

Abstract
The Grb2-related adaptor protein GADS plays a central role during the initial phases of signal transduction in T lymphocytes. GADS possesses N- and C-terminal Src homology 3 (SH3) domains flanking a central Src homology 2 (SH2) domain and a 126-residue region rich in glutamine and proline residues, presumed to be largely unstructured. The SH2 domain of GADS binds the adaptor protein LAT; the C-terminal SH3 domain pairs GADS to the adaptor protein SLP-76, whereas the function of the central region is unknown. High-resolution three-dimensional models are available for the isolated SH2 and C-terminal SH3 domains in complex with their respective binding partners, LAT and SLP-76. However, in part because of its intrinsic instability, there is no structural information for the entire GADS molecule. Here, we report the low-resolution structure of full-length GADS in solution using small-angle x-ray scattering (SAXS). Based on the SAXS data, complemented by gel filtration experiments, we show that full-length GADS is monomeric in solution and that its overall structural parameters are smaller than those expected for a protein with a long unstructured region. Ab initio and rigid body modeling of the SAXS data reveal that full-length GADS is a relatively compact molecule and that the potentially unstructured region retains a significant degree of structural order. The biological function of GADS is discussed based on its overall structure.
Introduction
T lymphocytes are key effector cells of the adaptive immune system that perform critical activities for the body's natural defense against infections and many tumors (1,2). T cells generally use a two-signal mechanism for activation: 1), the interaction between the T cell receptor (TCR) and specific peptide/MHC complexes on the antigen-presenting cells (s) delivers signals into the T cells; and 2), the costimulatory interaction between CD28 molecules expressed on T cells and B7 molecules on antigen presenting cells provides additional activating signals to T cells.
TCR signaling depends on coordinated interactions of multiple signaling proteins, which include adaptors or molecular scaffolds (3,4). Adaptor proteins lack any enzymatic activity or any transcription activation domains. Instead, they have discrete binding sites on distinct modules that bind to other proteins. Examples of these modules include SH2 and PTB domains (which bind phosphorylated tyrosine residues), SH3 and WW domains (which constitutively associate with proline-rich domains), and PH domains (which interact with phosphorylated membrane lipids) (5,6). T-cell adaptors include LAT (linker for activation of T cells), SLP-76 (SH2-domain-containing leukocyte protein of 76 kDa), ITK (T-cell protein tyrosine kinase), PLCγ1 (phosphatidyl inositol-specific phospholipase C1), NCK (adaptor protein containing SH2 and SH3 domain), VAV (guanine nucleotide exchange factor of the Rho family), GADS (Grb2-related adaptor downstream of Shc), ADAP (adhesion and degranulation promoting adaptor protein), GAB2 (GRB2-associated binding protein 2), and SHP-2 (Src homology 2-domain-containing tyrosine phosphatase 2) (Fig. 1 A).
Figure 1.

(A) Schematic representation of GADS-related signaling in T cells. TCR ligation induces side-specific phosphorylation of the intracellular chains of the TCR/CD3 complex. These initial events trigger phosphorylation of LAT and SLP-76. SLP-76 is subsequently recruited to LAT via GADS. This trimolecular complex (LAT/GADS/SLP-76) then recruits other modular proteins (VAV, NCK, ADAP, ITK, GAB2, PLCγ). The formation of this complex is required for the activation of the RAS-MAPK pathway and for the cytoskeleton reorganization. (B) Schematic representation of full-length GADS. GADS is composed of SH3-SH2 domains at the N-terminus and a C-terminal SH3 domain. These are separated by a long unstructured region composed of 126 amino acid residues. The SH2 domain of GADS binds phosphorylated LAT; the C-terminal SH3 domain binds the adaptor molecule SLP-76; the binding partner and the functional role of the N-terminal SH3 domain are still unknown.
