

Abstract
Translocation through the extracellular vestibule and binding of leucine in the leucine transporter (LeuT) have been studied with molecular dynamics simulations. More than 0.1 μs of all-atom molecular dynamics simulations have been performed on different combinations of LeuT, bound substrate, and bound structural Na+ ions to describe molecular events involved in substrate binding and in the formation of the occluded state and to investigate the dynamics of this state. Three structural features are found to be directly involved in the initial steps of leucine transport: a Na+ ion directly coordinated to leucine (Na-1), two aromatic residues closing the binding site toward the extracellular vestibule (Tyr-108 and Phe-253), and a salt bridge in the extracellular vestibule (Arg-30 and Asp-404). These features account for observed differences between simulations of LeuT with and without bound substrate and for a possible pathway for leucine binding and thereby formation of the occluded LeuT binding site.
Introduction
Neurotransmitter transporters have been attracting increasing attention because of their involvement in numerous physiological processes. Of particular importance are the monoamine (serotonin, dopamine, and norepinephrine) transporters, which are members of the neurotransmitter sodium symporter (NSS) family and responsible for reuptake of biogenic monoamines after signaling in the brain. They are involved in psychological disorders such as depression, obsessive-compulsive disorders, Parkinson's disease, and epilepsy (1–4). The transport of monoamines by NSSs is driven by the ionic gradient across the cellular membrane. All depend on symport of Na+, but some also symport Cl−, and a few antiport K+ (5). Experimental studies indicating different ion/substrate stoichiometries in the NSSs (5) suggest that, despite a high degree of similarity, the transport mechanism in NSSs is not entirely uniform. The molecular details of the transport mechanism and substrate/inhibitor selectivity are so far poorly understood, primarily because of lack of detailed structural information. Efforts were previously put into constructing models of the serotonin and dopamine transporter binding sites from structures of lactose permease and a Na+/H+ antiporter (6–9) even though these proteins belong to different transporter families. Other approaches have included developing ligand-based descriptors for the binding sites through, e.g., QSAR studies (10–14).
The first x-ray structure of the bacterial leucine transporter from Aquifex aeolicus (LeuT) (15) was solved in 2005. Another five structures, including bound tricyclic antidepressants (TCAs), have recently been published as well (16,17). LeuT belongs to the same family of NSSs as the monoamine transporters and, thus, presents a highly relevant model for investigation of the mechanism of transport in the family. Structural differences between LeuT and mammalian NSSs are primarily located in the loop regions, generally longer in the latter. The overall homology between human NSS proteins and LeuT is ∼20–25%, but up to ∼50% similarity can be detected in the transmembrane (TM) segments, clearly indicating the relevance of the mechanism of transport in LeuT to neurotransmitter transport in NSSs. Because of the high similarity, the structure of LeuT has recently been employed to improve and refine the amino acid sequence alignment of NSSs to LeuT (18) and to build homology models of the serotonin, dopamine, and norepinephrine transporters to study substrate and antidepressant binding and selectivity (19–24,72), providing new opportunities for the field of neurotransmitter research.
The crystal structure of LeuT (15) revealed a dimer with a crystallographic twofold axis. Each monomer has 12 TM α-helices (H1–H12) showing an inverted repeat motif between helices 1–5 and 6–10, respectively. A substrate molecule, Leu, and two structural Na+ ions (denoted Na-1 and Na-2) were bound in a closed binding site of each monomer. Two partially unwound helices, H1 and H6 (the N-terminal halves are denoted H1a and H6a, and the C-terminal halves H1b and H6b, respectively), make up the binding site together with residues from H3 and H8, which are slightly kinked around the binding site (Fig. 1). Of special interest here are the unwound segments of H1 and H6, where exposed backbone amide groups might interact with both the substrate and the structural Na+ ions. Unwound helices or half-helices with a common center have been found in a number of helical membrane transporters and channels and seem to present a common structural motif of functional importance in this family of proteins (15,25–29).
Figure 1.
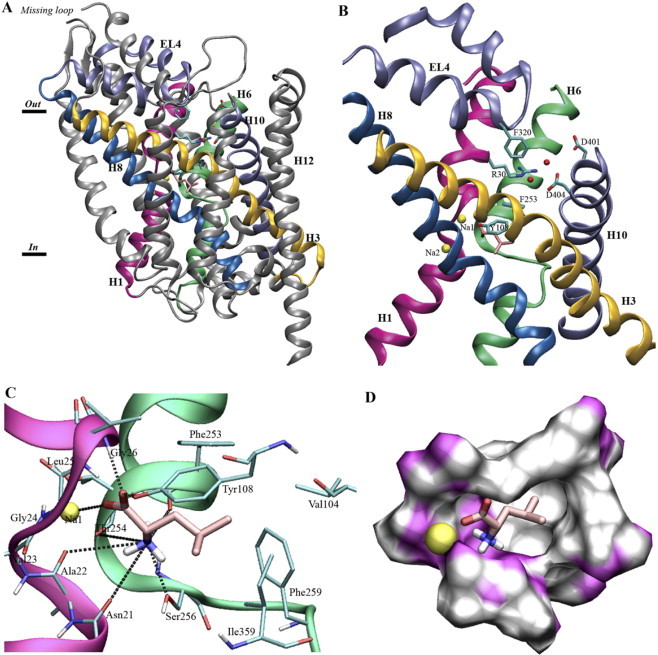
(A) The crystal structure of LeuT. The 12 TM α-helices in LeuT are bundled together in a unique spiral-like fold. (B) The two unwound helices, H1 (magenta) and H6 (green), as well as helices H3 (yellow) and H8 (blue) are important for substrate binding and transport. H10 (light blue) and the extracellular loop 4 (EL4) (light blue) provide residues close to the suspected pathway. The Arg-30–Asp-404 salt bridge is water (red balls) mediated. (C) Substrate binding in LeuT. Snapshot of Leu (pink) and interacting residues (cyan) together with Na-1 (yellow). Note how residues interacting with the substrate functional groups are located in the unwound parts of H1 (magenta) and H6 (green). Possible hydrogen bonds to Leu(N) are marked with heteroatom distance. (D) The substrate, Leu (pink), and Na-1 (yellow sphere) in a surface representation of the LeuT binding site. The front of the figure is cut away to clearly show the hydrophobic (white) and hydrophilic (magenta) duality of the binding site.
Aquifex aeolicus, the bacterium from which LeuT is isolated, is a thermophilic organism, exhibiting a strong binding affinity for Leu at room temperature (KD ≈ 20 nM) with a slow transport rate (kcat ≈ 1.2 h−1) (16). The Leu binding site is adapted to the amphiphilic structure of the substrate; highly hydrophobic regions are complemented by hydrophilic ones to optimally match the aliphatic side chain of the substrate and its charged groups, respectively (Fig. 1). A number of aromatic residues constitute an aromatic cage around the binding site with residues Tyr-108 (in H3) and Phe-253 (in H6) functioning as a “lid” covering the extracellular entrance of the binding pocket (Fig. 1). Above the aromatic lid, residues Arg-30 (in H1) and Asp-404 (in H10), positioned across the lumen from each other, form a water-mediated salt bridge. In the initial crystal structure of LeuT (15), this salt bridge and the above-mentioned aromatic lid close the binding site to the extracellular lumen, whereas the path to the intracellular side is closed by ∼20 Å of tightly packed protein residues. In the recently published structures of LeuT with TCAs bound in the extracellular vestibule, Arg-30 forms a direct (not water-mediated) salt bridge with Asp-404 (16,17). Arg-30, Asp-404, and Asp-401 are the only charged residues found in the extracellular vestibule of LeuT, suggesting their importance and possible interactions with the zwitterionic Leu substrate during transport. Indeed, mutational studies on the γ-aminobutyric acid and serotonin transporters have shown that these three residues are crucial for transport in NSSs (30,31).
The two structural Na+ ions, Na-1 and Na-2, are tightly coordinated in LeuT; Na-1 is positioned in the substrate binding site, directly coordinated by the substrate, whereas Na-2 is positioned somewhat farther away. Na-1 is found in an octahedral (six-liganded) coordination interacting with two backbone carbonyls, two side-chain amide carbonyls, and a hydroxyl group besides one of the oxygen atoms of the carboxylate group of the substrate. Na-2, which has no contact to the substrate, is bound in a trigonal bipyramidal (five-liganded) site composed of three backbone carbonyls and two hydroxyl groups. A similar picture of substrate and sodium binding is found in GltPh, an aspartate transporter from Pyrococcus horikishii, and a bacterial homolog of the mammalian glutamate transporters (25,26). The family of glutamate transporters is functionally related to the NSS family, although instead of 12, they include only 8 TM α-helices per monomer. The crystal structure of GltPh showed a trimer rather than a dimer as found for LeuT. Despite this structural difference, the substrate binding sites are organized in the same way and include free backbone carbonyl groups from unwound or half-helices and two Na+ binding sites in close proximity of the substrate (15,25,26).
