

Abstract
The methylation status of the CpG island located within the ribosomal RNA (rRNA) promoter in human hepatocellular carcinomas and pair-matched liver tissues was analyzed by bisulfite genomic sequencing. Significant hypomethylation of methyl-CpGs in the rRNA promoter was observed in the tumor samples compared with matching normal tissues, which was consistent with the relatively high level of rRNA synthesis in rapidly proliferating tumors. To study the effect of CpG methylation on RNA polymerase I (pol I)-transcribed rRNA genes, we constructed pHrD-IRES-Luc (human rRNA promoter-luciferase reporter). In this plasmid, Kozak sequence of the pGL3-basic vector was replaced by the internal ribosome entry site (IRES) of encephalomyocarditis viral genome to optimize pol I-driven reporter gene expression. Transfection of this plasmid into HepG2 (human) cells revealed reduced pol I-driven luciferase activity with an increase in methylation density at the promoter. Markedly reduced luciferase activity in Hepa (mouse) cells compared with HepG2 (human) cells showed that pHrD-IRES-Luc is transcribed by pol I. Site-specific methylation of human rRNA promoter demonstrated that methylation of CpG at the complementary strands located in the promoter (−9, −102, −347 with respect to the +1 site) inhibited luciferase activity, whereas symmetrical methylation of a CpG in the transcribed region (+152) did not affect the promoter activity. Immunofluorescence studies showed that the methyl-CpG-binding proteins, MBD1, MBD2, MBD3, and MeCP2, are localized both in the nuclei and nucleoli of HepG2 cells. Transient overexpression of MBD2 suppressed luciferase activity specifically from the methylated rRNA promoter, whereas MBD1 and MBD3 inhibited rRNA promoter activity irrespective of the methylation status. Chromatin immunoprecipitation analysis confirmed predominant association of MBD2 with the endogenous methylated rRNA promoter, which suggests a selective role for MBD2 in the methylation-mediated inhibition of ribosomal RNA gene expression.
The transcriptional regulation of ribosomal RNA (rRNA) genes is a control point in the complex process of ribosome biogenesis. Diploid somatic cells harbor 300–400 copies of the rRNA genes that code for the most abundant cellular RNA. Only a fraction of these genes is transcribed, which depends on the growth stage of the cells and extracellular stimuli (for a review, see Refs. 1 and 2). In general, multiple copies of rRNA are found as repeated clusters, usually arranged in a head-to-tail fashion. The core promoter region spanning from −50 to +20 bp with respect to the initiation site is necessary and sufficient for the initiation of basal transcription in most species (for a review, see Refs. 3-6). Another key element is the upstream control element (UCE)1 that extends 150–200 bp upstream of the transcription start site. Apart from core promoter and UCE, upstream enhancers and terminator also play a critical role in rRNA transcription. Whereas the transcription machineries of RNA polymerase II (pol II) and RNA polymerase III (pol III) are often compatible with genes from widely different species, RNA polymerase I (pol I) exhibits stringent (7) but not absolute (8) species specificity. This could result from very little sequence similarity between rRNA promoters from different species despite the general conservation of functional transactivation domains of the transcription factors from mice to humans (6, 9).
Although considerable advances have been made in the identification and characterization of factors that up-regulate rRNA gene expression, the factors controlling its down-regulation have not been fully characterized. Methylation of DNA at the 5-position of cytosine of CpG base pairs, particularly in the promoter region is the predominant epigenetic modification of DNA in mammals and is known to suppress many RNA polymerase II (pol II) genes (10-12). DNA methylation is essential for development (13, 14). It regulates inactivation of X chromosome in females, genomic imprinting and suppresses spurious transcription from promoters of retroviruses and transposable elements integrated with the genome (15). In addition, aberrations in DNA methylation cause activation of oncogenes, genomic instability, and silencing of a variety of tumor suppressor genes (e.g. P16, P15, P21, E-CAD, VHL, etc.), leading to uncontrolled cell proliferation (for a review, see Refs. 11 and 15-17). This modification is initiated by de novo DNA methyltransferases (DNMT3A and DNMT3B) and is propagated in successive cell divisions by the maintenance methyltransferase (DNMT1). DNMT1 transfers methyl group from S-adenosyl-l-methionine (AdoMet) to the newly replicated strand using hemimethylated strand as the template (for a review, see Refs. 18 and 19). Aberrations in DNA methylation lead to a variety of diseases. For example, ICF (immunodeficiency, centromeric instability, and facial anomaly) syndrome is caused by mutations in the DNMT3B gene (20-22). The drugs inhibiting DNMT, namely 5-deoxyazacytidine, 5-fluorocytidine, and zebularine, alone or in combination with histone deacetylase inhibitors are used clinically in certain types of cancer to activate methylated tumor suppressor or differentiation-inducing genes (23, 24).
DNA methylation can impede the transcriptional activity of a pol II gene (25) directly by blocking the access of a transcription factor (e.g. AP-2, NF-κB, E2F, and c-MYC) to their cognate sites (19). Most of the methylated promoters are, however, recognized by a group of proteins called methyl-CpG-binding proteins (MBDs) by virtue of their conserved methyl-CpG binding domain. Five such MBDs with highly conserved DNA binding domains have been identified (26). Among these proteins, MeCP2, MBD1, MBD2, and MBD4 can bind to methylated DNA. MBD4 is a uracil-DNA glycosylase involved in G-T mismatch repair (12, 27). MBDs repress transcription by recruiting a variety of proteins such as Sin3a, histone deacetylases, histone methyltransferases, and HP1α (heterochromatin protein 1α) as co-repressors (for reviews, see Refs. 10, 14, 28, and 29). Kaiso, a partner of β-catenin, lacking the signature methyl CpG binding domain can also bind to methyl-CpGs and repress methylated promoters (30). A defect in the functions of these proteins also leads to various abnormalities. Rett syndrome, a prevalent X-linked neurological disorder among Caucasian females, is caused by dominant negative mutations in the MECP2 gene (31). Adult MBD1 knock-out mice, like MeCP2 null mice, also exhibit neurological abnormalities (32), whereas MBD4 null mice are susceptible to cancer because of enhanced CpG to TpG mutation in their genome (33).
Most of the studies to date have focused on the up-regulation of rRNA promoter activity. Since methylation of DNA, particularly the promoter region, is known to silence many pol II genes (23-26), it was of interest to investigate whether the methylation status of rRNA promoter modifies pol I-directed rRNA transcription. In the present study, we explored the methylation status of the CpG island that spans the rRNA promoter in human primary hepatocellular carcinomas and corresponding normal liver tissues and elucidated a potential molecular mechanism for the methylation-mediated alteration in rRNA promoter activity in human cells.
MATERIALS AND METHODS
Construction of Plasmids
pIRES-Luc
The internal ribosome entry site (IRES; 16–518 bp) was amplified from pCITE vector (Novagen) using T3 (5′-AATTAACCCTCACTAAAGGG-3′) and T7 (5′-GTAATACGACTCACTATAGGGC-3′) oligonucleotides and digested with NcoI to generate the 503-bp IRES product. The Kozak sequence ((GCC)GCCRCCATGG, where R represents a purine) that directs translation of pol II-transcribed mRNAs was removed from pGL3-basic vector by digestion with NcoI and HindIII and was replaced by the amplified IRES fragment to generate the plasmid pIRES-Luc. The replacement of the Kozak sequence with IRES was essential to minimize pol II-directed spurious expression from human rRNA promoter-luciferase reporter constructs in transfected cells.
Human rRNA-luciferase Vector (pHrD-IRES-Luc)
Human rRNA promoter spanning −410 to +314 bp (accession number K01105) with respect to the transcription initiation site was amplified from ∼2-kb fragments of HeLa genomic DNA digested with EcoRI. The following primer pairs with KpnI (forward) and BglII (reverse) restriction sites at the 5′ ends were used for PCR: forward, 5′-gtggtacccCGCGATCCTTTCTGGAGAGTCCC-3′; reverse, 5′-ggagatctGACGAGAACGCCTGACACGCAC-3′.
The annealing temperature was 58 °C. The resultant 730-bp PCR product was treated with Pfu polymerase (Stratagene) to polish the ends following the manufacturer's protocol, digested with KpnI and BglII, and cloned into the same sites of pIRES-Luc to generate pHrD-IRES-Luc.
Expression Vectors for MBD1, MBD2, MBD3, and MeCP2
These plasmids were constructed in mammalian expression vectors pcDNA 3.1(+/−) (Invitrogen) to obtain pcMBD1, pcMBD2, pcMBD3, pcMBD4, and pcMeCP2. Briefly, rat MeCP2 cDNA in pBlueScript-SK(−) (a generous gift from Adrian Bird) was digested with NotI–EcoRV enzymes, and the resultant ∼1.8-kb fragment was cloned into the same sites in pcDNA3.1(−) to generate pcMeCP2. Similarly, human MBD1 cDNA from pET6H (a generous gift from Adrian Bird) was excised with NcoI, and the sticky 5′-end of the ∼2-kb insert fragment was filled in with dNTPs catalyzed by Klenow and cloned in the EcoRV site of pcDNA3.1(−). Amplification with the vector- and insert-specific primers determined the correct orientation of MBD1. The mouse MBD2 cDNA in pBSK vector (a generous gift from Brian Hendrich) was digested with EcoRI and XhoI and ligated to the same sites in pcDNA3.1(−) to generate the mammalian expression vector. MBD3 cDNA was amplified from a mouse cDNA library and ligated to BamHI-HindIII sites of pcDNA3.1(−) to generate the plasmid pcMBD3.