High-resolution structural information is available for isolated modules (i.e., SH2, SH3, WW, PH) of some of the above proteins, in free form or in complex with their minimal binding partners. However, there is no structural information for full-length molecules, and few biophysical studies have been published (6,7). This lack of information is primarily a result of the intrinsic instability and flexibility of full-length adaptor proteins. In fact, a common characteristic of these molecules is that they have a long region between their compact domains. These linker regions are proposed to be generally disordered and represent a major complication to studying the complete structure of adaptors (8,9).
We are interested in obtaining structural information on the adaptor protein GADS (10), as this macromolecule plays a pivotal role during the early events of signal transduction in T cells (Fig. 1 A). GADS is composed of N-terminal SH3 and SH2 domains and a C-terminus SH3 domain (11) (Fig. 1 B). These domains are connected by a long sequence that is rich in glutamine and proline residues (11) (Fig. 1 B). GADS binds several intracellular signaling proteins, including SLP-76, LAT, c-Cbl, HPK1, and Gab2 (Fig. 1 A) (12,13). Structural studies of the SH2 domain of GADS in complex with phosphorylated LAT peptides (14) and of the C-terminal SH3 domain in complex with atypical consensus motifs of SLP-76 (10,15,16) and HPK1 (17) (hematopoietic progenitor kinase 1) have been reported recently. However, no structural information is available for the full-length GADS molecule.
Here we report a low-resolution structural model of full-length GADS in solution obtained using small-angle x-ray scattering (SAXS). This method is well suited to studying multidomain proteins, such as GADS, that possess intrinsic flexibility and low compactness (18). SAXS can provide overall structural parameters as well as the shape and domain organization of proteins in solution. Analysis of GADS by SAXS reveals that this polypeptide is monomeric in solution and possesses a surprisingly compact overall structure. This picture is consistent with an ab initio model of GADS by a chain of pseudoresidues. In addition, the work reported here may serve as a guide for studying the overall shape of other modular proteins or protein complexes in T-cell signaling or, in general, for studying large proteins or protein complexes whose structure determination is not feasible using traditional techniques such as nuclear magnetic resonance spectroscopy or x-ray crystallography.
Methods
Protein expression and purification
Full-length GADS (residues 1–322; NCBI NM_010815) was expressed in E. coli and purified by size-exclusion chromatography as described by Houtman et al. (6).
SAXS data collection and processing
SAXS data were collected at the X33 beam line of the European Molecular Biology Laboratory (EMBL), Hamburg outstation at the storage ring DORIS III of the Deutsches Elektronen Synchrotron (DESY) synchrotron (19), as described by Dimasi et al. (20). Scattering data from GADS solutions with concentrations of 1.0, 2.4, 3.5, and 6.9 mg/ml were measured in a buffer containing 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 10 mM EDTA, 0.05% β-octylglucoside, and 2 mM DTT. All measurements were done at 15°C.
SAXS-based modeling
The low-resolution shape of GADS was reconstructed ab initio using the programs DAMMIN (21) and GASBOR (22). These programs represent the protein as an assembly of beads and dummy residues, respectively, inside a spherical search volume of diameter Dmax. Starting from a random assembly, DAMMIN and GASBOR employ simulated annealing to build scattering equivalent models fitting the experimental data Iexp(s) to minimize discrepancy:
where N is the number of experimental points, c a scaling factor, and Icalc(sj) and σ(sj) are the calculated intensity from the model and the experimental error at the momentum transfer sj, respectively. The two methods yielded consistent shapes for GADS. Ten independent GASBOR models were superposed and averaged using the programs DAMAVER (23) and SUBCOMP (24).
The topology of the protein domains inside the SAXS low-resolution GADS volume was examined by modeling the structure using the program BUNCH (25). The program employs a simulated annealing protocol to find the optimal positions and orientations of available high-resolution models of domains and the probable conformations of the dummy residue chains of the unknown regions to simultaneously fit the scattering data from all constructs. To construct the model with BUNCH, the structure of GADS was divided into four domains as displayed in Fig. 1 B: 1), the N-terminal SH3 domain (homology model based on the PDB entry code 1GBR; see below); 2), the SH2 domain (PDB code 1R1S chain A); 3), a 134-residue unstructured domain (Arg-154 to Gly-288) represented by a dummy residue chain; and 4), the C-terminal SH3 domain (PDB code 2D0N chain A). Multiple BUNCH runs were performed starting from random initial configurations (positions of domains and conformations of dummy residue linkers/fragments).