Membrane transporters are generally believed to operate by a mechanism known as the alternate access model described by Jardetzky in 1966 (32). In this model, the binding of substrate triggers a structural transition between two states, an inward open and an outward open, thereby inducing the translocation of the substrate across the membrane. Kinetic studies indicate the involvement of a third and possibly a fourth state in the transport facilitated by NSSs (33,34). The recent structures of LeuT (15–17) and GltPh (25,26) directly show the involvement of a so-called “occluded” state as a third structural state in the mechanism: a transporter conformation with a closed binding site but where the main parts of the transporter lumen are still exposed to the extracellular side.
Although the substrate/ion stoichiometry has been extensively studied for different NSSs (5), the sequence of binding and release of the ions and the substrate during transport in NSSs are poorly understood. Another important question concerns the conformational changes of the transporters during the transport mechanism. Because of the static nature of x-ray crystallography, the structure does not provide direct information about possible conformational changes to other states. For example, because of lack of a structural model for the outward open state, it is unknown if major conformational changes are involved in the formation of the occluded state after the binding of substrate from the extracellular lumen, or if primarily rearrangement of amino acid side-chain groups are involved. However, for the serotonin transporter, it has been suggested, based on cysteine-scanning experiments, that formation of the occluded state does not involve major conformational changes (35), as the accessibility of the tested residues did not change much between an apotransporter and a transporter with the substrate bound.
Complementary to static structures from x-ray crystallography, molecular dynamics (MD) simulations have proven very effective in providing dynamic information on biological systems (36–40). Despite time-scale limitations, previous studies have shown the usefulness of equilibrium MD simulations in describing physiologically relevant conformational states of proteins (41–45). Many processes, however, cannot be described by equilibrium MD simulations. For example, translocation of a substrate across the protein usually follows a longer time scale than sampled in presently accessible unbiased MD simulations. Biased methods, such as steered MD (SMD (46–48)), have been developed in which an external force or moving constraint is employed to accelerate certain molecular events. The application of the method to membrane proteins has been demonstrated, e.g., for aquaporins (49,50), lactose permease (51), and the outer membrane transporter BtuB (52). Although the resulting biased trajectories do not sample perfectly equilibriated structures and processes, numerous studies have revealed that careful application of such methods can provide relevant and useful information (46–51,53,54). In modeling of membrane transporters, MD and SMD simulations have been employed to directly study or deduce individual steps involved in the transport mechanism (45,52,55).
In this article we present the results of more than 0.1 μs of simulations of an all-atom membrane-embedded model of the outward-occluded conformation of the LeuT dimer. MD simulations have been conducted with different setups, systematically exploring the effect of the presence and absence of substrate and sodium ions on LeuT dynamics and disentangling the binding of Leu in LeuT. Furthermore, pulling simulations, employing different force profiles and pulling velocities to either Leu alone or Leu and Na-1 together, were performed to study the unbinding of the substrate from the binding site and to study the formation of the open state from the occluded state, thereby revealing the differences between an outward-open and the outward-occluded state as well as proposing a likely pathway for Leu binding to LeuT. This is the first such study of the NSS transport mechanism and can thus be utilized in further studies of monoamine transporters. Results show a very tight binding of Leu in the occluded binding site, highly influenced by the presence of free polar groups in the unwound backbones of H1 and H6. We have, furthermore, identified a possible binding pathway for Leu that has been well reproduced in pulling simulations employing different pulling schemes. This pathway especially involves the aromatic residues closing the binding site and a conserved salt bridge in the extracellular vestibule of LeuT.
Methods
Model building
The 1.65 Å resolution x-ray structure of LeuT (15) was obtained from the Protein Data Bank (56) (accession code 2A65). The structure includes the protein residues 5–132 and 135–515 from one of the monomers in the dimer. Furthermore, a substrate leucine (Leu), two sodium and one chloride ions, five detergent molecules, and 210 water molecules were included in the PDB file. The detergent molecules and the chloride ion were removed. Two missing residues, Asn-133 and Ala-134 located in extracellular loop 2 (EL2), were built as follows: first two glycine residues were introduced to complete the backbone, the residues were then mutated to alanine, and finally, the asparagine side chain was built for residue 133. Residues 132–135 were minimized after each step of the modeling process. Coordinates for missing atoms of side chains and hydrogen atoms were constructed with the psfgen plugin in VMD (57) employing the CHARMM32 topology for proteins and lipids (58–61). No information is available as to whether LeuT is functional in a monomeric or a dimeric form, but it was crystallized as a dimer (15). Other studies have shown that the biogenic amine transporters are functional as dimers (62,63). The dimer was constructed by applying a crystallographic twofold symmetry and space group using program O (64) to the monomer found in the pdb-file. As a further incentive to model LeuT as a dimer, we observed that the expected usual interactions between the lipid head groups and charged and polar residues would induce a deep depression of the lipid bilayer on one side of the transporter if it were modeled as a monomer. The inclusion of the dimer in the model further allowed us to double the statistics, as two trajectories, one from each monomer, are produced for every setup employed. As described below, to further improve the statistics, some SMD simulations were performed twice. Reproducibility of the observed molecular events in repeated simulations will provide additional reliability for the obtained results.
A preequilibrated and solvated 140 × 125 Å2 membrane bilayer consisting of palmitoyl-oleoyl phosphatidylethanolamine lipids was created using the membrane plugin in VMD (57). The LeuT dimer was embedded in the bilayer by aligning the lipid head groups with a ring of charged and aromatic residues at the surface of the dimer. The lipid bilayer was large enough to provide at least 15 Å of lipid padding around the dimer along any direction. Layers of water 20 Å thick were added to the two sides of the membrane to fully solvate the lipid-protein system. The system was then neutralized with sodium and chloride ions to a physiological concentration of 0.2 M. This resulted in a total atom count of 142,405, with dimensions of 130 × 113 × 90 Å3 after equilibration (Fig. 2).
Figure 2.
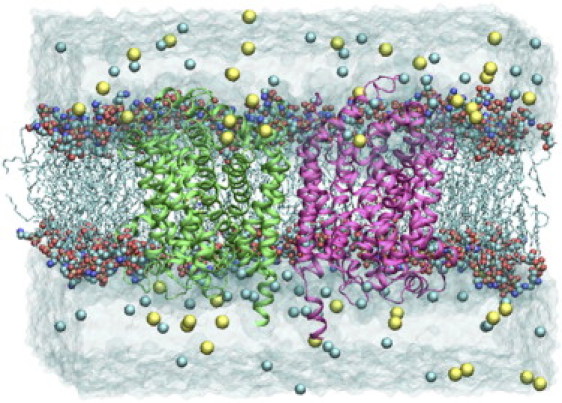
The simulated system from a side view. The two transporter monomers are depicted in green and magenta cartoons, lipid tails in cyan lines, lipid heads in CPK, water in transparent, and ions as van der Waals spheres in yellow (Na+) and cyan (Cl−). The limited interaction between the two monomers is through H11 and H12, which are not directly involved in substrate binding or transport, suggesting that the two monomers function independently.
The Leu substrate is modeled as a zwitterion, charged on both the amino and carboxylate groups. The two charged groups will be referred to as the ammonium and the carboxylate groups, respectively.
Assignment of protonation states
The titration states of ionizable amino acids (aspartate, glutamate, lysine, arginine, histidine, and tyrosine) were assigned based on a pKa calculation using the web application H++ from Virginia Technical University (65). Glutamate residues 112 (in H3), 287 (in H7), and 419 (in H10) exhibited high pKa values of 10.2, 13.9, and 23.1, respectively, and were thus modeled as neutral during the simulations. The three affected residues are all located facing other acidic residues (either an aspartate or a glutamate), presenting an opportunity for them to interact with each other. All other glutamate residues were modeled as charged. Of the seven histidine residues present in LeuT, His-74 and His-391 had high pKa values (13.2 and 7.3, respectively) and were thus modeled in their protonated form. His-7, His-377, and His-480 were modeled as neutral with the proton on Nɛ, whereas His-510 was modeled with a proton on Nδ to maximize favorable interactions with the local microenvironment. All aspartate, arginine, and lysine residues were modeled as charged, and tyrosines as neutral.
Simulation details
All simulations were performed using the CHARMM32 force field for proteins and lipids (58–60) including the CMAP corrections (61) with the MD program NAMD 2.6 (66,67). After an initial Conjugate Gradient minimization for 500 iterations, lipid tails were melted in a 500-ps NVT simulation at 310 K in which all atoms but the lipid tails were held fixed. The system was then equilibrated for a 2-ns constraint-free simulation with a time step of 1 fs. Equilibration was performed under periodic boundary conditions in the NPT ensemble employing the particle mesh Ewald algorithm (68) for calculation of long-range electrostatics. Constant temperature was achieved by employing Langevin dynamics with a damping constant of 0.1 ps−1 for melting of lipid tails and 5 ps−1 for the equilibration phase. The Langevin Piston method (69) was employed to maintain a constant pressure of 1 atm with a piston period of 100 fs and a piston decay of 50 fs. The van der Waals interactions were accounted for to a cut-off distance of 12 Å and gradually dampened by use of a switching function from 10 Å.
Production dynamics (MD and SMD) was performed in the NPT ensemble with a Langevin damping constant of 0.5 ps−1 and imposition of a constant area to the membrane patch. Coordinates for analysis were saved every picosecond. Four equilibrium MD simulations were set up to study the dynamics and stability of LeuT with or without substrate/sodium ions; setup 1 is the holo system represented by a dimer of LeuT; setup 2 is the apo transporter devoid of both substrate and the sodium ions; setup 3 is the apo transporter including sodium ions; and setup 4 included substrate but no sodium ions. Together these four simulations were used to study the dynamics of the substrate in the binding pocket as well as to deduce information about the sequence of sodium and substrate binding to the transporter.