Cell Culture and Transfection Assays
HepG2 cells were grown in Dulbecco's modified Eagle's medium with 10% fetal bovine serum. For transfection assay, 2.5 × 105 cells were plated onto 60-mm dishes 24 h prior to transfection and then transfected using calcium phosphate co-precipitation method (34, 35). Unless mentioned otherwise, each transfection mixture contains a maximum of 8.8 μg of total plasmid DNA that includes the reporter plasmid (0.5–1.0 μg), pRLTK (Renilla luciferase reporter driven by HSV-tk promoter (Promega)) (50 ng), as an internal control and the eukaryotic expression vectors (4 μg) harboring the gene of interest in a total volume of 500 μl. Briefly, DNA was dissolved in 220 μl of 0.1× TE (1 mm Tris·HCl, pH 8.0, 0.1 mm EDTA, pH 8.0) and mixed with 250 μl of 2× Hepes-buffered saline (280 mm NaCl, 10 mm KCl, 1.5 mm Na2HPO4, 12 mm dextrose, and 50 mm HEPES). Next, 31 μl of 2 m CaCl2 was added slowly to the DNA mixture and incubated for 20 min at room temperature before adding to the cell culture. The cells were allowed to incubate with the transfection reagent in complete medium for 16 h at 37 °C, followed by replacement with fresh medium. After 24–48 h in the fresh medium, the cells were harvested in the lysis buffer (Promega), and luciferase activity was measured using the Dual Luciferase Assay kit (Promega) in a Luminometer (Lumat LB 9507; EG&G Berthold, Oak Ridge, TN).
Bisulfite Genomic Sequencing
Genomic DNA isolated form hepatocellular carcinomas and matching liver tissues from the same individuals were treated with sodium bisulfite according to the protocol optimized in our laboratory (36, 37). The rRNA promoter spanning −377 to +51 bp was amplified using two sets of nested primers from the bisulfite-treated DNA. The primers used were as follows: hrRNA BF1, 5′-AATTTTTTTGGAGAGTTTTCGTG-3′; hrRNA BR1, 5′-GAGTCGGAGAGCGTTTTTTGAG-3′. The annealing temperature used was 50 °C.
The nested primers were hrRNA BF2 (5′-GAGTCGGAGAGCGTTTTTTGAG-3′) and hrRNA BR2 (5′-CATCCGAAAACCCAACCTCTCCAA-3′). The annealing temperature was 55 °C.
To confirm complete conversion of unmethylated cytosines to uracils, the PCR products were digested with TaqI, the restriction sites of which were generated only after bisulfite conversion. Completely converted PCR products were then cloned into TA cloning vector (Invitrogen). Ten clones selected at random from each DNA were sequenced in automated DNA sequencer.
DNA Methylation in Vitro with Bacterial Methylases
The KpnI-BglII fragment of pHrD-IRES-Luc was methylated with M.SssI methylase (New England Biolabs) or with M.HhaI methylase (New England Biolabs) in the presence (methylated) or absence (mock-methylated) of 160 μm AdoMet in a manufacturer-supplied buffer at 37 °C for 4 h. An additional 10 units of enzyme and AdoMet were added to the same mixture, and the methylation reaction was continued for an additional 4 h. The completion of the methylation reaction was determined by digestion of the fragment with BstUI, HpaII, or HhaI for M.SssI, M.HpaII, and M.HhaI methylases, respectively. These enzymes cannot cleave DNA if their cognate restriction sites are methylated. The methylated promoter fragment was then ligated to the same sites of pIRES-Luc. The ligated plasmid was separated on an agarose gel and purified using a gel extraction kit (Qiagen). Before transfection, the concentrations of methylated and mock-methylated plasmids were measured in a Beckman spectrophotometer at 260 nm.
Site-specific Methylation
Site-specific methylation was carried out following the methodology used for site-directed mutagenesis with some modifications. Single-stranded DNA was obtained by infection of XL1-blue-MRF′ bacteria harboring HrDNA plasmid with helper phage (R408; Stratagene). In a typical reaction, positive strand HrD-Luc plasmid (∼0.05 pmol) was annealed to 1.25 pmol of phosphorylated oligonucleotides (with a specific CpG either methylated denoted as “M” oligonucleotide or unmethylated control, depicted as “C” oligonucleotide) spanning different regions of the rRNA promoter or external transcribed spacer (external transcribed spacer region 1; ETS1). The annealing mixture (20 μl) contained 20 mm Tris·HCl (pH 7.5), 10 mm MgCl2, and 50 mm NaCl. The reaction was carried out in a thermocycler (PerkinElmer Life Sciences) programmed for a 5-min incubation at 96 °C followed by a slow descent (with a ramp of 3 min/degree Celsius) to 66 °C (annealing temperature of the oligonucleotide) and a 1-h incubation at 66 °C. Annealed oligonucleotide was then extended and ligated in a reaction mixture comprising 3 μl of 10× synthesis buffer (100 mm Tris·HCl, pH 7.5, 5 mm dNTPs, 10 mm ATP, and 20 mm dithiothreitol), 5 units of T4 DNA polymerase, and 1 unit of T4 DNA ligase in a total volume of 30 μl and was incubated at 37 °C for 90 min. The circularized plasmids were pooled, precipitated, and ligated overnight at 16 °C with a high concentration (10 units/μl) ligase (Invitrogen) in 10 μl of ligation mixture. These hemimethylated plasmids were methylated at the complementary CpG base pair with AdoMet catalyzed by DNMT1 (New England Biolabs) at 37 °C. Methylated (with AdoMet) or mock-methylated (without AdoMet), ligated plasmids were resolved by agarose gel electrophoresis and purified using gel extraction kit (Qiagen), and concentration was measured at 260 nm. Oligonucleotides used for site-specific methylation were C(−347) (5′-CGGCCAGGCCGCGACCTCTC-3′), M(−347) (5′-CGGCCAGGmCGCGACCTCTC-3′), C(−102) (5′-GCGCGACACGGACACCTGT-3′), M(−102) (5′-GCGCGACAmCGGACACCTGT-3′), C(−9) (5′-TCAGCAATAACCCGGCGGCC-3′), M(−9) (5′-TCAGCAATAACCmCGGCGGCC-3′), C(+152) (5′-CGGGAGTCGGGACGCTCGGA-3′), and M(+152) (5′-CGGGAGTCGGGACGCTmCGGA-3′).
Methylation at the respective sites were confirmed by sequencing the bisulfite-converted plasmids with the primers 5′-TGTGTGGAGTTGGAGAGTG-3′ and 5′-TTGGGGTTGATTAGAGGG-3′ (for the positive strand) and 5′-CCCTCCTACAACCAAAAC-3′ and 5′-TGTGTGGTTGTGATGGTG-3′ (for the negative strand).
Western Blot Analysis
Whole cell extract (200 μg) prepared from HepG2 cells overexpressing different MBDs were separated on a 7.5% SDS-polyacrylamide gel, transferred to nitrocellulose membrane, and blocked in 5% milk in Tris-buffered (pH 7.5) saline containing 0.1% Tween 20 (TBST). For detection of the overexpressed proteins, the membrane was subjected to immunoblot analysis with the respective antisera (37, 38) at appropriate dilution and the horseradish peroxidase-conjugated anti-rabbit IgG as the secondary antibody. The antigen-antibody complex was detected using the ECL™ kit (Amersham Biosciences), following the manufacturer's protocol.
Chromatin Immunoprecipitation
HepG2 cells at a density of 2.5 × 106 were plated on 150-mm tissue culture dish 24 h prior to the cross-linking. These cells were treated with formaldehyde (1%, final concentration) at 37 °C for 15 min to cross-link proteins to DNA. The reaction was stopped by the addition of 125 mm glycine. Soluble chromatin with an average size of 600–1000 bp was prepared following the protocol of Weinmann et al. (39). For chromatin immunoprecipitation analysis, antisera raised against the methyl binding proteins MBD1, MBD3, and MeCP2 in our laboratory were used (37). Anti-MBD2 antibody was from Upstate Biotechnology, Inc. (Lake Placid, NY). The chromatin was first precleared with preimmune sera coupled to protein A and protein G beads, followed by overnight incubation with preimmune or immune sera. The immune complex was then captured by protein G (for anti-MBD2) and protein A (for all other antibodies) beads, washed extensively as described (37, 39). Immunoprecipitated DNA-protein complex was eluted, decross-linked, treated with RNase A and proteinase K, and purified as described (37). The immunoprecipitated as well as the input DNA was digested with HpaII or MspI, and the digests along with an equal amount of the undigested immunoprecipitated DNA were amplified using the radiolabeled primers specific for the human rRNA promoter region: HrDChIP-F (5′-CTGCGATGGTGGCGTTTTTG-3′) and HrDChIP-R (5′-ACAGCGTGTCAGCAATAACC-3′). The primers used for the GAPDH promoter (accession number AY340484) are GAPDH-F (5′-GTGCCCAGTTGAACCAG-3′) and GAPDH-R (5′-AACAGGAGGAGCAGAGAGCGAAGC-3′). The annealing temperatures for rRNA and GAPDH primers were 58 and 60 °C, respectively. The sizes of the PCR products for rRNA and GAPDH are 152 and 223 bp, respectively. The amplicons were run on polyacrylamide gels (6% acrylamide), and the dried gels were subjected to autoradiography and PhosphorImager analysis. 32P-labeled PCR products were quantified using ImageQuant software (Amersham Biosciences), and results were depicted as the ratio of methylated and unmethylated DNA precipitated with the antibodies to the input methylated and unmethylated DNA, respectively.