Construction of the N-terminal GADS SH3 homology model
A homology model of the N-terminal SH3 domain of GADS (residues 1–54) was build using the coordinates of the Grb2 N-terminal SH3 domain (PDB entry code 1GBR Chain A) as a template. This choice was suggested by sequence alignment of the target/template with an overall identity of 51.8% in 54 overlapping residues. The three-dimensional structure was computed after alignment with the program Modeller (26). The geometry of the homology model was examined with the program PROCHECK using a 1.4 Å resolution cutoff (27). PROCHECK statistics showed that 83.0% of GADS-nSH3 residues were in the most favored regions of the Ramachandran plot; 14.9% were in the additional allowed regions, and 2.1% were in the generously allowed regions. The GADS-nSH3 homology model consisted of 54 total residues.
Sequence analysis
Protein disorder was estimated using a neural networks-based predictor, PROFbval (28), which detects flexibile regions with a high probability of forming disordered structures. The disposition to disorder for each domain of GADS was also estimated by the “unfoldability” index, as defined by Uversky and collaborators (29,30). The hydrophobicity, H, and the charge, R, were calculated with the program FoldIndex (31), and the “unfoldability” index was estimated as [H − (R + 1.151)/2.785]. Positive values indicate a structured polypeptide, whereas negative values indicate disordered proteins. Secondary structure predictions were carried out with the program JPred (32).
Results
Expression, folding, and purification of stable full-length GADS
Full-length GADS was expressed as inclusion bodies (Fig. 2 A) and was folded in vitro by dilution in a buffer containing arginine (Fig. 2 B). We have previously expressed and performed a biophysical characterization of the same full-length GADS, demonstrating that it is biochemically active (6). However, protein stability was a major problem that was encountered (N. Dimasi and R. A. Mariuzza, unpublished data). To improve protein stability, GADS was folded in an optimized folding buffer containing β-octylglucoside, and the protein was directly purified by size exclusion chromatography, avoiding a previously used affinity purification protocol (6).
Figure 2.

(A) Time course of protein expression. Protein expression was induced with 1 mM IPTG at 37°C for the time shown above the SDS-PAGE. Aliquots of bacteria were taken at the indicated intervals and directly loaded on the SDS-PAGE. The proteins were stained with colloidal Coomassie. The position of full-length GADS is shown with an arrow. As seen in the gel, GADS is the major protein present after induction, with an estimated molecular mass of 37 kDa. The protein molecular mass markers used were lysozyme (14 kDa), β-lactoglobulin (18 kDa), restriction endonuclease Bsp981 (25 kDa), carbonic anhydrase (37 kDa), ovalbumin (45 kDa), bovine serum albumin (66 kDa), and β-galactosidase (116 kDa). (B) Size-exclusion chromatography of folded full-length GADS. Elution volumes of the molecular mass standards are indicated with numbered arrows. The standards used were 1), albumin, 67 kDa; 2), ovalbumin, 43 kDa; 3), chymotrypsinogen A, 25 kDa; and 3), ribonuclease A, 14 kDa. Blue Dextran (2000 kDa) was used to measure the void volume of the column. The estimated mass of the full-length GADS (major peak) is 37 kDa.
As seen in Fig. 2 B, most folded GADS eluates from the gel filtration column as a major peak corresponding to a molecular weight that is in agreement with the expected molecular mass of 36.8 kDa calculated from the amino acid sequence. A second peak, corresponding to a molecular mass of 73.6 kDa, constitutes ∼6% of the total protein representing a dimeric form of GADS (Fig. 2 B). Gel filtration runs, systematically repeated for several weeks, revealed no major differences in the chromatographic profile, confirming that the protein was stable and amenable to structural characterization. The purified protein, corresponding to the monomeric peak of GADS, was concentrated by ultrafiltration and directly used for SAXS data collection without further purification.