In pulling simulations, a moving constraint is applied to the center of mass (COM) of a group of atoms. The chosen group of atoms will thus be forced to generally move along a specified direction while still being able to move freely along all other degrees of freedom (46–48). Constant-velocity SMD simulations were performed using the tclforces command in NAMD 2.6 (66,67). The force was applied to the COM of different groups of atoms (Table 1). The force constant employed was 500 pN/Å following the stiff spring protocol according to Park et al. (70) while different pulling velocities were systematically investigated (see Table 1). The system was oriented such that the applied force was along the membrane normal, i.e., along the putative transport pathway. To prevent net translation of the system that might arise as a result of the pulling force acting on the substrate, lipid phosphorus atoms as well as Cα atoms of residues Phe-51, Ile-204, and Phe-496 were constrained with a harmonic potential during the pulling simulations. In total, 14 pulling simulations were conducted to study substrate unbinding from LeuT. All performed simulations are summarized in Table 1. In setups 5–10 Leu was pulled out of the binding site at four different velocities, whereas in setups 11–18 both the substrate Leu and Na-1 were pulled simultaneously. As indicated in Table 1 several of the SMD simulations were repeated to improve the statistics at given velocities. In general, faster SMD simulations were initially used to explore the process and to identify events that might be of interest. This was then followed by increasingly slower simulations (as much as computational resources allowed) to verify the reproducibility of major events at slower pulling speeds.
Table 1.
Simulation setups reported in this study
| Setup | v/Å/ns* | t/ns† | Leu | Na-1 | Na-2 | Pulled group |
|---|---|---|---|---|---|---|
| 1 | N/A | 10.0 | + | + | + | N/A |
| 2 | N/A | 10.0 | − | − | − | N/A |
| 3 | N/A | 10.0 | − | + | + | N/A |
| 4 | N/A | 5.0 | + | − | − | N/A |
| 5 | 1.0 | 10.0 | + | + | + | Leu |
| 6 | 2.5 | 5.0 | + | + | + | Leu |
| 7 | 4.0 | 4.0 | + | + | + | Leu |
| 8 | 4.0 | 3.7 | + | + | + | Leu |
| 9 | 10.0 | 2.0 | + | + | + | Leu |
| 10 | 10.0 | 2.0 | + | + | + | Leu |
| 11 | 1.0 | 13.4 | + | + | + | Leu / Na-1 |
| 12 | 2.5 | 6.0 | + | + | + | Leu / Na-1 |
| 13 | 4.0 | 4.0 | + | + | + | Leu / Na-1 |
| 14 | 4.0 | 4.5 | + | + | + | Leu / Na-1 |
| 15 | 6.0 | 3.0 | + | + | + | Leu / Na-1 |
| 16 | 8.0 | 3.0 | + | + | + | Leu / Na-1 |
| 17 | 10.0 | 5.0 | + | + | + | Leu / Na-1 |
| 18 | 10.0 | 3.0 | + | + | + | Leu / Na-1 |
In the analysis the nomenclature 1A will refer to monomer A in setup 1. Note that some of the simulation setups are repeated under the same conditions to improve sampling.
Pulling velocity in SMD simulations.
Simulation time.
Data analysis
Analysis of the computed trajectories was performed with VMD 1.8.6 (57) and the included Tcl-scripting facility. All molecular images were rendered by VMD. The root mean-square deviation (RMSD) of protein Cα atoms in each simulation was calculated with respect to the initial minimized structure. Interaction energies were calculated via the NAMDEnergy plugin of VMD (57).
Results and discussion
Setups 1–4 were first made to study equilibrium dynamics of LeuT in the presence and absence of the Leu substrate and the two bound sodium ions. In addition to providing dynamic information about the structural stability of LeuT, these simulations might also shed light on the question of the sequence of binding of substrate and sodium ions to the transporter. From these four setups the specific binding of the substrate and the two sodium ions is studied and electrostatic interactions and the importance of the unwound helices are investigated. The binding of Leu to LeuT, a key step in the transport mechanism, is then studied by pulling simulations in setups 5–18 of the reverse unbinding event. Different pulling velocities are applied to either the COM of the substrate (setups 5–10) or to the COM of Leu and Na-1 together (setups 11–18), inducing substrate unbinding toward the extracellular lumen (see Table 1). Two different classes of setups, namely equilibrium MD simulations in 1–4 and pulling simulations in setups 5–18, are included to examine if unbinding of Leu is dependent on a simultaneous transport of Na-1 or if the two are independent of each other. Although the pulling speed in SMD simulations does not allow for the principle of microscopic reversibility to exactly apply, we expect to capture major steps and molecular events involved in initial association and binding of the substrate from the periplasm by simulating the reverse process, i.e., unbinding of the substrate from the binding pocket. In the following, specific atoms of individual amino acids are referred to as, e.g., Ala-22(O), Gly-26(N), and Ser-256(OH), respectively, for backbone amide O and N atoms and a side-chain hydroxyl group.
Protein stability and dynamics of the binding pocket
No major conformational changes of the protein backbone were observed throughout the performed MD and SMD simulations. RMSD values for LeuT Cα atoms were found to be between 1.5 Å and 2.2 Å for all modeled systems, and the overall structure of the transporter is conserved throughout (see Supplementary Material for average RMSD values for all simulations). Of particular interest are the unwound helices, which, despite their apparent structural flexibility, maintain their initial conformation and exhibit thermal fluctuations comparable to other parts of the protein and to the experimentally found B-factors in the x-ray diffraction experiment (15). Although no global protein conformational change is evident, we do observe a partial opening of the binding site in some of the MD simulations. This opening is furnished merely by rotation of the side chain (specifically χ1 and χ2 dihedral angles) of Tyr-108 and Phe-253, the two residues that form the aromatic lid of the binding site (see Figs. 3 and 4). Interestingly, there seems to be a correlation between this opening and the occupancy of the binding pocket; when Leu is present in the binding site (setup 1), these dihedral angles do not exhibit large fluctuations. Removal of either the substrate or the sodium ions, however, results in an increased flexibility of these side chains and, as a result, a partial opening of the binding site. In the SMD simulations we furthermore observe that release of Leu from the binding site only requires the aromatic lid to open, suggesting, along with the results of the MD simulations in setups 1 and 2, that no major conformational changes of the protein backbone are necessary for the initial binding of leucine in LeuT and formation of the occluded state. Because only side-chain movement is necessary for opening of the binding site, a close similarity between the outward open and occluded states is expected, a conclusion that is in line with what has been also hypothesized for the serotonin transporter (35).
Figure 3.
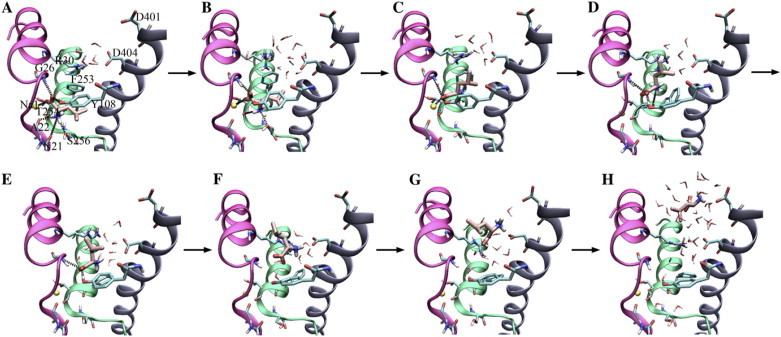
The mechanism and steps involved in substrate (un-)binding. (A) Leu bound in the closed binding site. (B) Hydrogen bonds to Asn-21 and Ala-22 are released, and the binding site starts to open. (C) Hydrogen bonds to Ser-256 and Thr-254 are released, and the Leu side chain protrudes from the aromatic lid. (D) The ionic interaction to Na-1 is broken. (E) The hydrogen bond to Phe-253 is released. (F) Hydrogen bonds to Gly-26 and Tyr-108 are released, and the substrate is free of the binding site. Instead, interactions are found to the extracellular salt bridge. (G) Leu is turning around in the vestibule as a result of ionic interactions. (H) Leu is outside LeuT and solvated.
Figure 4.
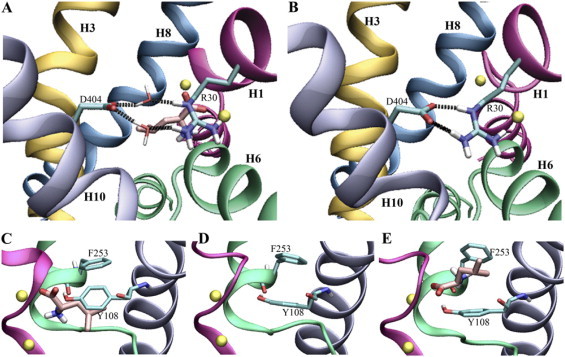
Water-mediated (A) and direct (B) interaction in the salt bridge between Arg-30 and Asp-404 in the extracellular vestibule. The aromatic lid on the extracellular side of the binding site can be either closed (C) or open (D), depending on the presence/absence of Leu. The open lid can also be seen during SMD simulation (E).