Indirect Immunofluorescence Assay
HepG2 cells were grown overnight on Labtek chamber slides (2 × 104 cells/chamber). Cells were fixed with 1:1 methanol and acetone mixture for 15 min at 4 °C, washed with PBS, and permeabilized with 0.3% Triton X-100 in PBS for 10 min at room temperature. Next the cells were incubated with 1% bovine serum albumin for 1 h to block nonspecific binding. Blocked chambers were subsequently washed and incubated overnight at 4 °C with a mixture of anti-nucleolin monoclonal antibody (anti-C23) and antibodies raised against recombinant MBD1, MBD3, and MeCP2 in our laboratory or MBD2 (Upstate Biotechnology). MBD1, MBD3, and MeCP2 antibodies were used at a dilution of 1:500 in PBS, whereas MBD2 antibody was used at a dilution of 1:20. Fluorescein isothiocyanate-conjugated anti-rabbit (for MBD1, MBD3, and MeCP2) and anti-sheep (for MBD2) were used for green channel detection, whereas monoclonal anti-C23 antibody was detected by TRITC-conjugated monoclonal anti-mouse antibody (Sigma) for detection in the red channel. Nuclei were stained using 4′,6-diamidino-2-phenylindole in the mounting fluid.
RESULTS
Ribosomal RNA Promoter Is Hypomethylated in Human Hepatocellular Carcinomas
The sequence analysis of rRNA transcriptional initiation region of the human rRNA gene showed that it is highly enriched in CpG and that the promoter harbors a CpG island encompassing 19 CpGs in the upstream control element (UCE) and six CpGs in the core promoter regions. On the other hand, the mouse and rat rRNA promoters contain only one and five CpGs, respectively (Fig. 1A). Most of the CpG islands of housekeeping genes transcribed by RNA polymerase II are located in the promoter and exon 1 regions that are methylation-free in normal somatic cells. Although ribosomal RNA genes are housekeeping genes, they are highly reiterated, and only a fraction of these genes is transcribed by pol I. Therefore, we sought to investigate the methylation status of each CpG within different cis elements (spanning from −200 to −9 bp) in the rRNA promoter in human livers and their potential alterations in hepatocellular carcinomas (HCCs). To determine the methylation status of individual CpGs in the human rRNA promoter region, we performed bisulfite genomic sequencing (40) of DNA isolated from hepatocellular carcinomas and the pair-matched liver tissues. Treatment of DNA with bisulfite reagent converts unmethylated cytosine residues to uracils that are amplified as thymine during subsequent PCR. On the other hand, the methylated cytosine residues remain unconverted during bisulfite reaction and amplify as cytosines during subsequent PCR (41). The rRNA promoter region from bisulfite-treated DNA was amplified with nested primers and the PCR product was cloned (for details, see “Materials and Methods”). To analyze quantitatively the methylation status of each CpGs spanning the core promoter and UCE, 8–10 randomly selected clones from each HCC as well as the matching liver were sequenced. Alignment and analysis of the methylation profiles of the rRNA promoter in HCC and liver from the same individual revealed a differential methylation profile of CpGs between the two tissues (Fig. 1B). Comparison of the methylation pattern of the six individual livers revealed variations among them, probably due to polymorphism. For example, the region spanning from −200 to −148 (with respect to the +1 site) was poorly methylated in individuals 3 and 6 but heavily methylated in individuals 1 and 5. However, the overall methylation density at this region was comparable among the tumors and livers (Fig. 1B). Hypomethylation at UCE between positions −136 and −58 was more pronounced in all six tumors. Most CpGs within this region (e.g. −136, −107, −102, −100, −98, −95, −86, −66, and −58 were significantly hypomethylated in tumors. The quantitative analysis of methylated and unmethylated cytosine at positions −58, −66, and −98 (Fig. 1C) showed the differential level of methylation in the liver DNA among the individuals. Further, in five of six samples, clones containing unmethylated cytosines were more abundant in HCCs than in corresponding livers. These data demonstrate that the rRNA promoter, particularly in the UCE, is heavily methylated in human liver and is significantly hypomethylated in hepatocellular carcinomas compared with the matched liver tissues.
Fig. 1.

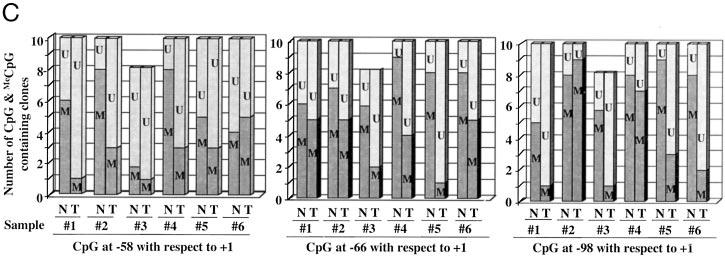
A, schematic depiction of the CpG dinucleotides on rRNA promoters in humans, rats, and mice (not drawn to scale) and the location of CpG dinucleotides (represented as lollipops) spanning the promoter region of human ribosomal RNA. B, methylation status of each CpG base pair spanning −200 to −9 bp of human rRNA promoter in six human hepatocellular carcinomas and matching livers. Ribosomal RNA promoter region was amplified from bisulfite-treated genomic DNA and cloned in TA vector (Invitrogen), and 8–10 clones, randomly selected from each sample, were subjected to automated sequencing. Each row represents the sequence of an individual clone, whereas each column depicts the position of the CpG. The filled and open circles denote methylated and unmethylated CpGs, respectively. C, quantitative analysis of methylation density at −58, −66, and −98 bp with respect to the +1 site of the rRNA promoter in individual tumors and matching livers. The number of clones methylated and unmethylated at these positions among 10 clones (eight for sample 3) is represented in this bar diagram. N and T denote livers and hepatocellular carcinomas, whereas U and M indicate unmethylated and methylated CpGs, respectively.
rRNA Promoter Activity Inversely Correlates with the Density of Methylation
To explore whether methylation of the CpG island in the human rRNA promoter indeed suppresses its expression, we cloned a region spanning from −410 to +314 bp comprising the upstream control element, core promoter, and part of ETS1 in pGL3-basic vector. The rRNA promoter-luciferase reporter (pHrD-IRES-Luc) was constructed by replacing the Kozak sequence of pGL3 vector with the internal ribosome entry site (IRES) (Fig. 2A) (for details, see “Materials and Methods”). This reporter plasmid was used in transient transfection studies. The rationale for eliminating the Kozak sequence was to minimize luciferase expression driven by spurious pol II promoters within the plasmid, since the 5′-trimethyl G-capped mRNAs (transcribed by pol II) exclusively require Kozak sequence for initial ribosome binding and translation (42). The IRES sequence was introduced immediately 5′ of the firefly luciferase cDNA for efficient translation of uncapped transcripts (pol I transcript and some viral RNAs), which requires IRES (43). To confirm further the authenticity of the pol I promoter activity in the transfection assay, we took advantage of the species specificity of pol I transcription (44). Accordingly, the human rRNA promoter will not be transcribed by mouse pol I transcription machinery and vice versa (7). For this purpose, we transfected both Hepa (mouse) and HepG2 (human) cells with either pIRES-Luc (promoterless) or pHrD-IRES-Luc vector. In HepG2 cells, the rRNA promoter-driven luciferase activity was 35-fold higher compared with pIRES-Luc, whereas only 3-fold higher activity of pHrD-IRES-Luc was observed in Hepa cells (Fig. 2, B and C). If the reporter gene were transcribed by pol II in HepG2 (human) cells transfected with pHrD-IRES-Luc, the luciferase activity would be comparable with that in Hepa (mouse) cells, since pol II transcription is not species-specific. Significantly higher (14-fold) activity of pHrD-IRES-Luc in HeG2 cells compared with that in Hepa cells clearly demonstrates that the rRNA promoter directs the luciferase expression from pHrD-IRES-Luc in HepG2 cells and is transcribed by pol I (Fig. 2C).