Sequence analysis of the long unstructured domain present in full-length GADS
It has been hypothesized that the long sequence connecting the SH3 and SH2 domains at the N-terminus to the C-terminal SH3 domain of GADS is disordered (11). In fact, analysis of this sequence by the protein-disorder predictor program PROFbval supports this hypothesis. The region between residues 150 and 262 is characterized by a high degree of flexibility (Fig. 3, upper panel), based on disorder indices from PROFbval, which predicts a disordered structure between residues 145 and 263 (Fig. 3). This analysis is consistent with a secondary structure prediction carried out with the program JPred, which did not assign any secondary structure to this domain (Fig. 3, lower panel).
Figure 3.
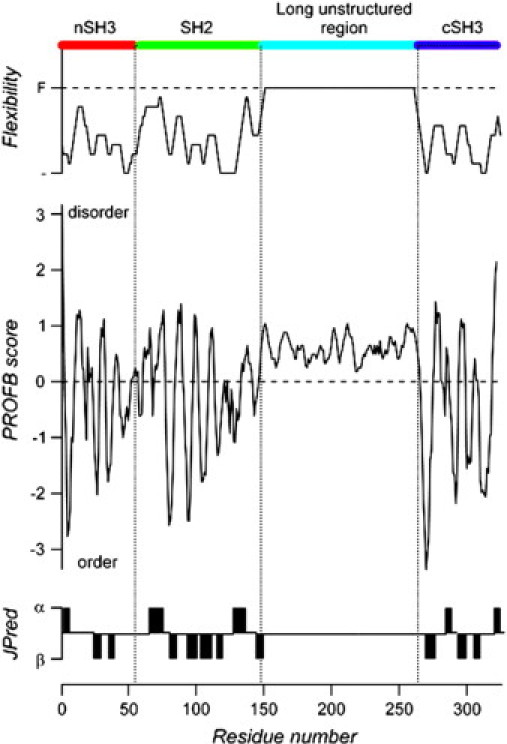
Sequence analysis of full-length GADS. Prediction of flexibility and scores for protein disorder were obtained with the program PROFbval. The prediction score (negative = order; positive = disorder) is plotted against the residue number. The overall domain organization of GADS is shown schematically at the top of the figure. (For domain organization see Fig. 1 B.) At the bottom of the figure is shown the secondary structure prediction of helices (α) and sheets (β) according to the program JPred. These independent analyses predict that the long central region of GADS (from residue 138 is to 263) is unstructured.
Further evidence of the disordered nature of the central region of GADS was obtained by calculation of the “unfoldability” index. The whole GADS sequence is characterized by a charge of R = 0.029 and a hydrophobicity index of H = 0.422, resulting in an unfoldability of −0.006, which indicates a disordered protein. This character is in sharp contrast to the well-“folded” regions of GADS-nSH3 (R = 0.098, H = 0.456), GADS-SH2 (R = 0.022, H = 0.433), and GADS-cSH3 (R = 0.068, H = 0.461), that yield “unfoldability” indices of 0.021, 0.032, and 0.064, respectively. By contrast, the central proline-rich long sequence has a negative “unfoldability” equal to −0.158 (R = 0.009, H = 0.360), clearly implying that this domain is highly disordered.