In setup 1 Leu is tightly bound to the LeuT binding site with five possible hydrogen bonds (not all present at the same time) to its ammonium group (Asn-21(O), Ala-22(O), Phe-253(O), Thr-254(O), and Ser-256(OH)) and two to its carboxylate group (Gly-26(N) and Tyr-108(OH)) as well as a salt bridge between its carboxylate group and Na-1 (Fig. 1). We also observe interactions between the Leu carboxylate group and backbone amide groups of residues Gly-24 and Leu-25 in the unwound part of H1. These interactions do not adopt a geometry that can be classified as hydrogen bonds, the amide N-H to Leu O angles are far from linear, but they do show highly favorable electrostatic interactions. The residues involved in hydrogen bonds to Leu via polar backbone groups are all found in the termini of the broken helices, H1 and H6, located near the center of the TM segment of LeuT, suggesting significance of these in substrate binding. Tyr-108 and Ser-256, which interact with the substrate through their polar side chains, are positioned in the middle of H3 and H6b, respectively. Average distances for direct interactions between Leu and the protein calculated from the 10-ns simulation of setup 1 are listed in Table 2. The observed binding of Leu in LeuT is similar to that observed for dopamine in an MD simulation of dopamine binding in a homology model of the dopamine transporter based on the structure of LeuT (23).
Table 2.
Direct interactions between Leu and the binding site of LeuT
| Interaction | Monomer A | Monomer B | Crystal |
|---|---|---|---|
| Leu(OT1)–Na-1 | 2.25 ± 0.11 | 2.24 ± 0.10 | 2.5 |
| Leu(OT1)–Gly-24(N) | 3.37 ± 0.23 | 3.82 ± 0.21 | 3.8 |
| Leu(OT1)–Leu-25(N) | 4.49 ± 0.22 | 4.55 ± 0.21 | 4.6 |
| Leu(OT1)–Gly-26(N) | 3.55 ± 0.20 | 3.60 ± 0.19 | 3.3 |
| Leu(OT2)–Gly-24(N) | 3.93 ± 0.21 | 3.96 ± 0.22 | 3.8 |
| Leu(OT2)–Leu-25(N) | 3.07 ± 0.16 | 3.08 ± 0.15 | 3.1 |
| Leu(OT2)–Gly-26(N) | 2.85 ± 0.12 | 2.83 ± 0.12 | 2.7 |
| Leu(OT2)–Tyr-108(OH) | 2.70 ± 0.12 | 2.69 ± 0.12 | 2.7 |
| Leu(N)–Asn-21(O) | 3.58 ± 0.24 | 3.44 ± 0.21 | 3.6 |
| Leu(N)–Ala-22(O) | 3.21 ± 0.25 | 3.29 ± 0.26 | 2.9 |
| Leu(N)–Phe-253(O) | 2.81 ± 0.13 | 2.80 ± 0.12 | 2.9 |
| Leu(N)–Thr-254(O) | 2.95 ± 0.16 | 2.96 ± 0.17 | 3.1 |
| Leu(N)–Ser-256(OH) | 2.81 ± 0.10 | 2.81 ± 0.10 | 2.8 |
Average distances and standard deviations from the equilibrium simulation of setup 1 are given in Å. Distances observed in the crystal structure are included for comparison.
Interaction energies between the substrate and various parts of the protein and the bound sodium ions are presented in Table 3. The interaction energy between the substrate and LeuT is primarily electrostatic in nature with EElect ≈ −100 kcal/mol, of which ∼75% is from interactions to H1 and H6. Helix H3 also has a large contribution, whereas a small unfavorable interaction energy is calculated between H8 and Leu. An additional EElect ≈ −40 kcal/mol can be found for interactions between Leu and Na-1. The computed high electrostatic energy between Leu and the protein is in accord with the experimentally measured very low rate of transport of leucine through LeuT (16). Furthermore, it can be speculated that the electrostatic repulsion between H8 and Leu can trigger major conformational changes needed for transitioning between the outward-facing and inward-facing states of LeuT. A similar reasoning was recently presented for the serotonin and dopamine transporters, where H8 is speculated to initiate the formation of the inward-facing form through interaction with the dopamine and serotonin substrates, respectively (71).
Table 3.
Average electrostatic interaction energies and standard deviations between the substrate and various parts of LeuT are calculated and compared for the 10 ns simulation of setup 1
| Leu to: | Monomer A, kcal/mol | Monomer B, kcal/mol |
|---|---|---|
| Full monomer | −99.6 ± 7.9 | −98.4 ± 8.0 |
| H1 | −55.8 ± 5.9 | −53.8 ± 5.8 |
| H3 | −13.6 ± 2.6 | −14.1 ± 2.7 |
| H6 | −42.6 ± 4.3 | −42.5 ± 4.4 |
| H8 | 5.4 ± 1.6 | 5.1 ± 1.4 |
| Na-1 | −36.9 ± 5.8 | −38.1 ± 5.7 |
| Na-2 | −2.4 ± 2.3 | −1.8 ± 2.3 |
| Asn-21 (H1) | −10.5 ± 1.6 | −10.6 ± 1.7 |
| Ala-22 (H1) | 1.8 ± 2.5 | 2.6 ± 2.3 |
| Val-23 (H1) | −4.1 ± 0.6 | −4.0 ± 0.6 |
| Gly-24 (H1) | −8.5 ± 1.2 | −8.3 ± 1.2 |
| Leu-25 (H1) | −4.2 ± 1.6 | −3.8 ± 1.4 |
| Gly-26 (H1) | −15.0 ± 1.5 | −15.1 ± 1.5 |
| Arg-30 (H1) | −7.6 ± 1.1 | −7.2 ± 1.0 |
| Tyr-108 (H3) | −13.2 ± 2.6 | −13.5 ± 2.6 |
| Phe-253 (H6) | −18.1 ± 2.0 | −18.2 ± 2.0 |
| Thr-254 (H6) | −4.7 ± 2.5 | −4.7 ± 2.4 |
| Leu-255(H6) | 0.0 ± 0.6 | 0.0 ± 0.6 |
| Ser-256 (H6) | −14.1 ± 1.9 | −14.1 ± 1.9 |
Ala-22 and Thr-254 are also coordinated to Na-1 and have smaller or unfavorable contributions to the binding of Leu.
Unwound helices present an interesting structural feature in LeuT. It can be expected that a bound substrate in a binding site composed of unwound helices can be, at least partly, stabilized by the helical macrodipoles. Furthermore, the backbone peptide groups of the unwound segments can directly participate in hydrogen bonding to the substrate. To investigate the contribution of these effects in stabilization of the substrate in the binding pocket, we decomposed the computed electrostatic interaction energies between Leu and LeuT into contributions from individual residues. Very high electrostatic interaction energies are detected, showing the significance of direct interactions between the charged groups of the substrate and the exposed polar backbone groups of the unwound helices in the binding site. Further analysis of the results, however, also shows that the total electrostatic interaction energy between each helix and Leu is slightly higher than the sum of interaction energies of residues directly interacting with Leu, indicating participation of other elements of the unwound helices in stabilization of the substrate in the occluded binding site (see Table 3). Therefore, we conclude that, although the binding energy seems to be primarily caused by the numerous direct hydrogen bonds to charged groups of Leu, the macrodipole of the unwound helices may also play a role in stabilizing the bound substrate.
Coordination of the two structural sodium ions during MD simulations likewise presents a picture of tight binding. For both ions average coordination distances are within 0.1 Å of those observed in the crystal structure (average distances and standard deviations for sodium coordination in setups 1 and 3 are tabulated in the Supplementary Material). The presence (setup 1) or absence (setup 3) of the substrate does not affect the Na-1 coordination significantly; indeed, in setup 3 (in which Leu is removed), the missing substrate coordination is taken up by a water molecule entering from the bulk, thereby maintaining the octahedral coordination. Before water penetrates into the binding site, the five interactions to LeuT remain in a square pyramidal coordination, similar to the one found in setup 1, leaving the sixth coordination site unoccupied. This observation, along with the strong binding of the Na+ ion in the Na-1 binding site as found in our pulling simulations, might suggest that the binding of Na-1 to the transporter precedes substrate association. This Na+ ion most likely enters an empty binding pocket (substrate-free) with a few water molecules comprising its first hydration shell, though some of these water molecules will be gradually replaced by protein side chains as the ion settles in its binding site. A complete dehydration would occur when the substrate binds in the binding pocket. Although our results can be interpreted consistently with a model in which Na-1 binding precedes that of the substrate, we note that the possibility of simultaneous binding of the sodium ion and the substrate should also be kept in mind.
No significant electrostatic interaction, either direct or long range, can be identified between the substrate and Na-2. It might be inferred that the binding of Leu is independent of Na-2, at least during the initial association of the substrate to the transporter, and that Na-2 might not be involved in the transport mechanism but merely have a structural role. It should be kept in mind, however, that subsequent steps in the transport mechanism might involve major protein conformational changes and substrate translocation that would bring the substrate and Na-2 close to each other; such effects, however, can only be speculated at this time.