Fig. 2.
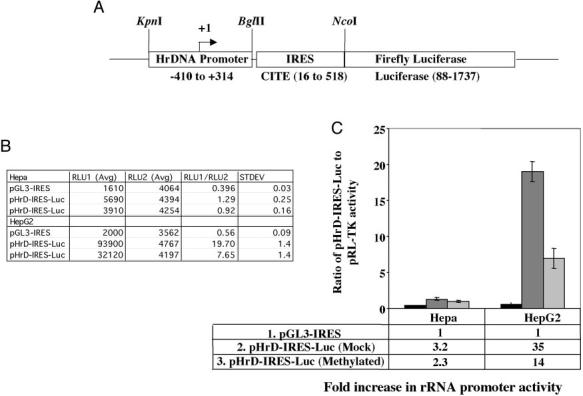
A, schematic representation of pHrD-IRES-Luc. A segment of the rRNA gene (−410 to +314 bp with respect to the transcriptional start site) was amplified using corresponding oligonucleotides and was cloned into modified pGL3-basic vector to generate pHrD-IRES-Luc (for details, see “Materials and Methods”). B and C, luciferase activity driven by pHrD-IRES-Luc is directed by pol I. Human rRNA promoter was methylated by M.HhaI or mock-methylated and ligated to pIRES-Luc to generate pHrD-IRES-Luc. The mock-methylated or methylated plasmid (500 ng) along with the internal control, pRLTK (50 ng), was transfected into HepG2 or Hepa cells by the calcium phosphate-DNA precipitation method. pRL-TK was used as internal control to measure transfection efficiency. After 24 h, both firefly and Renilla luciferase activity was measured using the Dual Luciferase Assay kit (Promega). Table B represents the firefly (RLU1) and Renilla (RLU2) luciferase activities and their ratios among different transfectants. C, graphical representation of the data.
Next, we investigated the effect of methylation of the rRNA promoter on luciferase activity. To address this issue, HepG2 and Hepa cells were transfected with pHrD-IRES-Luc harboring methylated or mock-methylated (with M.HhaI) rRNA promoter or pIRES-Luc. Luciferase activity was measured 24 h post-transfection. The rRNA promoter activity was significantly inhibited in HepG2 cells when methylated by M. HhaI, compared with the mock-methylated control (Fig. 2, B, rows 5 and 6, and C). The inhibitory effect of methylation was also robust in HepG2 cells (10-fold) compared with only 2-fold reduction in Hepa cells (Fig. 2, B, rows 2 and 3, and C), attributing the minimal promoter activity observed in Hepa cells to some leaky expression. These results further confirmed that the rRNA promoter-driven luciferase activity in HepG2 cells was sensitive to methylation. These results also demonstrate that rRNA promoter activity in human system is sensitive to CpG methylation.
We next investigated whether the pol I-driven pHrD-IRES-Luc activity was sensitive to the density of methylation at the promoter. This was accomplished by methylating the promoter and the ETS1 region (−410 to +314) with bacterial methylases that methylate densely (M.SssI), moderately (M.HpaII), or sparsely (M.HhaI) (Fig. 3A). The promoter activity was indeed dependent on the extent of methylation (Fig. 3, B and C). Methylation of only seven sites (five in the upstream of UCE and two in UCE) with M.HhaI resulted in significant (78%) inhibition of the luciferase activity compared with the mock-methylated promoter (Fig. 3C). Methylation with HpaII (which methylates 13 CpGs in the promoter region) resulted in almost complete (94%) inhibition in rRNA transcription compared with the mock-methylated promoter. As expected, the promoter activity was abolished when all of the CpGs located in the region spanning −410 to +314 were methylated with M.SssI (Fig. 3C). These results were highly reproducible in different batches of transfected cells. These data clearly demonstrate that the methylation density at the CpG island plays an important role in repression of human rRNA transcription.
Fig. 3.
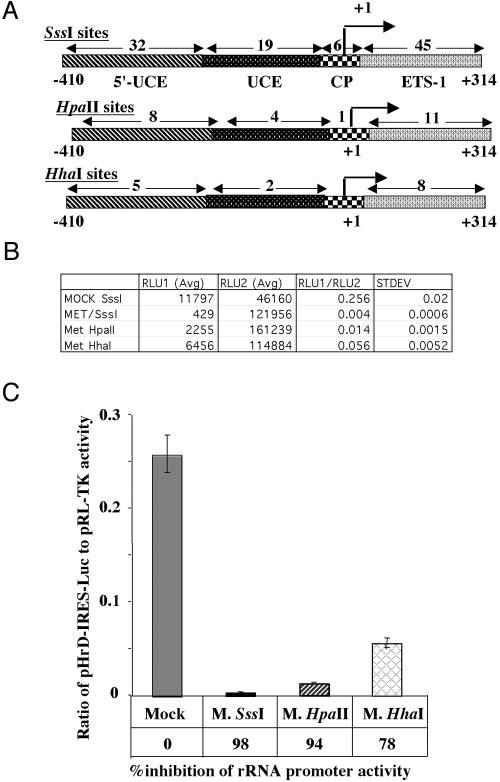
A, location of the recognition sites of different methylases on human rRNA promoter. B, human rRNA promoter activity in HepG2 cells transfected with pHrD-IRES-Luc, where the rRNA promoter region was methylated with M.SssI, M.HpaII, or M.HhaI and AdoMet. 1.5 × 105 cells were transfected with 500 ng of mock-methylated or methylated plasmid DNA and promoter activity was analyzed as described in the legend to Fig. 2B. Table B represents the average value of the luciferase activities and the ratio of RLU1 (pHrD-IRES-Luc) to RLU2 (pRL-TK) from each set of transfected cells. C, graphical representation of the data.
Methylation at Single Sites Located on the Promoter but Not on ETS1 of the Human rRNA Gene Down-regulates the Promoter Activity
The bacterial methylases can methylate specific sites not only in the promoter but also in ETS1. The effect of methylation in the ETS1 on rRNA promoter activity cannot be addressed by the previous experiment. To investigate whether methylation at specific sites in the promoter or the coding region had a differential effect on rRNA promoter activity, we developed site-specific methylation technique. For this purpose, we designed oligonucleotides containing either a methylated cytosine or unmethylated cytosine (as a control) at positions −347, −102, −9, and +152 (Fig. 4B) of the rRNA gene and generated mock-methylated, hemimethylated, and fully methylated circular pHrD-IRES-Luc plasmids (Fig. 4A; for details, see “Materials and Methods”). The promoter activity was measured in HepG2 cells 24 h post-transfection, and the result is expressed as the ratio of human rRNA promoter-directed firefly luciferase activity to the internal control, thymidine kinase promoter-driven Renilla luciferase activity (pRL-TK). When CpG at position −347 (upstream UCE) was symmetrically methylated on both strands (Meth M), the promoter activity was inhibited by ∼50%, whereas methylation on individual strands (Meth C, Mock M) had no significant effect (Fig. 4, C, compare rows 2–4 with row 1 and D, lanes 2–4 with lane 1). A comparable level of inhibition of luciferase activity was attained when pHrD-IRES-Luc was methylated at −102 (located in UCE) or at −9 (located in the core promoter) on both strands (Fig. 4, C, rows 8 and 12, and D, lanes 8 and 12, respectively). In contrast, methylation at +152 located on ETS1 has no inhibitory effect on the promoter activity (Fig. 4, C, rows 13–16, and D, lanes 13–16). These data clearly demonstrate that only symmetrical methylation of CpG at single sites located within the promoter but not in the ETS1 down-regulates human rRNA promoter activity.
Fig. 4.
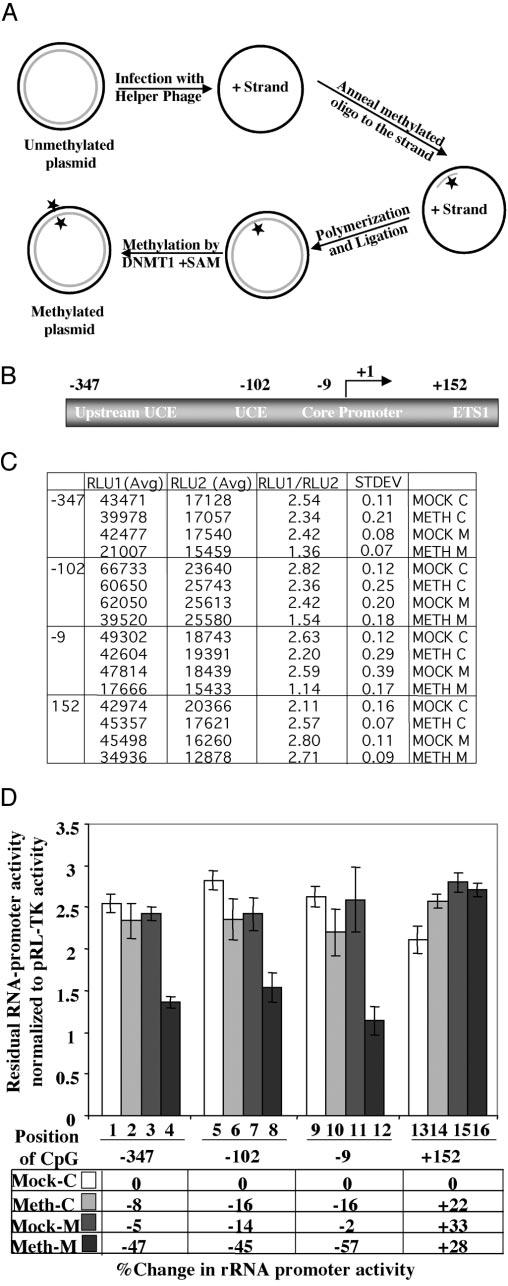
A, schematic depiction of site-specific methylation technique. B, location of CpGs on human rRNA gene subjected to site-specific methylation to study their effect on the promoter activity. These sites were selected because of the ability to generate primers spanning these regions. The construction of pHrD-IRES-Luc methylated at specific sites is described under “Materials and Methods.” C and D, plasmids (500 ng) methylated in both strands (Meth M), methylated in the (−)-strand (Mock M), or nonmethylated controls (Mock C and Meth C) along with pRL-TK (50 ng) were transfected into 2 × 105 HepG2 cells, and both firefly (RLU1) and Renilla (RLU2) luciferase activities were measured 24 h post-transfection using the Dual Luciferase Assay kit. The data of a representative experiment are tabulated in C and graphically represented in D.
