with upstream sequence in the Core-p7 region or the N-terminal part of NS2. This is in accordance with findings in a study on H77/JFH1 recombinants (10). Inclusion of additional genotype 4 sequence in the recombinant genome could contribute to an even better understanding of the minimal JFH1 region/properties required for cell culture viability (20).
In contrast to J6/JFH that did not show any changes after three serial passages, putative adaptive mutations were identified in the ED43/JFH1 viruses. In reverse genetic studies, pED43/JFH1-β with the T827A amino acid change as well as pED43/JFH1-γ with T827A and T977S yielded immediate production of infectious viruses in Huh7.5 cells without further need for adaptation. The ED43 consensus T, one of various aliphatic and hydroxylated amino acids at position 827 in isolates of different genotypes, was found in the majority of deposited genotype 4 sequences and in JFH1, whereas the adapted A was found in few genotype 4 sequences.†† Because position 827 changed completely in both ED43/JFH1 constructs, and an M827V mutation was seen in an experiment with an H77/JFH1 β-junction virus (10), this position seems important for the function of intergenotypic JFH1 recombinants. Placed in the NS2 transmembrane domain, this mutation might facilitate intramembranous interaction between HCV proteins. In addition, ED43/JFH1-γ viability depended on the T977S mutation in a loop near the catalytic site of the NS2 protease domain.†† Because JFH1 (and most other non-genotype 4 sequences) encodes S at this position, whereas all deposited genotype 4 sequences encode T,†† this suggests a need for adaptation to a residue similar to that of JFH1. Although the function of NS2 is still unclear, its importance at an early stage of viral morphogenesis has been demonstrated (21). The E1 mutation T329S did not confer increased viral fitness. It reversed to the wild-type sequence and was probably a result of coselection. The adaptation patterns found for ED43/JFH1-β and -γ were not likely to be caused by suboptimal consensus sequences inserted during construction, because the patient ED43 sequence (12) and all clones from chimpanzee ED43 plasma at these positions were identical to the consensus. Hence, we identified adaptive NS2 mutations that were vital for production of infectious ED43/JFH1 viruses.
H77C/JFH1 also depended on adaptive mutations. Although Q1247L or R1408W in NS3 conferred efficient growth in Huh7.5 cells, combination of Q1247L with V787A led to accelerated kinetics in Huh7.5 cells. The R1408W mutation has not been reported previously. In contrast, acquisition of V787A or Q1247L mutations was found in viable H77/JFH1 recombinants (10). However, in this earlier study, the sequence of viruses resulting from transfection experiments with recombinants containing these mutations was not confirmed. In a study on 3a/JFH1 viruses, we similarly found that a single mutation in NS3 allowed production of infectious particles (11). Although NS3 mutations do not seem important for viability of our genotype 4 recombinants, this emphasizes the importance of NS3 mutations in JFH1-based recombinants of various genotypes.
This study demonstrates that HCV genotype 4 viruses, as demonstrated for genotype 1–3 (6, 9–11, 22), use CD81 for entry into host cells. This finding is in agreement with earlier observations for genotype 4 in the HCVpp system (18). It underlines the biological relevance of the developed systems and suggests further investigations of genotype specific entry involving receptors such as SR-BI (23) and Claudin-1 (24).
We conducted cross-genotype neutralization studies in HCV cell culture systems recapitulating the entire viral life cycle using JFH1-based viruses with envelope sequences of all six major genotypes, which has previously not been possible. HCV E1/E2 assembled on HCVpp, used in previous neutralization studies could show an unphysiological confirmation, glycosylation pattern and/or lipoprotein association because of the nature of the HCVpp and the nonhepatic producer cell-lines used. We found that viruses of genotype 1a, 4a, 5a, and 6a but not 2a and 3a were efficiently neutralized by H06 genotype 1a serum derived from reference Patient H. Neutralization of the ancestral H77C/JFH1 virus, whose sequence originates from acute phase Patient H serum, is in agreement with an extensive longitudinal study on neutralizing antibodies in Patient H carried out in the HCVpp system showing neutralization by serum samples taken later but not concurrently or earlier than the envelope sequence used for HCVpp (25). Our results in the cell culture systems compare well with neutralization experiments using Patient H serum from year 26 (H03) carried out in HCVpp systems with envelope proteins of the same prototype isolates of all six HCV genotypes as used in the present study, and heterogeneity between the genotypes is thus as reported (15). In addition, we found that cross-genotype neutralization extended to a genotype 4a antiserum, which efficiently neutralized genotype 4a, 5a, and 6a. Interestingly however, whereas 4a virus was efficiently neutralized by 1a serum, 1a virus was neutralized by 4a serum only at the lowest dilution tested (1:50), emphasizing the complexity of HCV neutralization. Although our data might imply a closer serological relationship among genotypes 1a, 4a, 5a, and 6a than among these genotypes and genotypes 2a and 3a, neutralization failure could be associated with the specific isolates used (26). In vivo, cross-genotype protection is possible also to genotype 2 and 3 (27), although protection is not always observed (28), and could be due mainly to cellular immunity (J.B., unpublished data). Overall however, our results indicate that it might be possible to identify and raise cross-neutralizing antibodies, which is important for the development of active and passive immunization strategies. The availability of cell culture-grown HCV particles bearing envelope proteins of different genotypes theoretically enables the development of inactivated whole-virus vaccines and comprehensive virus-neutralization studies. Hereby, the availability of cell culture-grown recombinant genotype 4a viruses is of particular importance, because this genotype is found in the majority of HCV-infected individuals in Egypt, where a relatively high incidence of HCV (5) creates a potential region for future vaccine trials.