After removal of the two sodium ions (setup 4), water from the solvation box penetrates into the Na-1 binding site and coordinates the Leu ammonium group. At the same time the aromatic lid opens slightly with a rotation of the two aromatic rings, allowing additional water molecules to interact with the Leu carboxylate group. In the absence of sodium ions, the substrate binding site shows larger thermal fluctuations than when sodium ions are present. For example, in setup 4A, where these fluctuations are particularly large, hydrogen bonds between the Leu ammonium group and Asn-21(O) and Ser-256(OH) are broken, and the ammonium group is positioned closer to the water molecule located in the Na-1 site. Meanwhile, a new interaction is established between Ser-355(OH), located in H8, and the Leu carboxylate group. Together, these events result in a rotation of the substrate side chain from a configuration approximately parallel to the membrane plane to an almost perpendicular one, with the charged carboxylate group pointing toward the extracellular vestibule and the side chain toward the cytoplasm. This new conformation is observed for more than 2 ns and may indicate a destabilization of substrate binding in the absence of Na-1, providing further support for the key role of Na-1 in Leu binding.
Pathway for substrate unbinding in LeuT
A total of 69 ns of pulling simulations in 14 setups (setups 5–18) were conducted to investigate the unbinding of substrate from the binding pocket (see Table 1). That gives a total of 28 unbinding trajectories because of the dimeric structure of the system.
The majority, 18, of the trajectories resulted in a pathway that appears physiologically relevant and is consistent with data from unbiased simulations of setups 1–4. This pathway is reproducible in simulations with pulling velocities ranging from 1 to 10 Å/ns, strongly suggesting that the observed pathway and the mechanism represent highly relevant features of the natural unbinding process. During the unbinding process interactions to the binding site residues are broken one by one as Leu passes the aromatic lid, but new interactions are formed along the extracellular vestibule, leading to a specific path where Leu is passed along from one set of specific interactions to another. Throughout this identified translocation mechanism Na-1 stays in its original binding pocket. It should be noted that the large degree of reproducibility is remarkable for the pulling simulations, thereby indicating that the collected trajectories are of biological relevance. Because the sampled pathway was observed to be largely independent of the pulling velocity applied on either Leu or Leu/Na-1, it is very likely that even slower pulling will result in a similar trajectory.
In another five of the SMD trajectories, where either a higher pulling velocity is employed or when both Leu and Na-1 are pulled, Na-1 shows a different behavior, namely, it moves along with the substrate. However, this does not affect the sampled pathway or the translocation steps involved in Leu unbinding; the only difference is that Na-1 travels tightly coordinated to Leu along the same pathway. This gives a total of 23 of 28 trajectories sampling the same unbinding pathway for Leu. We note that in several trajectories, although Na-1 was being pulled along with Leu, Na-1 did not leave its binding site despite the applied force, indicating a strong tendency of Na-1 to stay bound to the protein even after Leu has left the binding site.
A rare event, which was observed only in the last five fast-pulling trajectories, was translocation of Na-1 and Leu on opposite sides of helices H1 and H6. This pathway exposes Na-1 to a highly hydrophobic environment and is concluded to be an artifact of the fast-pulling protocols employed in these few simulations.
As in the equilibrium MD simulations, Na-2 is highly stable in all SMD simulations, further demonstrating that, most likely, there is no connection between the bound Na-2 and the initial translocation of Leu in the extracellular lumen of LeuT (average distances and standard deviations for Na-2 coordination during the SMD simulations are tabulated in the Supplementary Material). This observation is consistent with the notion that the nature of Na-2 is mostly structural, and Na-1 is the only sodium ion directly involved in initial association of Leu to LeuT.
Transfer pathway for substrate unbinding
Under the assumptions that binding and unbinding are reverse processes and, as discussed above, that the nonequilibrium pulling simulations very closely sample the equilibrium pathway, a binding pathway for Leu to LeuT can be extracted from the reverse unbinding pathway found in SMD simulations. The unbinding pathway of Leu from LeuT, as observed in our SMD simulations, is illustrated with a cartoon in Fig. 3. (A movie of the unbinding mechanism is provided in the Supplementary Material. The reader is strongly encouraged to examine the movie because it is a powerful visual aid in depicting the rather complex series of molecular events described in the following section.) Beginning with Leu in its binding site (Fig. 3 A), step 1 of unbinding involves the release of hydrogen bonds between the Leu ammonium group and free carbonyl groups in H1 (Asn-21(O) and Ala-22(O)). This allows the Leu side chain to penetrate halfway through the aromatic lid and to be sandwiched between hydrophobic residues Tyr-108 and Phe-253 (Fig. 3 B). The particularly weak electrostatic interaction between Leu and Ala-22 as found in equilibrium simulations (setups 1–4) is in line with the ease of disrupting this interaction during the first step of unbinding.
Step 2 in the relay of events involves breaking of hydrogen bonds between the Leu ammonium group and H6 in the lower part of the binding site (Thr-254(O) and Ser-256(OH); see Fig. 3, C and D). The remaining interactions between Leu and residues in the binding pocket at this point are to residues located in the upper part of the binding site. This partial release allows the substrate to travel farther out between the two aromatic residues and to establish a new interaction, highly resembling a cation–π interaction, between the Leu ammonium group and the aromatic ring of Phe-253 (Fig. 3 D). Concurrently a water-mediated interaction is formed between the Leu ammonium group and Asp-404 (H10) located in the extracellular vestibule. Complete release of the Leu ammonium group from hydrogen bonding to Phe-253(O) takes place in step 3. This results in a flip in the backbone conformation of Phe-253 moving the carbonyl group back down into the binding site, thereby closing the aromatic lid by an accompanying side-chain rotation of Phe-235 (Fig. 3 E). Simultaneously a direct salt bridge is formed between Leu(N) and Asp-404.
Na-1 is affected by the translocation of the substrate from the binding pocket. During the initial phase of the induced unbinding of the substrate, Na-1 is partially, but only transiently, displaced from its binding site (Fig. 3 C). The displacement seems to coincide with the formation of a strong double interaction with both carboxylate oxygen atoms of the substrate, which transiently forms along the dislocation of the substrate in the binding pocket. However, because of an even stronger interaction of Na-1 with its binding site, it soon falls back into its original binding site (EElect ≈ −160 kcal/mol to LeuT, Fig. 3 D where the sodium ion is located behind the unwound part of H1). In some of the simulations the interaction between Na-1 and the substrate is broken during step 2, in others at a later time, mostly just before or during step 3. The disruption of this interaction coincides with a large drop in the applied force; for setup 7 this appears after 1.5 ns, when the COM of Leu has moved <5 Å (see figures in the Supplementary Material), indicating that Na-1 contributes a major interaction energy to the binding of the substrate. A possible scenario could be that the binding of Na-1 precedes that of Leu. In this case, a bound Na-1 will contribute to electrostatic attraction of the substrate toward the binding pocket during its translocation from the vestibule into the binding pocket, and in particular during its final orientation in the binding pocket.
After step 3 only one of the Leu carboxylate oxygen atoms remains coordinated to the binding site through hydrogen bonds to Gly-26(N) and Tyr-108(OH) in the aromatic lid (Fig. 3 E). These bonds are subsequently broken in step 4, allowing Leu to slide between the two salt-bridge-forming residues in the extracellular vestibule, Arg-30 and Asp-404 (Fig. 3 F).
During the initial stages of unbinding, the Leu side chain moves ahead of the charged backbone. This is primarily because of the strong electrostatic interaction between the residues in the binding pocket and the Leu backbone, which makes it the last part to leave the binding site. Later along the permeation pathway, the Leu side chain is caught by aliphatic and aromatic residues, Val-29, Leu-33, Ala-319, and Phe-320, in the extracellular vestibule. Additional water-mediated interactions to Asp-401 pull the substrate out of the extracellular vestibule in an orientation where the charged backbone of Leu is first (Fig. 3, C–H). After the release of the side chain (step 5), the substrate is almost fully solvated in the entrance of the extracellular lumen of LeuT (Fig. 3 H).
The maximum buildup force during the SMD simulations is ∼1500 pN with a single peak developed during the first three steps. The force decreases slightly after step 2 but does not drop significantly until after the interaction between the substrate and Na-1 is broken. A second force peak (of ∼800 pN) is found around step 4, although no force is needed to relinquish the interactions to the salt-bridge residues. Graphs of a representative force profile are available in the Supplementary Material.
After release of the substrate, up to three water molecules enter the binding site to replace the interactions of the substrate head groups with the protein and Na-1. This is similar to what is observed in the equilibrium simulation of the apo system (setup 3). This observation is a further indicative of the fact that the SMD simulations are highly relevant, as they converge toward molecular assemblies that are highly similar to those seen in the equilibrium simulations.
Formation of the occluded state
The crystal structure of LeuT is suggested to represent the occluded state of the transporter, in which the aromatic lid separates the substrate from the extracellular vestibule leading to the extracellular lumen (15). During the simulation of setup 1 this lid remains tightly closed (average distances between closest carbon atoms of ∼4.3 Å in both monomers). This is also found in one of the monomers in setup 2 (2B), whereas monomer 2A and setups 3 and 4 show an average distance of ∼5–7 Å, thus showing a partially opened lid. The opening is facilitated by rotation of the two aromatic side chains of the lid. In setup 1 the dihedral angles of these side chains are stable at either ∼90° (both residues, χ2) or ∼200° (both residues, χ1). In setups 2, 3, and 4, a higher degree of flexibility is observed, and either one of the residues changes conformation into a different rotamer state to stabilize the lid in its new, partly opened, conformation. The partly open binding site is also observed in the SMD simulations after release of the Leu(N)–Phe-253(O) interaction, resulting in a partially open lid, similar to the one found in setup 3, also with a binding site with both Na+ ions, but no Leu present.