Methyl-CpG-binding Protein MBD2 Specifically Represses Methylated rRNA Promoter Activity in HepG2 Cells, whereas MBD1, MBD3, and MeCP2 Inhibit the Promoter Activity Irrespective of Its Methylation Status
Methylation density-dependent inhibition of rRNA promoter activity implicates the role of MBDs in the repression of the methylated rRNA promoter. Transcription of rRNA precursor; its processing to mature 28 S, 18 S, and 5.8 S rRNAs; and their assembly into ribosomal subunits occur in the nucleolus. As a first step to explore the role of the MBDs in rRNA gene transcription in humans, we determined whether any of these MBDs are localized in the nucleoli. To address this issue, we studied co-localization of MBDs with nucleolin, a nucleolar marker protein (45). As expected, all of the MBDs tested are localized in the nucleus as demonstrated by their overlap with 4′,6-diamidino-2-phenylindole-stained nuclei (Fig. 5, panels 2 and 4). Merging of MBD signals at two spots (panel 3) with that of nucleolin indicated that MBDs are localized also in the nucleolus. These results provided the impetus to explore the role of MBDs in rRNA expression.
Fig. 5. Colocalization of methyl-binding proteins with nucleolin in HepG2 cells.
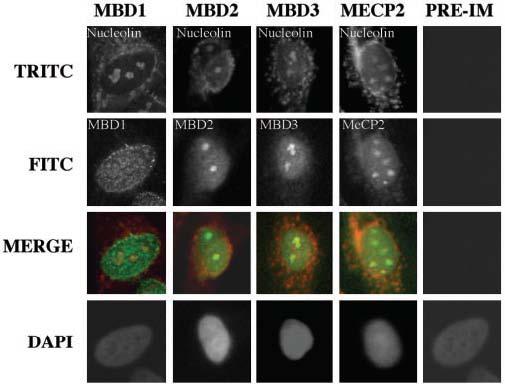
Co-localization studies were performed with antibodies specific for methyl-binding proteins raised in rabbit (except MBD2, which was raised in sheep) and monoclonal antibody (C23) against nucleolin, a nucleolus-specific protein. MBDs were recognized by fluorescein isothiocyanate-conjugated secondary antibody (green fluorescence), and nucleolin was recognized by TRITC-conjugated secondary antibody (red fluorescence). The yellow signal indicates co-localization of MBDs with nucleolin.
Next, we co-transfected the methylated (M.HhaI) or mock-methylated rRNA-promoter in pIRES-Luc into HepG2 cells, along with the expression vectors encoding different MBDs. Overexpression of MBDs in HepG2 cells was confirmed by Western blot analysis with antibodies specific for MBD1 to -3 and MeCP2. These antibodies were raised against C-terminal recombinant proteins that lack highly homologous N-terminal MBD domains and do not cross-react (37, 38). The results showed that MBD2 is the most abundant among these MBDs in HepG2 cells, and its level increased 2–3-fold after overexpression (Fig. 6A). In contrast, endogenous levels of MBD1, MBD3, and MeCP2 are low and increased at least 5–10-fold upon overexpression. Five different alternatively spliced variants of MBD1 have been identified in different mammalian cells (46). The endogenous variants of MBD1 expressed in HepG2 cells are ∼55 and 60 kDa, whereas that of the overexpressed MBD1 is ∼65 kDa. Overexpressed MeCP2 generated two polypeptides of ∼75 and 66 kDa detected by MeCP2-specific antibody. The lower polypeptide may arise either due to proteolysis or initiation from an internal ATG site. Overexpressed MBD3 co-migrated with the larger of the two closely migrating endogenous polypeptides.
Fig. 6.
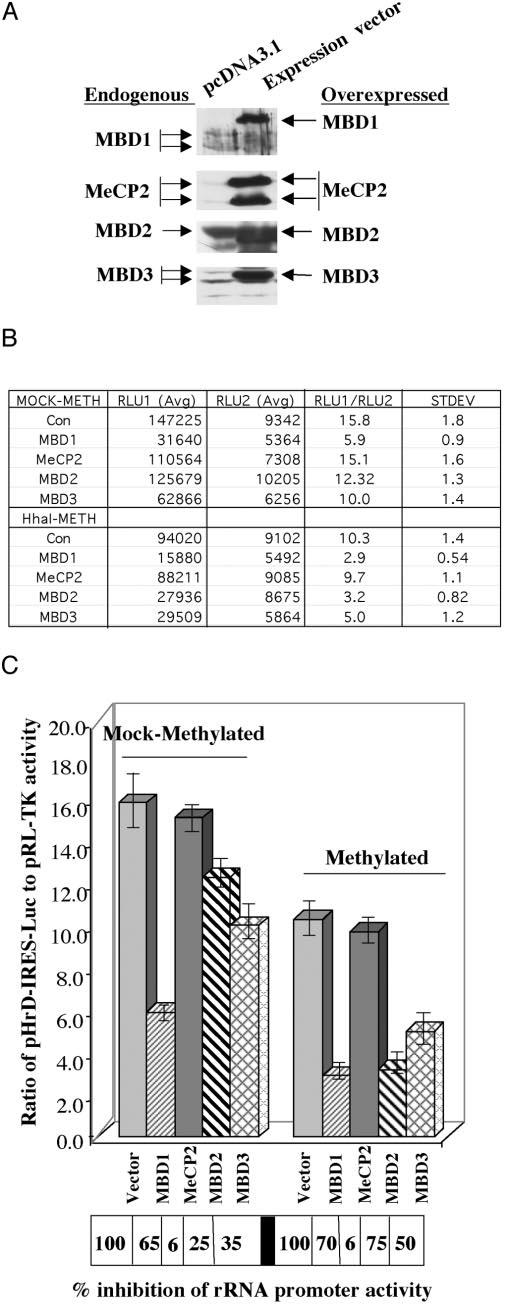
A, Western blot analysis of whole cell extracts overexpressing different MBDs. HepG2 cells were transfected with 4.0 μg of expression vectors harboring cDNA for specific MBDs or empty vector, and the whole cell extract (200 μg of protein) prepared 48 h post-transfection was subjected to Western blot analysis with specific antibodies. B and C, effect of overexpression of MBDs on rRNA promoter activity. HepG2 cells were cotransfected with pHrD-IRES-Luc with (4 μg) of the empty vector or pcMBD1, pcMBD2, pcMBD3, or pcMeCP2. After 48 h of transfection, firefly luciferase activity was measured in the cell extracts. Results are represented as pHrD-IRES-Luc (RLU2)/pRL-TK (RLU1) activity. B, average values of the luciferase activities and the ratio of pHrD-IRES-Luc to pRL-TK from each set of transfection are presented in the table. C, the left panel depicts cotransfection experiments with mock-methylated pHrD-IRES-Luc, whereas the right panel shows the results from cotransfection experiments using methylated (M.HhaI) pHrD-IRES-Luc.
We then determined the effect of MBDs on rRNA promoter activity 48 h post-transfection in HepG2 cells. MBD2 specifically suppressed the activity of the methylated (M.HhaI) promoter (75%) compared with unmethylated promoter (25%). In contrast, MBD1 had a profound repressive effect on the activity of both methylated (70%) and mock-methylated rRNA promoter (65%) (Fig. 6, B and C). A relatively small but reproducible repression (35–40%) of both promoters was observed in cells overexpressing MBD3. On the other hand, overexpression of MeCP2 had no significant effect on the rRNA promoter activity. This experiment was repeated at least three times with different batches of cells, and each transfection reaction was performed in triplicate. These results demonstrate that methyl-CpG-binding proteins indeed differentially modulate human rRNA promoter-driven reporter activity.