We have developed JFH1-based cell culture systems recapitulating the full viral life cycle of HCV genotype 4a intergenotypic recombinants. Infectivity depended on the site of the intergenotypic NS2 junction and specific adaptive mutations in NS2. These observations might aid the development of full-length cell culture systems for this important genotype. The developed systems allow genotype 4-specific functional studies of the structural genes, p7 and NS2 and of anti-viral drugs and neutralizing antibodies in a true cell culture system. Importantly, we report a comprehensive analysis of cross-genotype neutralization made possible by the development of JFH1-based virus systems for all six major HCV genotypes.
Materials and Methods
Source of HCV and Patient Serum.
The source of the genotype 4a virus was an acute-phase plasma pool of strain ED43 from an experimentally infected chimpanzee (J.B., unpublished work). Strain ED43 originated from an Egyptian patient; virus recovered from this patient was originally sequenced by Chamberlain et al. (12). pJFH1 and pJFH1/GND used in recombinant construction was provided by T. Wakita (Tokyo Metropolitan Institute for Neuroscience, Tokyo) (6). pCV-H77C (16) was used for construction of pH77C/JFH1. C. Rice (Rockefeller University, New York) provided pFL-J6/JFH and pFL-J6/JFH-GND (8). For neutralization experiments, we used sera from persistently infected Patient H (2006, year 29 after infection, genotype 1a) and an Egyptian Patient [AA, 1994, genotype 4a provided by D. Thomas (Johns Hopkins University School of Medicine, Baltimore)]. Analysis of AA E1 sequence encoding amino acid residues 213–383 demonstrated a relatively close relationship to the ED43 sequence, with only 4% amino acid differences (data not shown).
Construction of JFH1-Based Intergenotypic Recombinants.
The ED43 sequence was amplified by RT-PCR using SuperScriptII reverse transcriptase (Invitrogen) and Advantage 2 PCR Enzyme System (Clontech) and cloned by using TOPO-XL Cloning kit (Invitrogen). From 4 ED43 clones and the ED43 HCVpp E1/E2 expression vector, pCMV-ED43(4a-1) (15), we assembled an ED43 sequence deviating from the nucleotide consensus at two noncoding positions only (A2458G and A2593G). pED43/JFH1-α, -β and -γ were constructed by inserting a fusion PCR product containing the ED43 region and the respective genotype junctions directly into pJFH1. Prefusion PCRs were done on pJFH1 and the ED43 consensus sequence with overlapping primers yielding the correct ED43/JFH1 junctions. After fusion PCR, the obtained product was digested by AgeI and SpeI in the JFH1 5′ UTR and NS3, respectively, and ligated into pJFH1. pH77C/JFH1 was constructed similarly from pCV-H77C and pJFH1. As a negative control, a viral polymerase mutant pED43/JFH1-GND was constructed in analogy to pED43/JFH1-γ by ligating into pJFH1/GND. For reverse genetic studies, mutations were introduced by using mutated primers in fusion PCRs. All PCRs were done by using Pfu polymerase (Stratagene). DNA stocks of final plasmids were prepared by using Qiagen Endo Free Plasmid Maxi kit. The complete HCV sequence of final plasmid preparations was confirmed.
Culturing, Transfection, and Infection of Huh7.5 Cells.