SAXS experiments
The SAXS pattern from GADS, after subtraction of the solvent scattering and extrapolation to infinite dilution, is displayed in Fig. 4 A. The Guinier plot of these data (33) yields an effective molecular mass of the protein of 39 ± 1 kDa, compatible with the value predicted from the primary amino acid sequence (36.8 kDa) and the gel filtration experiments. This confirms that GADS remains monomeric in solution under the experimental conditions used for carrying out the SAXS experiments. The gyration radius (Rg) of GADS calculated from the SAXS data is equal to 3.53 ± 0.3 nm. Surprisingly, the Kratky plot of s2I(s) versus s (Fig. 4 A, inset) shows a bell-shaped form typical of globular proteins (34), indicating that full-length GADS is rather compact in solution. The corresponding distance distribution function p(r) is displayed in Fig. 4 B. Analysis of these data yields a Rg of 3.93 ± 0.1 nm and a maximum size of the protein Dmax of 14 ± 1 nm. The overall SAXS parameters thus suggest that GADS has an elongated shape, which is also confirmed by the p(r) function having a skewed appearance characteristic of elongated globular particles.
Figure 4.
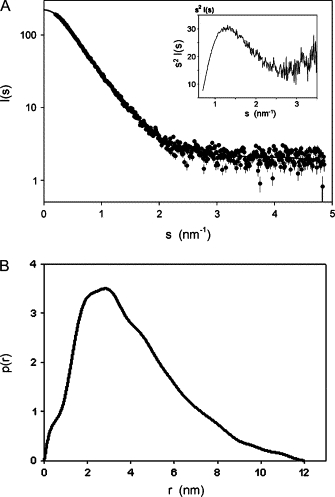
(A) Solution x-ray scattering patterns of full-length GADS extrapolated to zero concentration (●). The continuous line represents the smooth interpolation of the experimental data, extrapolated to s = 0; inset displays the normalized Kratky's plot s2I(s) versus s. (B) Distance distribution function of the experimental data calculated by the program GNOM.
Low-resolution modeling of full-length GADS
The low-resolution models of full-length GADS generated independently by the ab initio programs DAMMIN and GASBOR displayed superimposable overall shapes fitting the experimental data with no significant discrepancy. The averaged model of 10 independent GASBOR reconstructions displayed in Fig. 5 A, although elongated, still shows a relatively compact shape, with principal axis dimensions of 5.6 × 9.7 × 3.5 nm. In an attempt to obtain information on the distribution of the different domains of the protein in the low-resolution SAXS model, the structure of GADS was represented using the atomic and homology models of individual domains connected by the linkers, and the program BUNCH was used to fit this arrangement to the experimental data. Fifty independent BUNCH runs, starting from random initial positions of domains and configurations of the linkers, fitted the data (χ < 1.4). The resulting models were classified according to their topology. The first class, shown in Fig. 5 B, was represented by 21 models that showed the cSH3 domain at one extreme of the molecular volume and the nSH3 at the other extreme, whereas SH2 was in between, near to the nSH3 domain. A second class, composed by 20 models, was basically similar to the first, but the localization of nSH3 and SH2 domains was inverted, such that it presented the SH2 at the extreme of the molecular volume (Fig. 5 C). The other nine models presented the three structured domains in different topologies in the wider extreme of the volume (lower part of the models presented in Fig. 5; see Supplementary Material). The two more conspicuous classes were quite well reproducible, but when starting from random positions, we have about the same probability (0.4) of obtaining either of these two models.
Figure 5.

(A) Averaged ab initio rigid model of full-length GADS obtained using the program GASBOR (violet beads). Two representative models obtained by the optimization of positions and orientations of the GADS known domains obtained using the program BUNCH (B and C). The model was obtained using three-dimensional structural information from the GADS N-terminal SH3 homology model, the SH2 domain (PDB entry 1R1S chain A), and the C-terminal SH3 domain (PDB 2D0N chain A). The N-terminal SH3, the SH2, and C-terminal SH3 domains of GADS are schematically labeled and are colored in blue, red, and yellow, respectively. The binding site regions are labeled and colored in green.
Discussion
Among the physicochemical techniques that can be used to gain structural information on adaptor proteins, information obtained in solution using SAXS is particularly useful. We have carried out an ab initio structure determination of the adaptor protein GADS. Because GADS plays a pivotal role during the initial phases of signaling in T lymphocytes, knowledge of the GADS full-length structure is important for understanding the structural basis of intracellular signaling. Consequently, important questions can be answered by analyzing the full-length structure of GADS. For example: 1), Is GADS monomeric in solution? 2), Is GADS extended and therefore “natively” unfolded? 3), How are the compact SH3 and SH2 domains spatially arranged? 4), Does their spatial arrangement block their binding function?