ChIP Assay Demonstrates Association of MBD2 Specifically with the Endogenous Methylated rRNA Promoter in HepG2 Cells
To confirm that the inhibitory effect of MBDs indeed occurs in vivo in the chromatin context, as observed in transient transfection assay, we analyzed the association of the MBDs with endogenous rRNA promoter by a ChIP assay. In the first step, formaldehyde cross-linked chromatin from growing HepG2 cells was immunoprecipitated with antibodies against specific MBDs. To distinguish between the methylated and unmethylated promoter, the input and ChIP-DNAs were digested with the isoschizomers MspI (methylation-insensitive) or HpaII (methylation-sensitive). The human rRNA promoter was then amplified from the undigested and digested DNAs. The region of the rRNA promoter amplified by the promoter-specific primers harbors four HpaII/MspI sites (Fig. 7A). The HpaII-resistant PCR product generated from the input DNA measures the level of the rRNA promoter methylated at all four sites in HepG2 cells, whereas the difference between mock-digested and HpaII-digested signal reflects the level of the rRNA promoters unmethylated in at least one of the four HpaII sites. Similarly, the HpaII-resistant PCR product obtained from the DNA immunoprecipitated by antibody for a specific MBD indicates its association with the rRNA promoter methylated at all four sites in HepG2 cells. The difference in the signal between the amplified product generated from the undigested and HpaII-digested DNA measures the association of the MBD with the rRNA promoter unmethylated at least in one of the four HpaII sites. As expected, the input and ChIP DNAs were completely digested with the methylation-insensitive enzyme MspI (3, 6, 9, 12, 15, 18, 21, 24). Quantification of the signal from the input DNA showed methylation of approximately half of the 300–400 copies of rRNA promoters at all four HpaII sites within the amplified region (Fig. 7A) in HepG2 cells (Fig. 7B, lanes 1, 2, 4, and 5). All MBDs were associated with both methylated and unmethylated rRNA promoters, albeit to different extents (lanes 13 and 14, lanes 16 and 17, lanes 19 and 20, and lanes 22 and 23). The results suggest that MBD2 is predominantly associated with the methylated promoter, since the rRNA promoter immunoprecipitated by anti-MBD2 antibody is mostly HpaII-resistant (Fig. 7B (lanes 19 and 20), C, and D). In contrast, MBD3 bound to both promoters equally well (Fig. 7, B (lanes 16 and 17), C, and D). The amount of the 32P-labeled, amplified rRNA promoter significantly decreased after HpaII digestion of the DNA immunoprecipitated with MBD1 (Fig. 7, B (lanes 22 and 23), C, and D), MBD3 (Fig. 7, B (lanes 16 and 17), C, and D) and MeCP2 (Fig. 7, B (lanes 13 and 14), C, and D) antibodies. Quantitative analysis demonstrated that MBD1 and MeCP2 have higher affinity for the methylated rRNA promoter, whereas MBD3 bound to both promoters equally well (Fig. 7, C and D). As expected, acetylated histone H4 was predominantly associated with the unmethylated promoter (Fig. 7B, lanes 10 and 11). As a control, we also amplified the promoter region of the GAPDH gene, a highly expressed, unmethylated, single copy gene, from the input and ChIP DNA. The amplicon of GAPDH harbors two HpaII sites. Amplification of GAPDH from the mock-digested, but not the HpaII-digested input DNA confirmed that this promoter is indeed methylation-free in HepG2 cells. Among the proteins tested, only acetylated histone H4 and MeCP2 were associated with the GAPDH promoter (Fig. 7B, lanes 10 and 13, respectively). The interaction of MeCP2 with unmethylated rRNA or GAPDH promoter is probably mediated through a nonspecific DNA binding domain that resides in the transcriptional repressor domain located at the C terminus of MeCP2 (47). The lack of association of MBD2 with the unmethylated GAPDH promoter further reinforces the notion that it is specifically targeted to the methylated promoters. This experiment was performed at least twice with different chromatin preparations, and the results were reproducible. These results demonstrate the association of MBDs with the rRNA promoter in the chromatin context and correlate well with their inhibitory effect on transcriptional activity of the promoter.
Fig. 7. Analysis of methyl-binding protein association with methylated/unmethylated human rRNA promoter by ChIP analysis.
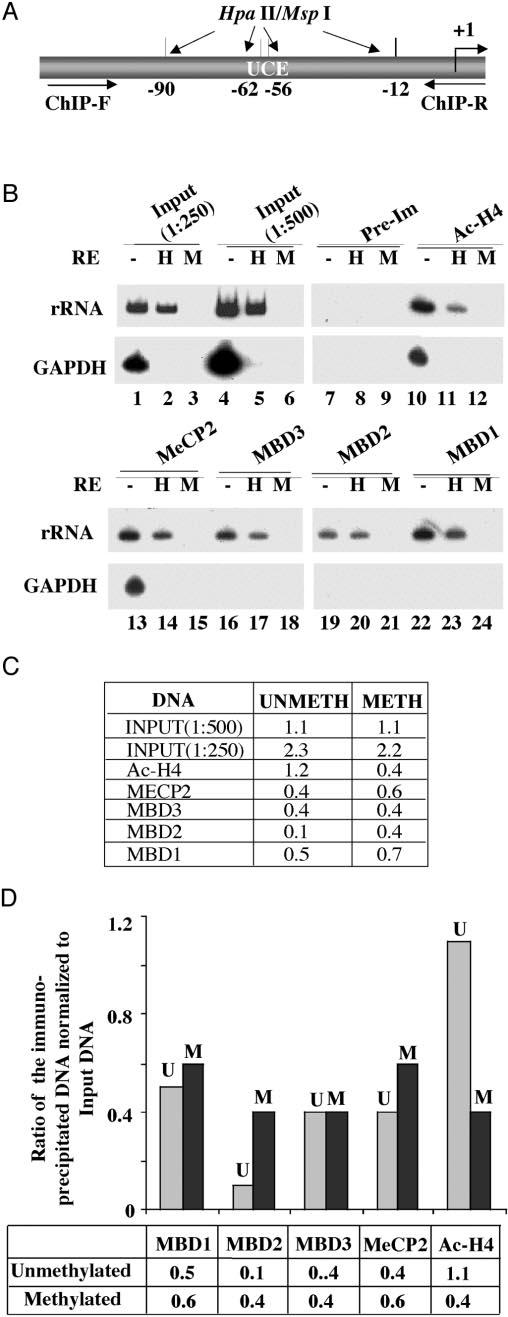
A, schematic presentation of the rRNA promoter region amplified and the locations of HpaII sites. B, formaldehyde cross-linked chromatin was precleared and immunoprecipitated overnight with antisera specific for MBDs, acetylated histone H3, or preimmune sera. The immune complexes were pulled down by protein A/G beads, washed with different buffers, eluted, and de-cross-linked. DNAs pulled down by different antibodies as well as input DNA were divided into three identical fractions that were either mock-digested or digested with HpaII (H) or MspI (M). An aliquot of the product was subjected to semiquantitative PCR with 32P-labeled forward primer specific for rRNA or GAPDH promoters. The reaction products were separated on polyacrylamide (8% for rRNA and 6% for GAPDH), and the dried gel was subjected to autoradiography (2-h exposure for rRNA and overnight exposure for GAPDH) and PhosphorImager analysis. C and D, quantitative analysis and graphical representation of the association of different MBDs with rDNA promoter. The Volume Analysis program of the ImageQuant software was used to quantify the 32P signal. Association with methylated promoter = HpaII signal in ChIP DNA/HpaII signal in input (1:500 dilution). Association with the unmethylated promoter = signal in undigested minus signal in HpaII-digested ChIP DNA/input signal in undigested minus HpaII-digested DNA (1:500 dilution). U and M, methylated and unmethylated rRNA promoters, respectively.
DISCUSSION
The molecular mechanisms of the methylation-mediated alteration in gene expression associated with neoplastic transformation have not been completely elucidated. Multistage hepatocarcinogenesis is associated with regional hypermethylation of tumor suppressor genes (e.g. hMLH1 (48), RASSF1A (49), P16 (50), O-6-MGMT (51), and PTPRO (52)) and genome-wide hypomethylation of repetitive elements (53) and oncogenes (e.g. RAS (54) and C-MYC (52)). Recent studies with Dnmt1-deficient mice have shown that global hypomethylation causes propensity toward tumorigenesis (55). A hallmark of cancer cells is the augmented transcription of rRNA genes to meet the demand for increased production of ribosomes and for protein synthesis. We hypothesized that the higher level of rRNA synthesis in the tumors could also result from hypomethylation of rRNA promoters in addition to up-regulation of pol I-specific transcription factor(s) (e.g. upstream binding factor (56)). To our knowledge, the present study is the first report on the analysis of the methylation status of each CpG in the promoter region of rRNA genes in human primary tumors and its relationship to the rRNA promoter activity. Comparison of the assembled genomic sequences from seven different species has shown that the diversity among animals lies not in the protein coding region but rather in the cis regulatory elements, implicating more complex regulation of gene expression in higher organisms (57). The existence of CpG island in human, but not in rodent, rRNA promoters suggested that the underlying molecular mechanism of methylation-mediated silencing among these two species may be distinct. This prompted us to explore whether the CpG island, located on the rRNA promoter, is differentially methylated in human hepatocellular carcinomas and the potential role of its methylation on rRNA expression. Indeed, the present data have revealed an inverse relationship between human rRNA promoter activity and CpG methylation status. Bisulfite genomic sequencing also demonstrated polymorphic variations in this epigenetic modification in the liver DNA among the six individuals analyzed. We have observed polymorphic variations in the liver DNA among the rats of the same strain (Fisher 344) at the NotI site of a CpG island on a protein-tyrosine phosphatase (PTPRO) promoter (52). Like the rRNA promoter, the PTPRO promoter also showed tumor-specific methylation (52).