Culturing of Huh7.5 cells was done as described (11). One day before transfection or infection, naïve Huh7.5 cells were plated at 3 × 105 per well in six-well plates. In vitro transcription was carried out for 2 h with T7 RNA polymerase (Promega) on 5 μg of plasmid linearized with XbaI and treated with Mung Bean Nuclease (New England Biolabs) to yield the exact HCV 3′ end (16). For transfection, 2.5 μg of unpurified RNA transcripts were incubated with 5 μl of Lipofectamine2000 (Invitrogen) in 500 μl of Opti-MEM (Invitrogen) for 20 min at room temperature. RNA-Lipofectamine2000 transfection complexes were left on cells for 16–24 h before washing. For infection, virus-containing supernatant was left on cells for 6–24 h. Supernatants collected during experiments were sterile filtered and stored at −80°C.
Evaluation of Infected Cultures.
Anti-Core immunostaining was done with mouse anti-HCV Core protein monoclonal antibody (B2) (Anogen) as primary antibody and Alexa Fluor 594 goat anti-mouse IgG (H+L) (Invitrogen) as secondary antibody (11). HCV RNA titers were determined by a TaqMan real-time PCR assay (11). Infectivity titers were determined by using an earlier described protocol (8). Naïve Huh7.5 cells (6 × 103) were plated per well in a poly-d-lysine-coated 96-well plate (Nunc) the day before inoculation with 10-fold dilutions of cell culture supernatants in replicates of six for 2–3 days. Primary Ab for development was anti-NS5A 9E10 (a gift from C. Rice). Secondary Ab was ECL anti-mouse IgG HRP-linked whole antibody (GE Healthcare Amersham). Staining was developed by using DAB substrate kit (DAKO). Wells were scored positive if one or more cells were infected, and the TCID50 value was calculated (11). Sequence analysis of recovered viruses was done as described in SI Text and SI Tables 5 and 6.
Inhibition of Infection by Blocking of CD81.
Naïve Huh7.5 cells (6 × 103) were plated per well in a poly-d-lysine-coated 96-well plate the day before a 1-hour incubation with anti-CD81 (JS-81; BD Pharmingen) or isotype matched control anti-HIV p24, clone Kal-1 (DAKO) in 50 μl in concentrations as indicated in Fig. 4. Approximately 100 TCID50 ED43/JFH1 in 150 μl were added for 4 h. Cells were washed once, supplemented with fresh media, and incubated for 2 days before staining as described for infectivity titration. Infection was scored by counting the number of FFUs per well. One FFU was defined as one or more infected cells, separated from other infected cells by at least two uninfected cells.
Neutralization of Virus by Patient Sera.
Approximately 100 TCID50 viruses were incubated for 1 h at 37°C with 2-fold dilutions of heat-inactivated (56°C for 30 min) patient sera or a mixture of sera from four healthy controls in final dilutions as indicated. The virus–serum mixture was incubated for 3 h at 37°C with 6 × 103 plated Huh7.5 cells in a poly-d-lysine-coated 96-well plate. Cells were washed once, supplemented with fresh media, and left for 2 days before staining as described for infectivity titration. FFUs were scored as above.
Supplementary Material
ACKNOWLEDGMENTS.
We thank B. Landt and A.-L. Soerensen for technical assistance; J. O. Nielsen, O. Andersen, G. Lisby, and B. Lundgren (Copenhagen University Hospital) and R. Purcell (National Institutes of Health) for their support of the project; and C. Rice, T. Wakita, and D. Thomas for reagents. This work was supported by The Danish Medical Research Council, the Lundbeck Foundation, and the Novo Nordisk Foundation (J.B.). T.K.H.S. was in part supported by the Novo Scholarship Program in Biotechnology and Pharmaceutical Sciences and the Carlsberg Memorial Scholarship for Brewer J. C. Jacobsen.
Footnotes
The authors declare no conflict of interest.
Data deposition: The nucleotide sequences of pED43/JFH1-α, -β, and -γ and pH77C/JFH1 have been deposited in the GenBank database (accession nos. EU363758, EU363759, EU363760, and EU363761, respectively).
This article contains supporting information online at www.pnas.org/cgi/content/full/0711044105/DC1.
The Los Alamos hepatitis C sequence database.