We have shown that full-length GADS is monomeric in solution and retains a compact overall structure despite the fact that it possess a long unstructured region. We estimated the gyration radius of full-length GADS, and for each of its domains, from its chemical composition using the algorithm of Tcherkasskaya et al. (35), which was developed to predict the value of Rg of folded and unfolded globular proteins. The measured Rg of GADS is 3.53 nm, which does not fit to the predicted Rg for compact globular proteins, relaxed proteins (molten globule), or completely denaturated protein (1.79 nm, 2.06 nm, and 4.88 nm, respectively). Measurements of Rg done for the two domains of known atomic structure, SH2 (1.23 nm) and cSH3 (1.08 nm), and for the homology-predicted structure of domain nSH3 (0.9 nm) are very near to the Rg values predicted for globular proteins (1.22 nm, 1.06 nm, and 1.03 nm, respectively). By simple geometry, given an arrangement of objects, the minimum Rg of the set will be the sum of the Rg values of the two biggest ones. Therefore, assuming that the three structured domains of GADS remain compact in the native protein to satisfy this condition, it is very unlikely that they would assemble with an unfolded long unstructured domain, which would be expected to have a Rg ≥ 3 nm, because it would largely exceed the measured Rg of GADS of 3.5 nm. It follows that, most probably, the unfolded long unstructured domain is neither fully compact nor completely unfolded.
We attempted to determine the domain arrangement of GADS using the available SAXS data. However, the optimization of positions and orientations of the available high-resolution models of domains and the probable conformations of the dummy residue chains done by BUNCH do not provide an unequivocal solution. After repeating the calculations numerous times, starting from random positions, and without assuming any particular conformation for the long unstructured domain, we obtained two main classes of models, as represented in Fig. 5, B and C. We do not have any objective criteria for deciding between these two possible topologies.
The binding sites of the GADS domains have been directly identified from the crystallographic study of the receptor-ligand interaction of cSH3 (10 and references therein), SH2 (14), and by homology on nSH3. In most SAXS models the putative binding sites are exposed, as indicated in Fig. 5, B and C. These observations supports the idea that when GADS is in solution, the binding sites are available to their respective substrates and are not blocked by the back-folding of the molecule.
In conclusion, we have obtained information regarding the overall architecture of GADS, a modular protein involved in T-lymphocyte intracellular signaling. We propose that structural analysis using the SAXS approach is an effective methodology to study other modular proteins or complexes participating in similar processes.
Acknowledgments
We thank Dmitri I. Svergun (European Molecular Biology Laboratory, Hamburg Outstation, Hamburg, Germany) for assistance with SAXS data collection and processing.
We acknowledge the support of the European Community-Research Infrastructure Action under the FP6 “Structuring the European Research Area Program contract number RII/CT/2004/5060008” to the European Molecular Biology Laboratory (EMBL), Hamburg outstation that covered the travel and accommodation expenses of Nazzareno Dimasi at the EMBL Hamburg. The authors declare no conflict of interest.
Footnotes
Editor: Jill Trewhella.
Contributor Information
Oscar Moran, Email: moran@ge.ibf.cnr.it.
Nazzareno Dimasi, Email: ndimasi@gmail.com.
Appendix A.