The significant hypomethylation specifically in the UCE of the rRNA promoter merits some explanation. This region is probably less accessible to DNA methyltransferases during replication in the rapidly proliferating tumor tissues. Alternatively, CpGs in the UCE region may undergo active demethylation by MBD2b, a variant of MBD2 that has been reported to exhibit demethylase activity in vitro and in vivo (58). In this context, it is noteworthy that differential methylation of rRNA repeats has been observed in other diseases, such as ATRX (59) and Werner syndrome (60). In ATRX syndrome, an X-linked disorder leading to α-thalassaemia, rRNA arrays are hypomethylated due to mutation in the ATRX gene (59). On the contrary, rRNA repeats are hypermethylated in fibroblasts from patients with Werner syndrome (60). These observations show that methylation of rRNA repeats is tightly regulated in normal cells.
In mouse cells, pol I transcription could be inhibited by methylation of a single CpG located at −133 in the UCE region. This forms a repressive chromatin structure inhibiting access of the key transcription factor UBF (61). The relatively larger number of CpGs in the human rRNA promoter region compared with the rodent, promoter prompted us to investigate whether methylation density plays any role in repression of rRNA gene transcription. To address this issue, we generated a rRNA-promoter-luciferase reporter plasmid where the Kozak sequence was replaced by IRES to facilitate efficient translation of pol I-transcribed luciferase mRNA. Minimal luciferase activity in mouse (Hepa) cells compared with that in human (HepG2) cells confirmed pol I-driven transcription of the reporter gene. The same strategy was used earlier to link pol I promoter to chloramphenicol acetyltransferase reporter (43). Inverse correlation of pol I promoter activity with methylation density (Fig. 3) suggests that the mechanism of methylation-mediated silencing in humans may be distinct from that in mice. We cannot, however, rule out the potential role of MeCP2 that can bind to a symmetrically methylated CpG (31) in silencing the rRNA promoter in mice, since methylation at −133 does not directly impede upstream binding factor binding (61).
To study the effect of methylation at a single site within the promoter or coding region, we developed a site-specific methylation technique by modifying site-directed mutagenesis protocol. Here, a combination of a methylated oligonucleotide (in place of a mutant oligonucleotide) and hemimethylase activity of Dnmt1 generated a plasmid DNA with a single site methylated in one or both strands (Fig. 4A). Transfection studies using these plasmids have shown that symmetrical methylation at single sites located within the core promoter, UCE, or sequence upstream of UCE, but not in the transcribed (ETS1) region, represses rRNA promoter activity. Inhibition of promoter activity upon methylation of the cytosine residue at −347 indicates that methylation at sites outside of upstream binding factor binding site can modulate rRNA promoter activity, probably by recruiting MBDs. The unabated activity of the hemimethylated rRNA promoter also indicates the involvement of MBDs in the repression of RNA genes, since recruitment of MBDs to the DNA requires symmetrically methylated CpGs (Fig. 4B). The technically demanding site-specific methylation analysis, used in the present study, provided the most conclusive answer regarding the effect of methylation at a single site on gene transcription.
Although all MBD proteins have highly homologous methyl-CpG binding domains, immunolocalization studies demonstrated that in vivo they are targeted to different regions of the genome by yet unidentified mechanisms. Co-localization of MBDs with nucleolin implicated their role in rRNA expression. We used two approaches to identify the MBDs regulating the rRNA promoter: (i) transient overexpression and its effect on rRNA-luciferase activity and (ii) ChIP assay to identify their interactions with the endogenous rRNA promoter. Among these MBDs, MBD2 specifically repressed the methylated rRNA promoter activity, whereas MeCP2 inhibited promoter activity irrespective of its methylation status only when expressed at a high level (data not shown). MBD1 also repressed the rRNA promoter irrespective of its methylation status. This observation is in agreement with its ability to bind both methylated and unmethylated DNA (26, 46). The ChIP data corroborate well the effect of MBDs on rRNA promoter activity in transient transfection assays. MBDs associate with a variety of corepressors that include mSin3a, histone deacetylases, and histone methyltransferases (29, 62, 63). These repressor-corepressor complexes are responsible for the ultimate suppression of pol II-directed gene expression. Identification of the corepressors that are recruited by different MBDs to repress rRNA gene expression will be of considerable interest. Further studies are needed to address this issue. Nevertheless, the present study has clearly demonstrated hypomethylation of the rRNA promoter in human hepatocellular carcinomas and elucidated the role of promoter methylation in the ribosomal RNA gene expression and the potential role of the methyl-CpG-binding protein MBD2 in the methylation-mediated silencing of the rRNA promoter.
Acknowledgments
We thank Drs. Adrian Bird and Brian Hendrich for MBD1, MeCP2, and MBD2 cDNAs, respectively, Ram Reddy for anti-nucleolin antibody, Arthur Burghes and Jill Rafael-Fortney for generosity with the use of the fluorescence microscope, and Dennis Summers for excellent technical assistance.
Footnotes
This work was supported, in part, by National Institutes of Health Grants ES 10874, CA 81024, and CA 86978.
The abbreviations used are: UCE, upstream control element; ETS1, external transcribed spacer region 1; pol I, II, and III, RNA polymerase I, II, and III, respectively; MBD, methyl-CpG-binding protein; DNMT, DNA methyltransferase; ChIP, chromatin immunoprecipitation; AdoMet, S-adenosyl-l-methionine; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IRES, internal ribosome entry site; TRITC, tetramethylrhodamine isothiocyanate; HCC, hepatocellular carcinoma.