References
- 1.Simmonds P, et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology. 2005;42:962–973. doi: 10.1002/hep.20819. [DOI] [PubMed] [Google Scholar]
- 2.Abdo AA, Lee SS. Management of hepatitis C virus genotype 4. J Gastroenterol Hepatol. 2004;19:1233–1239. doi: 10.1111/j.1440-1746.2003.03325.x. [DOI] [PubMed] [Google Scholar]
- 3.Ray SC, Arthur RR, Carella A, Bukh J, Thomas DL. Genetic epidemiology of hepatitis C virus throughout Egypt. J Infect Dis. 2000;182:698–707. doi: 10.1086/315786. [DOI] [PubMed] [Google Scholar]
- 4.Frank C, et al. The Role of parenteral antischistosomal therapy in the spread of hepatitis C virus in Egypt. Lancet. 2000;355:887–891. doi: 10.1016/s0140-6736(99)06527-7. [DOI] [PubMed] [Google Scholar]
- 5.Mohamed MK, et al. Intrafamilial transmission of hepatitis C in Egypt. Hepatology. 2005;42:683–687. doi: 10.1002/hep.20811. [DOI] [PubMed] [Google Scholar]
- 6.Wakita T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhong J, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci USA. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindenbach BD, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 9.Pietschmann T, et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci USA. 2006;103:7408–7413. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yi M, Ma Y, Yates J, Lemon SM. Compensatory mutations in E1, P7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J Virol. 2007;81:629–638. doi: 10.1128/JVI.01890-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gottwein JM, et al. Robust hepatitis C genotype 3a cell culture releasing adapted intergenotypic 3a/2a (S52/JFH1) viruses. Gastroenterology. 2007;133:1614–1626. doi: 10.1053/j.gastro.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 12.Chamberlain RW, Adams N, Saeed AA, Simmonds P, Elliott RM. Complete nucleotide sequence of a type 4 hepatitis C virus variant, the predominant genotype in the Middle East. J Gen Virol. 1997;78:1341–1347. doi: 10.1099/0022-1317-78-6-1341. [DOI] [PubMed] [Google Scholar]
- 13.Kalinina O, Norder H, Mukomolov S, Magnius LO. A natural intergenotypic recombinant of hepatitis C virus identified in St. Petersburg. J Virol. 2002;76:4034–4043. doi: 10.1128/JVI.76.8.4034-4043.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhong J, et al. Persistent hepatitis C virus infection in vitro: Coevolution of virus and host. J Virol. 2006;80:11082–11093. doi: 10.1128/JVI.01307-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meunier JC, et al. Evidence for cross-genotype neutralization of hepatitis C virus pseudo-particles and enhancement of infectivity by apolipoprotein C1. Proc Natl Acad Sci USA. 2005;102:4560–4565. doi: 10.1073/pnas.0501275102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yanagi M, Purcell RH, Emerson SU, Bukh J. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc Natl Acad Sci USA. 1997;94:8738–8743. doi: 10.1073/pnas.94.16.8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lavillette D, et al. Characterization of host-range and cell entry properties of the major genotypes and subtypes of hepatitis C virus. Hepatology. 2005;41:265–274. doi: 10.1002/hep.20542. [DOI] [PubMed] [Google Scholar]
- 18.McKeating JA, et al. Diverse hepatitis C virus glycoproteins mediate viral infection in a CD81-dependent manner. J Virol. 2004;78:8496–8505. doi: 10.1128/JVI.78.16.8496-8505.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noppornpanth S, et al. Identification of a naturally occurring recombinant genotype 2/6 hepatitis C virus. J Virol. 2006;80:7569–7577. doi: 10.1128/JVI.00312-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murayama A, et al. The NS3 Helicase and NS5B-to-3′X regions are important for efficient hepatitis C virus strain JFH-1 replication in Huh7 cells. J Virol. 2007;81:8030–8040. doi: 10.1128/JVI.02088-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM. Hepatitis C virus P7 and NS2 proteins are essential for production of infectious virus. J Virol. 2007;81:8374–8383. doi: 10.1128/JVI.00690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yi M, Villanueva RA, Thomas DL, Wakita T, Lemon SM. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc Natl Acad Sci USA. 2006;103:2310–2315. doi: 10.1073/pnas.0510727103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scarselli E, et al. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002;21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans MJ, et al. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 25.von Hahn T, et al. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology. 2007;132:667–678. doi: 10.1053/j.gastro.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 26.Logvinoff C, et al. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc Natl Acad Sci USA. 2004;101:10149–10154. doi: 10.1073/pnas.0403519101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lanford RE, et al. Cross-genotype immunity to hepatitis C virus. J Virol. 2004;78:1575–1581. doi: 10.1128/JVI.78.3.1575-1581.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prince AM, et al. Immunity in hepatitis C infection. J Infect Dis. 1992;165:438–443. doi: 10.1093/infdis/165.3.438. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}