Data Supplement 1
References
- 1.Rosenberg S.A., Yang J.C., Restifo N.P. Cancer immunotherapy: moving beyond current vaccines. Nat. Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shankaran V., Ikeda H., Bruce A.T., White J.M., Swanson P.E., Old L.J., Schreiber R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 3.Jordan M.S., Singer A.L., Koretzky G.A. Adaptors as central mediators of signal transduction in immune cells. Nat. Immunol. 2003;4:110–116. doi: 10.1038/ni0203-110. [DOI] [PubMed] [Google Scholar]
- 4.Samelson L.E. Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annu. Rev. Immunol. 2002;20:371–394. doi: 10.1146/annurev.immunol.20.092601.111357. [DOI] [PubMed] [Google Scholar]
- 5.Mayer B.J. Protein-protein interactions in signaling cascades. Mol. Biotechnol. 1999;13:201–213. doi: 10.1385/MB:13:3:201. [DOI] [PubMed] [Google Scholar]
- 6.Houtman J.C.D., Higashimoto Y., Dimasi N., Cho S., Yamaguchi H., Bowden B., Regan C., Malchiodi E.L., Mariuzza R., Schuck P., Appella E., Samelson L.E. Binding specificity of multiprotein signaling complexes is determined by both cooperative interactions and affinity preferences. Biochemistry. 2004;43:4170–4178. doi: 10.1021/bi0357311. [DOI] [PubMed] [Google Scholar]
- 7.Houtman J.C.D., Yamaguchi H., Barda-Saad M., Braiman A., Bowden B., Appella E., Schuck P., Samelson L.E. Oligomerization of signaling complexes by the multipoint binding of GRB2 to both LAT and SOS1. Nat. Struct. Mol. Biol. 2006;13:798–805. doi: 10.1038/nsmb1133. [DOI] [PubMed] [Google Scholar]
- 8.Bhalla J., Storchan G.B., MacCarthy C.M., Uversky V.N., Tcherkasskaya O. Local flexibility in molecular function paradigm. Mol. Cell. Proteomics. 2006;5:1212–1223. doi: 10.1074/mcp.M500315-MCP200. [DOI] [PubMed] [Google Scholar]
- 9.Dunker A.K., Brown C.J., Obradovic Z. Identification and functions of usefully disordered proteins. Adv. Protein Chem. 2002;62:25–49. doi: 10.1016/s0065-3233(02)62004-2. [DOI] [PubMed] [Google Scholar]
- 10.Dimasi N. Crystal structure of the C-terminal SH3 domain of the adaptor protein GADS in complex with SLP-76 motif peptide reveals a unique SH3–SH3 interaction. Int. J. Biochem. Cell Biol. 2007;39:109–123. doi: 10.1016/j.biocel.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 11.Liu S.K., McGlade C.J. Gads is a novel SH2 and SH3 domain-containing adaptor protein that binds to tyrosine-phosphorylated Shc. Oncogene. 1998;17:3073–3082. doi: 10.1038/sj.onc.1202337. [DOI] [PubMed] [Google Scholar]
- 12.Berry D.M., Nash P., Liu S.K.W., Pawson T., McGlade C.J. A high-affinity Arg-X-X-Lys SH3 binding motif confers specificity for the interaction between Gads and SLP-76 in T cell signaling. Curr. Biol. 2002;12:1336–1341. doi: 10.1016/s0960-9822(02)01038-2. [DOI] [PubMed] [Google Scholar]
- 13.Yoder J., Pham C., Iizuka Y.M., Kanagawa O., Liu S.K., McGlade J., Cheng A.M. Requirement for the SLP-76 adaptor GADS in T cell development. Science. 2001;291:1987–1991. doi: 10.1126/science.1057176. [DOI] [PubMed] [Google Scholar]
- 14.Cho S., Velikovsky C.A., Swaminathan C.P., Houtman J.C., Samelson L.E., Mariuzza R.A. Structural basis for differential recognition of tyrosine-phosphorylated sites in the linker for activation of T cells (LAT) by the adaptor Gads. EMBO J. 2004;23:1441–1451. doi: 10.1038/sj.emboj.7600168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Q., Berry D., Nash P., Pawson T., McGlade C.J., Li S.S.-C. Structural basis for specific binding of the Gads SH3 domain to an RxxK motif-containing SLP-76 peptide: a novel mode of peptide recognition. Mol. Cell. 2003;11:471–481. doi: 10.1016/s1097-2765(03)00046-7. [DOI] [PubMed] [Google Scholar]