REFERENCES
- 1.McStay B, Paule M, Schultz MC, Willis I, Pikaard CS. Gene Expr. 2002;10:263–269. doi: 10.3727/000000002783992415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leary DJ, Huang S. FEBS Lett. 2001;509:145–150. doi: 10.1016/s0014-5793(01)03143-x. [DOI] [PubMed] [Google Scholar]
- 3.Jacob ST. Biochem. J. 1995;306:617–626. doi: 10.1042/bj3060617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hannan KM, Hannan RD, Rothblum LI. Front. Biosci. 1998;3:376–398. doi: 10.2741/a282. [DOI] [PubMed] [Google Scholar]
- 5.Grummt I. Prog. Nucleic Acids Res. Mol. Biol. 1999;62:109–154. doi: 10.1016/s0079-6603(08)60506-1. [DOI] [PubMed] [Google Scholar]
- 6.Paule MR, White RJ. Nucleic Acids Res. 2000;28:1283–1298. doi: 10.1093/nar/28.6.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heix J, Grummt I. Curr. Opin. Genet. Dev. 1995;5:652–656. doi: 10.1016/0959-437x(95)80035-2. [DOI] [PubMed] [Google Scholar]
- 8.Ghosh AK, Niu H, Jacob ST. Biochem. Biophys. Res. Commun. 1996;225:890–895. doi: 10.1006/bbrc.1996.1268. [DOI] [PubMed] [Google Scholar]
- 9.Reeder RH. Cell. 1984;38:349–351. doi: 10.1016/0092-8674(84)90489-6. [DOI] [PubMed] [Google Scholar]
- 10.Bird AP, Wolffe AP. Cell. 1999;99:451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- 11.Baylin SB, Esteller M, Rountree MR, Bachman KE, Schuebel K, Herman JG. Hum. Mol. Genet. 2001;10:687–692. doi: 10.1093/hmg/10.7.687. [DOI] [PubMed] [Google Scholar]
- 12.Wade PA. BioEssays. 2001;23:1131–1137. doi: 10.1002/bies.10008. [DOI] [PubMed] [Google Scholar]
- 13.Okano M, Bell DW, Haber DA, Li E. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 14.Jaenisch R, Bird A. Nat. Genet. 2003;33:245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 15.Robertson KD, Wolffe AP. Nat. Rev. Genet. 2000;1:11–19. doi: 10.1038/35049533. [DOI] [PubMed] [Google Scholar]
- 16.Jones PA, Baylin SB. Nat. Rev. Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 17.Plass C. Hum. Mol. Genet. 2002;11:2479–2488. doi: 10.1093/hmg/11.20.2479. [DOI] [PubMed] [Google Scholar]
- 18.Bestor TH. Hum. Mol. Genet. 2000;9:2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 19.Jeltsch A. ChemBioChem. 2002;3:274–293. doi: 10.1002/1439-7633(20020402)3:4<274::AID-CBIC274>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 20.Hansen RS, Wijmenga C, Luo P, Stanek AM, Canfield TK, Weemaes CM, Gartler SM. Proc. Natl. Acad. Sci. U. S. A. 1999;96:14412–14417. doi: 10.1073/pnas.96.25.14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ehrlich M, Buchanan KL, Tsien F, Jiang G, Sun B, Uicker W, Weemaes CM, Smeets D, Sperling K, Belohradsky BH, Tommerup N, Misek DE, Rouillard JM, Kuick R, Hanash SM. Hum. Mol. Genet. 2001;10:2917–2931. doi: 10.1093/hmg/10.25.2917. [DOI] [PubMed] [Google Scholar]
- 22.Xu GL, Bestor TH, Bourc'his D, Hsieh CL, Tommerup N, Bugge M, Hulten M, Qu X, Russo JJ, Viegas-Pequignot E. Nature. 1999;402:187–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- 23.Zhu WG, Otterson GA. Curr. Med. Chem. Anti-cancer Agents. 2003;3:187–199. doi: 10.2174/1568011033482440. [DOI] [PubMed] [Google Scholar]
- 24.Cheng JC, Matsen CB, Gonzales FA, Ye W, Greer S, Marquez VE, Jones PA, Selker EU. J. Natl. Cancer Inst. 2003;95:399–409. doi: 10.1093/jnci/95.5.399. [DOI] [PubMed] [Google Scholar]
- 25.Holliday R. Cancer Surv. 1996;28:103–115. [PubMed] [Google Scholar]
- 26.Hendrich B, Bird A. Mol. Cell Biol. 1998;18:6538–6547. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. Nature. 1999;401:301–304. doi: 10.1038/45843. [DOI] [PubMed] [Google Scholar]
- 28.Hendrich B, Bird A. Curr. Top. Microbiol. Immunol. 2000;249:55–74. doi: 10.1007/978-3-642-59696-4_4. [DOI] [PubMed] [Google Scholar]
- 29.Wade PA. Oncogene. 2001;20:3166–3173. doi: 10.1038/sj.onc.1204340. [DOI] [PubMed] [Google Scholar]
- 30.Prokhortchouk A, Hendrich B, Jorgensen H, Ruzov A, Wilm M, Georgiev G, Bird A, Prokhortchouk E. Genes Dev. 2001;15:1613–1618. doi: 10.1101/gad.198501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nan X, Bird A. Brain Dev. 2001;23(suppl):S32–S37. doi: 10.1016/s0387-7604(01)00333-3. [DOI] [PubMed] [Google Scholar]
- 32.Zhao X, Ueba T, Christie BR, Barkho B, McConnell MJ, Nakashima K, Lein ES, Eadie BD, Willhoite AR, Muotri AR, Summers RG, Chun J, Lee KF, Gage FH. Proc. Natl. Acad. Sci. U. S. A. 2003;100:6777–6782. doi: 10.1073/pnas.1131928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Millar CB, Guy J, Sansom OJ, Selfridge J, MacDougall E, Hendrich B, Keightley PD, Bishop SM, Clarke AR, Bird A. Science. 2002;297:403–405. doi: 10.1126/science.1073354. [DOI] [PubMed] [Google Scholar]
- 34.Ghosh AK, Kermekchiev M, Jacob ST. Gene (Amst.) 1994;141:271–275. doi: 10.1016/0378-1119(94)90584-3. [DOI] [PubMed] [Google Scholar]
- 35.Majumder S, Ghoshal K, Gronostajski RM, Jacob ST. Gene Expr. 2001;9:203–215. doi: 10.3727/000000001783992588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Majumder S, Ghoshal K, Li Z, Jacob ST. J. Biol. Chem. 1999;274:28584–28589. doi: 10.1074/jbc.274.40.28584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghoshal K, Datta J, Majumder S, Bai S, Dong X, Parthun M, Jacob ST. Mol. Cell Biol. 2002;22:8302–8319. doi: 10.1128/MCB.22.23.8302-8319.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Majumder S, Ghoshal K, Datta J, Bai S, Dong X, Quan N, Plass C, Jacob ST. J. Biol. Chem. 2002;277:16048–16058. doi: 10.1074/jbc.M111662200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Weinmann AS, Bartley SM, Zhang T, Zhang MQ, Farnham PJ. Mol. Cell Biol. 2001;21:6820–6832. doi: 10.1128/MCB.21.20.6820-6832.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clark SJ, Harrison J, Paul CL, Frommer M. Nucleic Acids Res. 1994;22:2990–2997. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghoshal K, Majumder S, Jacob ST. Methods Enzymol. 2002;353:476–486. doi: 10.1016/s0076-6879(02)53070-6. [DOI] [PubMed] [Google Scholar]
- 42.Kozak M. Proc. Natl. Acad. Sci. U. S. A. 1986;83:2850–2854. doi: 10.1073/pnas.83.9.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palmer TD, Miller AD, Reeder RH, McStay B. Nucleic Acids Res. 1993;21:3451–3457. doi: 10.1093/nar/21.15.3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heix J, Zomerdijk JC, Ravanpay A, Tjian R, Grummt I. Proc. Natl. Acad. Sci. U. S. A. 1997;94:1733–1738. doi: 10.1073/pnas.94.5.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valdez BC, Henning D, Busch RK, Srivastava M, Busch H. Mol. Immunol. 1995;32:1207–1213. doi: 10.1016/0161-5890(95)00093-3. [DOI] [PubMed] [Google Scholar]
- 46.Nakao M, Matsui S, Yamamoto S, Okumura K, Shirakawa M, Fujita N. Brain Dev. 2001;23:S174–176. doi: 10.1016/s0387-7604(01)00348-5. [DOI] [PubMed] [Google Scholar]
- 47.Cross SH, Charlton JA, Nan X, Bird AP. Nat. Genet. 1994;6:236–244. doi: 10.1038/ng0394-236. [DOI] [PubMed] [Google Scholar]
- 48.Matsukura S, Soejima H, Nakagawachi T, Yakushiji H, Ogawa A, Fukuhara M, Miyazaki K, Nakabeppu Y, Sekiguchi M, Mukai T. Br. J. Cancer. 2003;88:521–529. doi: 10.1038/sj.bjc.6600743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schagdarsurengin U, Wilkens L, Steinemann D, Flemming P, Kreipe HH, Pfeifer GP, Schlegelberger B, Dammann R. Oncogene. 2003;22:1866–1871. doi: 10.1038/sj.onc.1206338. [DOI] [PubMed] [Google Scholar]
- 50.Pogribny IP, James SJ. Cancer Lett. 2002;187:69–75. doi: 10.1016/s0304-3835(02)00408-1. [DOI] [PubMed] [Google Scholar]
- 51.Yu J, Ni M, Xu J, Zhang H, Gao B, Gu J, Chen J, Zhang L, Wu M, Zhen S, Zhu J. BMC Cancer. 2002;2:29–42. doi: 10.1186/1471-2407-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Motiwala T, Ghoshal K, Das A, Majumder S, Weichenhan D, Wu YZ, Holman K, James SJ, Jacob ST, Plass C. Oncogene. 2003;22:6319–6331. doi: 10.1038/sj.onc.1206750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin CH, Hsieh SY, Sheen IS, Lee WC, Chen TC, Shyu WC, Liaw YF. Cancer Res. 2001;61:4238–4243. [PubMed] [Google Scholar]
- 54.Zapisek WF, Cronin GM, Lyn-Cook BD, Poirier LA. Carcinogenesis. 1992;13:1869–1872. doi: 10.1093/carcin/13.10.1869. [DOI] [PubMed] [Google Scholar]
- 55.Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R. Science. 2003;300:489–492. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 56.Huang R, Wu T, Xu L, Liu A, Ji Y, Hu G. FASEB J. 2002;16:293–301. doi: 10.1096/fj.01-0687com. [DOI] [PubMed] [Google Scholar]
- 57.Levine M, Tjian R. Nature. 2003;424:147–151. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- 58.Detich N, Bovenzi V, Szyf M. J. Biol. Chem. 2003;278:27586–27592. doi: 10.1074/jbc.M303740200. [DOI] [PubMed] [Google Scholar]
- 59.Gibbons RJ, McDowell TL, Raman S, O'Rourke DM, Garrick D, Ayyub H, Higgs DR. Nat. Genet. 2000;24:368–371. doi: 10.1038/74191. [DOI] [PubMed] [Google Scholar]
- 60.Machwe A, Orren DK, Bohr VA. FASEB J. 2000;14:1715–1724. doi: 10.1096/fj.99-0926com. [DOI] [PubMed] [Google Scholar]
- 61.Santoro R, Grummt I. Mol. Cell. 2001;8:719–725. doi: 10.1016/s1097-2765(01)00317-3. [DOI] [PubMed] [Google Scholar]
- 62.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 63.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Nat. Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]