Arg-30, Asp-401, and Asp-404 are the only charged residues found in the extracellular vestibule of LeuT and must therefore be responsible for the electrostatic properties of the presumed binding pathway. Arg-30 and Asp-404 have been shown to be vital for transport in NSSs (15,30,31). The observed water-mediated contact of these residues in the crystal structure of LeuT is preserved during our equilibrium simulation (setup 1). These two residues were proposed to constitute a second gate, which would be closed through water-mediated interactions once the substrate is bound to the binding site (15). The water-mediated interactions are also found in the beginning of the SMD simulations, i.e., while the substrate is still in the binding pocket, and even in setup 4, where the sodium ions are excluded from the simulation. Removal of the substrate from the binding site, both in setup 3 and during later stages of the SMD simulations (when the substrate has left the binding site), induces the formation of a direct, bifurcated, salt bridge between Arg-30 and Asp-404 similar to the one found in structures of LeuT with bound TCA (16,17), the two situations are depicted in Fig. 4, A and B. Apart from the directly bound and water-bridged configurations, SMD simulations revealed a third mode of interaction between Arg-30 and Asp-404 arising during the substrate translocation, in which Leu is positioned between the two residues while forming salt bridges to both (Fig. 3 F).
Because of the strong interaction between Na-1 and the substrate in its bound form, as observed in our equilibrium and SMD simulations (above), Na-1 is most likely needed to mediate the displacement of Leu from the salt bridge site (between Arg-30 and Asp-404) into the binding site. Combined with the results from MD simulations of setup 4, we propose that both Leu and Na-1 need to be present in the binding site for formation and stabilization of the occluded state. Furthermore, in addition to playing a gating role, we propose that Arg-30 and Asp-404 facilitate the substrate binding by coordinating the substrate in the extracellular vestibule of LeuT, then positioning and orienting it properly for interactions to Na-1 and residues located in the binding site. It has to be noted that, despite the computational effort of this study, the limited time scale of MD and SMD simulations in general does not allow one to exclude the possibility of large protein conformational changes being involved in the formation of the occluded state. However, the consistency found between the results of our simulations and the conclusions reached in various studies of the serotonin transporter (35) provides a strong support for the significance of the observed phenomena in this study.
Implications for binding and transport of leucine in LeuT
Based on the crystal structures of LeuT and GltPh, Gouaux and co-workers (15,25,26) suggested a transport mechanism including a minimum of three states: outward open, outward occluded, and inward open states. Our simulations of the apo system with and without sodium ions (setups 2 and 3) indicate that LeuT does not necessarily undergo large conformational changes to open toward the extracellular cavity; instead, a few water molecules might penetrate into the binding site to occupy available coordination sites in the transporter while the hydrophobic part of the binding pocket remains empty. Residues Arg-30 and Asp-404 have been suggested to be part of a gating mechanism in the NSS family (30,31). In the crystal structure of the outward occluded conformation they interact through two water molecules, whereas in the simulation of setup 3 (sodium ions present) they become directly connected, a configuration also found after substrate release in our pulling simulations. Together with the observed interaction with Asp-401, this suggests these three charged residues play a role in attracting the substrate, via electrostatic interactions, into the extracellular vestibule, where it is then guided into the binding site. Asp-401 is located close to the surface of the transporter. Once an interaction between Leu and Asp-401 is established, the substrate can readily form an additional hydrogen bond to Asp-404. From this point, the substrate can then establish an interaction to Arg-30.
After insertion between Arg-30 and Asp-404, the substrate can be guided into the binding site by establishing stepwise interactions with various parts of the binding pathway, as observed in the reverse from the SMD simulations. Electrostatic attraction between Leu and Na-1 (−40 kcal/mol to −60 kcal/mol) are more favorable than those to Arg-30 (∼−25 kcal/mol) and would initially draw the carboxylate group toward the binding site, where interactions to Gly-26(N) and Tyr-108(OH) could be formed on the way. As a result, the interaction from Leu(N) to Asp-404 would be reduced and formation of a hydrogen bond to Phe-253(O) be more favorable, ensuring a rotation of its aromatic ring and thus opening of the “lid” to allow for full binding of the substrate in a highly orchestrated mechanism.
The initial stabilizing interactions between Leu and the extracellular vestibule are also important in desolvation of the highly charged substrate and likely in orienting the substrate for an optimal insertion into the binding pocket. These two factors may explain the experimentally observed requirements for the three charged residues for transport (15,30,31).
The binding of the substrate and the two sodium ions reveals a clear role for the unwound helices, that is, to provide free carbonyl groups for binding of the transported entities. An additional role can be found for H6. The large loop in the middle of H6 interacts with the hydrophobic side chain of the substrate. Furthermore, the interaction between Leu(N) and Phe-253 is relevant for the opening and closing of the aromatic lid.
The proposed pathway for substrate binding is similar to the recently suggested pathway for dopamine binding in the dopamine transporter (23). Furthermore, the observation that no major conformational changes take place during occlusion is consistent with experimental observations made on the serotonin transporter (35). Because of the rather scarce information on LeuT in the literature, it is difficult, at this time, to provide additional experimental evidence supporting the binding pathway suggested for Leu. One possible way to validate our results would be solid-state NMR measurements with labeled Leu and selectively labeled LeuT, which would enable one to identify long-lived interaction sites during the transport. Another possibility is to perform single-point mutagenesis on LeuT and study binding and transport in the mutants. Some mutagenesis studies have appeared in the literature showing that residues corresponding to Arg-30 and Asp-404 in other NSSs are essential for activity (30,31). Other interesting residues to study would be the hydrophobic residues interacting with the Leu side chain during the transport process, namely Leu-29, Val-33, Ala-319, and Phe-320. The first two of these residues are located in the C-terminal part of H1 and become close to Leu during the unbinding; they might be the ones holding the side chain during the rotation of the molecule. The two other residues are located in the EL4 loop (the position of Phe-320 is depicted in Fig. 1 B) and pointing down into the extracellular vestibule. As observed for the structure of LeuT with bound TCA (16,17), the EL4 loop is flexible, and Ala-319 and Phe-320 are in close contact with Leu during the end of the unbinding pathway.
The next step in the transport mechanism will be to proceed from the outward-occluded state to an inward-facing conformation. Based on the present structures of LeuT, this will require considerable conformational changes of the protein, which present a much more challenging problem to simulation methodologies and beyond the scope of this study.
Conclusion
Reuptake of neurotransmitters is a physiologically important process, but, until recently, the lack of structural knowledge had hampered structural studies including atomic details of the transport mechanism. In this study, using a structure of a bacterial homolog, the LeuT, we report extensive simulations in which several relevant aspects of the transport mechanism have been investigated. By simulating the process of substrate unbinding toward the extracellular vestibule, we provide a detailed view of a possible binding pathway along with the steps involved in the process, thus proposing a mechanism for initial substrate association to LeuT. The binding process involves a salt bridge in the extracellular lumen of LeuT (Arg-30 and Asp-404) to which an incoming zwitterionic substrate initially binds. Sliding through the opened space between these two residues, the substrate establishes multiple interactions with other residues along the pathway, thus penetrating deeper into the center of LeuT. Investigation of the pH dependence of Leu association and the effect of mutation of either of the salt bridge residues would provide a strong test for examining the recruiting and/or gating role of this residue pair. Access of the substrate to the binding pocket is possible only through an opening of the aromatic lid on top of the binding site. The substrate then moves into the binding site and participates in several interactions, primarily to one of the bound sodium ions (Na-1) and to residues in the unwound parts of H1 and H6. Side-chain conformational changes, namely the opening and closing of the aromatic lid, seem to be one of the main steps involved in the transition of LeuT between the outward open and occluded states. This proposal could be tested by site-directed mutagenesis introducing a tryptophan residue instead of Phe-253 in the aromatic lid. If there is no major functional effect of LeuT on this mutation, FRET studies aimed at probing movements in the aromatic lid should be able to provide information in this regard. One of the structurally bound sodium ions in the LeuT binding site (Na-1) is found to play a major role in substrate binding, whereas the other sodium ion (Na-2) interacts with the substrate in neither the outward open nor the occluded state. Therefore, we propose that during the transport cycle, the binding of Na-1 precedes that of the substrate or accompanies substrate binding. This can be measured experimentally by studying the association rates of Leu as a function of Na+ concentration. A strong dependence of the association rate of Leu is expected if binding requires either initial or simultaneous sodium ion binding. Experiments along these lines are in progress in our collaborators’ laboratory.
Acknowledgments
We thank Anne-Marie Lund Winther and Prof. Poul Nissen, Department of Molecular Biology, University of Aarhus, for providing the LeuT dimer and Dr. Steffen Sinning for inspiring discussions.