- 16.Harkiolaki M., Lewitzky M., Gilbert R.J.C., Jones E.Y., Bourette R.P., Mouchiroud G., Sondermann H., Moarefi I., Feller S.M. Structural basis for SH3 domain-mediated high-affinity binding between Mona/Gads and SLP-76. EMBO J. 2003;22:2571–2582. doi: 10.1093/emboj/cdg258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewitzky M., Harkiolaki M., Domart M.-C., Jones E.Y., Feller S.M. Mona/Gads SH3C binding to hematopoietic progenitor kinase 1 (HPK1) combines an atypical SH3 binding motif, R/KXXK, with a classical PXXP motif embedded in a polyproline type II (PPII) helix. J. Biol. Chem. 2004;279:28724–28732. doi: 10.1074/jbc.M402745200. [DOI] [PubMed] [Google Scholar]
- 18.Svergun D.I., Koch M.H. Advances in structure analysis using small-angle scattering in solution. Curr. Opin. Struct. Biol. 2002;12:654–660. doi: 10.1016/s0959-440x(02)00363-9. [DOI] [PubMed] [Google Scholar]
- 19.Roessle M.W., Klaering R., Ristau U., Robrahn B., Jahn D., Gehrmann T., Konarev P., Round A., Fiedler S., Hermes C., Svergun D. Upgrade of the small-angle x-ray scattering beamline X33 at the European Molecular Biology Laboratory, Hamburg. J. Appl. Cryst. 2007;40:s190–s194. [Google Scholar]
- 20.Dimasi N., Roessle M., Moran O., Candiano G., Svergun D.I., Biassoni R. Molecular analysis and solution structure from small-angle x-ray scattering of the human natural killer inhibitory receptor IRp60 (CD300a) Int. J. Biol. Macromol. 2007;40:193–200. doi: 10.1016/j.ijbiomac.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 21.Svergun D.I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Svergun D.I., Petoukhov M.V., Koch M.H. Determination of domain structure of proteins from x-ray solution scattering. Biophys. J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Volkov V.V., Svergun D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Cryst. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kozin M.B., Svergun D.I. Automated matching of high- and low-resolution structural models. J. Appl. Cryst. 2001;34:33–41. [Google Scholar]
- 25.Petoukhov M.V., Svergun D.I. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 2005;89:1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 27.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 28.Schlessinger A., Rost B. Protein flexibility and rigidity predicted from sequence. Proteins. 2005;61:115–126. doi: 10.1002/prot.20587. [DOI] [PubMed] [Google Scholar]
- 29.Uversky V.N. What does it mean to be natively unfolded? Eur. J. Biochem./FEBS. 2002;269:2–12. doi: 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 30.Uversky V.N., Gillespie J.R., Fink A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 31.Prilusky J., Felder C.E., Zeev-Ben-Mordehai T., Rydberg E.H., Man O., Beckmann J.S., Silman I., Sussman J.L. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21:3435–3438. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- 32.Cuff J.A., Clamp M.E., Siddiqui A.S., Finlay M., Barton G.J. JPred: a consensus secondary structure prediction server. Bioinformatics. 1998;14:892–893. doi: 10.1093/bioinformatics/14.10.892. [DOI] [PubMed] [Google Scholar]
- 33.Guinier A., Fournet G. Wiley; New York: 1955. Small angle scattering of x-rays. [Google Scholar]
- 34.Kratky O., Pilz I. Recent advances and applications of diffuse x-ray small-angle scattering on biopolymers in dilute solutions. Q. Rev. Biophys. 1972;5:481–537. doi: 10.1017/s0033583500001049. [DOI] [PubMed] [Google Scholar]
- 35.Tcherkasskaya O., Davidson E.A., Uversky V.N. Biophysical constraints for protein structure prediction. J. Proteome Res. 2003;2:37–42. doi: 10.1021/pr025552q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Supplement 1