This work was supported by grants from the National Institutes of Health, R01-GM067887 and P41-RR05969, and from the Lundbeck Foundation, the Danish Natural Science Research Council, and iNANO. The authors acknowledge supercomputer time provided by a TeraGrid grant (MCA06N060) at the National Center for Supercomputing Applications and Indiana University as well as the computer time provided by the University of Illinois at Urbana-Champaign Turing cluster and the Danish Center for Scientific Computing.
Footnotes
Editor: Gregory A. Voth.
Supplementary material
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Appendix A.
Tables S1 through S6
The unbinding pathway of Leu from LeuT
References
- 1.Torres G.E., Gainetdinov R.R., Caron M.G. Plasma membrane monoamine transporters: structure, regulation and function. Nat. Rev. Neurosci. 2003;4:13–25. doi: 10.1038/nrn1008. [DOI] [PubMed] [Google Scholar]
- 2.Hahn M.K., Blakely R.D. Monoamine transporter gene structure and polymorphisms in relation to psychiatric and other complex disorders. Pharmacogenomics J. 2002;2:217–235. doi: 10.1038/sj.tpj.6500106. [DOI] [PubMed] [Google Scholar]
- 3.Masson J., Sagne C., Hamon M., Mestikawy S.E. Neurotransmitter transporters in the central nervous system. Pharmacol. Rev. 1999;51:439–464. [PubMed] [Google Scholar]
- 4.Owens M.J., Nemeroff C.B. Role of serotonin in the pathophysiology of depression: focus on the serotonin transporter. Clin. Chem. 1994;40:288–295. [PubMed] [Google Scholar]
- 5.Rudnick G. Mechanisms of biogenic amine neurotransmitter transporters. In: Reith M.E.A., editor. Neurotransmitter transporters: structure, function, and regulation. 2nd edition. Humana Press; Totowa, NJ: 2002. pp. 25–52. [Google Scholar]
- 6.Ravna A.W., Sylte I., Dahl S.G. Molecular model of the neural dopamine transporter. J. Comput. Aided Mol. Des. 2003;17:367–382. doi: 10.1023/a:1026116017725. [DOI] [PubMed] [Google Scholar]
- 7.Ravna A.W., Sylte I., Dahl S.G. Molecular mechanism of citalopram and cocaine interactions with neurotransmitter transporters. J. Pharmacol. Exp. Ther. 2003;307:34–41. doi: 10.1124/jpet.103.054593. [DOI] [PubMed] [Google Scholar]
- 8.Dahl S.G., Sylte I., Ravna A.W. Structures and models of transporter proteins. J. Pharmacol. Exp. Ther. 2004;309:853–860. doi: 10.1124/jpet.103.059972. [DOI] [PubMed] [Google Scholar]
- 9.Ravna A.W., Sylte I., Kristiansen K., Dahl S.G. Putative drug binding conformations of monoamine transporters. Bioorg. Med. Chem. 2006;14:666–675. doi: 10.1016/j.bmc.2005.08.054. [DOI] [PubMed] [Google Scholar]
- 10.Gundertofte K., Bøgesø K.P., Liljefors T. A stereoselective pharmacophoric model of the serotonin reuptake site. In: vad de Waterbeemd H., Testa B., Folkers G., editors. Computer-assisted lead finding and optimization. Verlag Helvetica Chimica Acta; Basel: 1997. [Google Scholar]
- 11.Pratuangdejkul J., Schneidier B., Jaudon P., Rosilio V., Baudoin E., Loric S., Conti M., Launay J.M., Manivet P. Definition of an uptake pharmacophore of the serotonin transporter through 3D-QSAR analysis. Curr. Med. Chem. 2005;12:2393–2410. doi: 10.2174/0929867054864813. [DOI] [PubMed] [Google Scholar]
- 12.Hoffman B.T., Kopajtic T., Katz J.L., Newman A.H. 2D QSAR modeling and preliminary database searching for dopamine transporter inhibitors using genetic algorithm variable selection of molconn Z descriptors. J. Med. Chem. 2000;43:4151–4159. doi: 10.1021/jm990472s. [DOI] [PubMed] [Google Scholar]
- 13.Kulkarni S.S., Newman A.H., Houlihan W.J. Three-dimensional quantitative structure-activity relationships of mazindol analogues at the dopamine transporter. J. Med. Chem. 2002;45:4119–4127. doi: 10.1021/jm0102093. [DOI] [PubMed] [Google Scholar]
- 14.Christensen H., Boye S., Thinggaard J., Sinning S., Wiborg O., Schiøtt B., Bols M. QSAR studies and pharmacophore identification for arylsubstituted cycloalkenecarboxylic acid methyl esters with affinity for the human dopamine transporter. Bioorg. Med. Chem. 2007;15:5262–5274. doi: 10.1016/j.bmc.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 15.Yamashita A., Singh S.K., Kawate T., Jin Y., Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 16.Singh S.K., Yamashita A., Gouaux E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature. 2007;448:952–956. doi: 10.1038/nature06038. [DOI] [PubMed] [Google Scholar]
- 17.Zhou Z., Zhen J., Karpowich N.K., Goetz R.M., Law C.J., Reith M.E.A., Wang D.-N. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science. 2007;317:1390–1393. doi: 10.1126/science.1147614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beuming T., Shi L., Javitch J.A., Weinstein H. A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol. Pharmacol. 2006;70:1630–1642. doi: 10.1124/mol.106.026120. [DOI] [PubMed] [Google Scholar]
- 19.Ravna A.W., Jaronczyk M., Sylte I. A homology model of SERT based on the LeuTAa template. Bioorg. Med. Chem. Lett. 2006;16:5594–5597. doi: 10.1016/j.bmcl.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 20.Jørgensen A.M., Tagmose L., Jørgensen A.M.M., Topiol S., Sabio M., Gundertofte K., Bøgesø K.P., Peters G.H. Homology modeling of the serotonin transporter: Insights into the primary escitalopram-binding site. ChemMedChem. 2007;2:815–826. doi: 10.1002/cmdc.200600242. [DOI] [PubMed] [Google Scholar]
- 21.Jørgensen A.M., Tagmose L., Jørgensen A.M.M., Bøgesø K.P., Peters G.H. Molecular dynamics simulations of Na+/Cl−-dependent neurotransmitter transporters in a membrane-aqueous system. ChemMedChem. 2007;2:827–840. doi: 10.1002/cmdc.200600243. [DOI] [PubMed] [Google Scholar]
- 22.Paczkowski F.A., Sharpe I.A., Dutertre S., Lewis R.J. Chi-conopeptide and tricyclic antidepressant interactions at the norepinephrine transporter define a new transporter model. J. Biol. Chem. 2007;282:17837–17844. doi: 10.1074/jbc.M610813200. [DOI] [PubMed] [Google Scholar]
- 23.Huang X., Zhan C. How dopamine transporter interacts with dopamine: insights from molecular modeling and simulation. Biophys. J. 2007;93:3627–3639. doi: 10.1529/biophysj.107.110924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Indarte M., Madura J.D., Surratt C.K. Dopamine transporter comparative molecular modeling and binding site prediction using the LeuTAa leucine transporter as a template. Proteins Struct. Funct. Bioinformatics. 2007;70:1033–1046. doi: 10.1002/prot.21598. [DOI] [PubMed] [Google Scholar]
- 25.Yernool D., Boudker O., Jin Y., Gouaux E. Structure of a glutamate transporter homologue from pyrococcus horikoshi. Nature. 2004;431:811–818. doi: 10.1038/nature03018. [DOI] [PubMed] [Google Scholar]
- 26.Boudker O., Ryan R.M., Yernool D., Shimamoto K., Gouaux E. Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature. 2007;445:387–393. doi: 10.1038/nature05455. [DOI] [PubMed] [Google Scholar]
- 27.Doyle D.A., Cabral J.M., Pfuetzner R.A., Kuo A., Gulbis J.M., Cohen S.L., Chait B.T., MacKinnon R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 28.Hunte C., Screpanti E., Venturi M., Rimon A., Padan E., Michel H. Structure of a Na+/H+ antiporter and insights into mechanism of action and regulation by pH. Nature. 2005;435:1197–1202. doi: 10.1038/nature03692. [DOI] [PubMed] [Google Scholar]
- 29.Sui H., Han B., Lee J.K., Walian P., Jap B.K. Structural basis of water-specific transport through the AQP1 water channel. Nature. 2001;414:872–878. doi: 10.1038/414872a. [DOI] [PubMed] [Google Scholar]
- 30.Pantanowitz S., Bendahan A., Kanner B.I. Only one of the charged amino acids located in the transmembrane α-helices of the γ-aminobutyric acid transporter (subtype A) is essential for its activity. J. Biol. Chem. 1993;268:3222–3225. [PubMed] [Google Scholar]
- 31.Cao Y., Li M., Mager S., Lester H.A. Amino acid residues that control pH modulation of transport-associated current in mammalian serotonin transporters. J. Neurosci. 1998;18:7739–7749. doi: 10.1523/JNEUROSCI.18-19-07739.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jardetzky O. Simple allosteric model for membrane pumps. Nature. 1966;211:969–970. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- 33.Jones S.R., Joseph J.D., Barak L.S., Caron M.G., Wightman R.M. Dopamine neuronal transport kinetics and effects of amphetamine. J. Neurochem. 1999;73:2406–2414. doi: 10.1046/j.1471-4159.1999.0732406.x. [DOI] [PubMed] [Google Scholar]
- 34.Schenk J. The functioning neuronal transporter for dopamine: kinetic mechanisms and effects of amphetamines, cocaine and methylphenidate. Prog. Drug Res. 2002;59:111–131. doi: 10.1007/978-3-0348-8171-5_4. [DOI] [PubMed] [Google Scholar]
- 35.Rudnick G. Serotonin transporters—structure and function. J. Membr. Biol. 2006;213:101–110. doi: 10.1007/s00232-006-0878-4. [DOI] [PubMed] [Google Scholar]
- 36.Adcock S.A., McCammon J.A. Molecular dynamics: Survey of methods for simulating the activity of proteins. Chem. Rev. 2006;105:1589–1615. doi: 10.1021/cr040426m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karplus M., Kuriyan J. Chemical theory and computation special feature: molecular dynamics and protein function. Proc. Natl. Acad. Sci. USA. 2005;102:6679–6685. doi: 10.1073/pnas.0408930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tajkhorshid E., Cohen J., Aksimentiev A., Sotomayor M., Schulten K. Towards understanding membrane channels. In: Martinac B., Kubalski A., editors. Bacterial ion channels and their eukaryotic homologues. American Society of Microbiology Press; Washington, DC: 2005. pp. 153–190. [Google Scholar]
- 39.Gumbart J.C., Wang Y., Aksimentiev A., Tajkhorshid E., Schulten K. Molecular dynamics simulations of proteins in lipid bilayers. Curr. Opin. Struct. Biol. 2005;15:423–431. doi: 10.1016/j.sbi.2005.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tajkhorshid E., Aksimentiev A., Balabin I., Gao M., Isralewitz B., Phillips J.C., Zhu F., Schulten K. Large scale simulation of protein mechanics and function. Adv. Protein Chem. 2003;66:195–247. doi: 10.1016/s0065-3233(03)66006-7. [DOI] [PubMed] [Google Scholar]
- 41.Celik L., Lund J.D.D., Schiøtt B. Conformational dynamics of the estrogen receptor α: molecular dynamics simulations of the influence of binding site structure on protein dynamics. Biochemistry. 2007;46:1743–1758. doi: 10.1021/bi061656t. [DOI] [PubMed] [Google Scholar]
- 42.Lindorff-Larsen K., Best R.B., DePristo M.A., Dobson C.M., Vendruscolo M. Simultaneous determination of protein structure and dynamics. Nature. 2005;433:128–132. doi: 10.1038/nature03199. [DOI] [PubMed] [Google Scholar]
- 43.Hornak V., Okur A., Rizzo R.C., Simmerling C. HIV-1 protease flaps spontaneously open and reclose in molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 2006;103:915–920. doi: 10.1073/pnas.0508452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hornak V., Okur A., Rizzo R.C., Simmerling C. HIV-1 protease flaps spontaneously close to the correct structure in simulations following manual placement of an inhibitor into the open state. J. Am. Chem. Soc. 2006;128:2812–2813. doi: 10.1021/ja058211x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yin Y., Jensen M.Ø., Tajkhorshid E., Schulten K. Sugar binding and protein conformational changes in lactose permease. Biophys. J. 2006;91:3972–3985. doi: 10.1529/biophysj.106.085993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Israelewitz B., Gao M., Schulten K. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 2001;11:224–230. doi: 10.1016/s0959-440x(00)00194-9. [DOI] [PubMed] [Google Scholar]
- 47.Gao M., Sotomayor M., Villa E., Lee E., Schulten K. Molecular mechanisms of cellular mechanics. Phys. Chem. Chem. Phys. 2006;8:3692–3706. doi: 10.1039/b606019f. [DOI] [PubMed] [Google Scholar]
- 48.Sotomayor M., Schulten K. Single-molecule experiments in vitro and in silico. Science. 2007;316:1144–1148. doi: 10.1126/science.1137591. [DOI] [PubMed] [Google Scholar]
- 49.Jensen M.Ø., Park S., Tajkhorshid E., Schulten K. Energetics of glycerol conduction through aquaglyceroporin GlpF. Proc. Natl. Acad. Sci. USA. 2002;99:6731–6736. doi: 10.1073/pnas.102649299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y., Schulten K., Tajkhorshid E. What makes an aquaporin a glycerol channel? A comparative study of AqpZ and GlpF. Structure. 2005;13:1107–1118. doi: 10.1016/j.str.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 51.Jensen M.Ø., Yin Y., Tajkhorshid E., Schulten K. Sugar transport across lactose permease probed by steered molecular dynamics. Biophys. J. 2007;93:92–102. doi: 10.1529/biophysj.107.103994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gumbart J.C., Wiener M.C., Tajkhorshid E. Action at a distance: mechanics of force propagation in TonB-dependent outer membrane transport. Biophys. J. 2007;93:496–504. doi: 10.1529/biophysj.107.104158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gao M., Craig D., Lequin O., Campbell I., Vogel V., Schulten K. Structure and functional significance of mechanically unfolded fibronectin type III1 intermediates. Proc. Natl. Acad. Sci. USA. 2003;100:14784–14789. doi: 10.1073/pnas.2334390100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marszalek P.E., Lu H., Li H., Carrion-Vazquez M., Oberhauser A.F., Schulten K., Fernandez J.M. Mechanical unfolding intermediates in titin modules. Nature. 1999;402:100–103. doi: 10.1038/47083. [DOI] [PubMed] [Google Scholar]
- 55.Arkin I.T., Xu H., Jensen M.Ø., Arbely E., Bennett E.R., Bowers K.J., Chow E., Dror R.O., Eastwood M.P., Flitman-Tene R., Gregersen B.A., Klepeis J.L., Kolossvary I., Shan Y., Shaw D.E. Mechanism of Na+/H+ antiporting. Science. 2007;317:799–803. doi: 10.1126/science.1142824. [DOI] [PubMed] [Google Scholar]
- 56.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 58.MacKerell A.D.J., Bashford D., Bellott M., Dunbrack R.L., Jr., Evanseck J.D., Field M.J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F.T.K., Mattos C., Michnick S., Ngo T., Nguyen D.T., Prodhorn B., Reiher W.E., III, Roux B., Schlenkrich M., Smith J.C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 59.MacKerell A.D.J. Atomistic models and force fields. In: Becker O.M., MacKerell A.D.J., Roux B., Watanabe M., editors. Computational biochemistry and biophysics. Marcel Dekker; New York: 2001. pp. 7–38. [Google Scholar]
- 60.MacKerell A.D.J. Empirical force fields for biological macromolecules: Overview and issues. J. Comput. Chem. 2004;25:1584–1604. doi: 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]
- 61.MacKerell A.D., Feig M., Brooks C.L., III Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 62.Kocabas A.M., Rudnick G., Kilic F. Functional consequences of homo- but not hetero-oligomerization between transporters for the biogenic amine neurotransmitters. J. Neurochem. 2003;85:1513–1520. doi: 10.1046/j.1471-4159.2003.01793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Just H., Sitte H.H., Schmid J.A., Freissmuth M., Kudlacek O. Identification of an additional interaction domain in transmembrane domains 11 and 12 that supports oligomer formation in the human serotonin transporter. J. Biol. Chem. 2004;279:6650–6657. doi: 10.1074/jbc.M306092200. [DOI] [PubMed] [Google Scholar]
- 64.Jones T.A., Zou J.-Y., Cowan S.W., Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 65.Onufriev A., Case D.A., Ullmann G.M. A novel view of pH titration in biomolecules. Biochemistry. 2001;40:3413–3419. doi: 10.1021/bi002740q. [DOI] [PubMed] [Google Scholar]
- 66.Kalé L., Skeel R., Bhandakar M., Brunner R., Gursoy A., Krawetz N., Phillips J., Shinozaki A., Varadarajan K., Schulten K. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 1999;151:283–312. [Google Scholar]
- 67.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kalé L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Darden T., York D., Pedersen L. Particle mesh Ewald: An N*log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 69.Feller S.E., Zhang Y., Pastor R.W., Brooks B.R. Constant pressure molecular dynamics simulation: The langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 70.Park S., Khalili-Araghi F., Tajkhorshid E., Schulten K. Free energy calculation from steered molecular dynamics simulations using Jarzyinski's equality. J. Chem. Phys. 2003;119:3559–3566. [Google Scholar]
- 71.Guptaroy, B., M. Zhang, F. Binda, E. Bowton, L. Johnson, A. Galli, J. Javitch, and M. Gnegy. 2006. An N-terminal mutation of the dopamine transporter results in an efflux promoting conformation. Program No. 35.13. 2006 Neuroscience meeting planner. Society for Neuroscience, Atlanta, GA. http://www.sfn.org/am2006/.
- 72.Celik, L., S. Sinning, K. Severinsen, C. G. Hansen, M. S. Møller, M. Bols, O. Wiborg, and B. Schiøtt. 2008. Binding of serotonin to the human serotonin transporter. Molecular modelling and experimental validation. J. Am. Chem. Soc. In press. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1 through S6
The unbinding pathway of Leu from LeuT