

Summary
Germinal centers (GCs) are important sites of antibody affinity maturation that are induced during immune responses. They are organized into two major zones: dark and light zones. In the classical model, the dark zone contains large centroblasts that are rapidly proliferating and undergoing somatic mutation of their antibody variable region genes. These cells are suggested to give rise to smaller non-proliferating centrocytes in the light zone that compete for binding antigen on follicular dendritic cells (FDCs) and then depend on receiving signals from helper T cells to survive and differentiate. Recently, the approach of real-time imaging of GCs by two-photon microscopy of intact lymph nodes has provided new insights into GC dynamics that both support and challenge fundamental aspects of this model. Here we review recent and older findings on cell migration, proliferation, and interaction dynamics in the GC and discuss a model where dark and light zone cells are morphologically similar, where proliferation occurs in both zones, and where GC B cells compete for T cell help as well as antigen.
Role of the GC in Antibody Responses
The GC was first described in 1884 by Walther Flemming, who observed a site of large lymphocytes undergoing mitosis in the follicles of lymph nodes and other secondary lymphoid organs and proposed this site to be a major source of all lymphocytes in the body (reviewed in Nieuwenhuis and Opstelten, 1984). Flemming’s work prompted intense study of the GC that continues today, although his original proposed function for the GC was eventually disproven. The GC is now known to be associated with T-dependent antibody responses and experimental evidence indicates that it is the main site in which high affinity antibody-secreting plasma cells and memory B cells are generated.
T-dependent antibody responses are initiated when rare B and T cells specific for an incoming antigen cluster at the boundary between B cell follicles and T cell zones and engage in cognate interactions (Garside et al., 1998; MacLennan et al., 1997; Okada et al., 2005). The activated B cells then can adopt one of two fates: 1) movement into extrafollicular areas followed by proliferation and terminal differentiation into short-lived plasma cells that secrete antibody or 2) movement into B cell follicles followed by proliferation and the establishment of GCs (Jacob et al., 1991a; Liu et al., 1991b). The mechanisms responsible for this fate decision remain poorly understood, although various studies suggest that the affinity of the B cell receptor (BCR) for the foreign antigen, the amount of antigen-receptor engagement and the costimulatory signals received from T cells may all be involved (Benson et al., 2007; Dal Porto et al., 2002; Dal Porto et al., 1998; Paus et al., 2006; Shih et al., 2002; Vinuesa et al., 2000).
Several decades ago, it was observed that the average affinity of serum antibody for a given foreign antigen increases over time after immunization (Eisen and Siskind, 1964). This process was later termed affinity maturation and shown to be due to somatic mutations in the antibody variable region genes in antibody-secreting plasma cells (reviewed in Tarlinton and Smith, 2000). Subsequent work showed that most short-lived plasma cells in primary extrafollicular foci lacked somatic mutations, whereas a high frequency of somatic mutations was evident in GC B cells, suggesting that a hypermutation mechanism is activated in GC B cells (Berek et al., 1991; Jacob et al., 1991b). These somatic mutations were clustered in the regions of antibodies that form the interface with the antigens. A high ratio of amino acid replacement to silent mutations was observed, indicating that selection had taken place. In addition, many of the GC B cells picked out from individual GCs appeared to be clonally related, and indeed some GCs appeared to be dominated by cells derived from a single clone. These observations suggest that the affinity maturation of the antibody response occurs in GCs, through the processes of clonal proliferation, somatic hypermutation and selection. GCs do not appear to be absolutely required for affinity maturation, however, as mice deficient in GCs do exhibit measurable affinity maturation with certain types of immunization (Futterer et al., 1998; Koni and Flavell, 1999; Matsumoto et al., 1996; Wang et al., 2000). Therefore, the GC is thought to be a site specialized to support the events required for efficient affinity maturation of the antibody response.
As the immune response progresses, the extrafollicular foci of plasma cells wane, whereas long-lived plasma cells and memory B cells begin to appear (reviewed in Tarlinton and Smith, 2000). Many of the long-lived plasma cells are found in the bone marrow, where they secrete antibody for several weeks or longer. Notably, the antibody variable genes in most long-lived plasma cells and memory B cells exhibit a high degree of somatic mutations that show evidence of selection, suggesting that these cells were derived from GCs (Blink et al., 2005; McHeyzer-Williams et al., 2006; Tarlinton and Smith, 2000)
GC Kinetics and Organization
The precise kinetics of GC development vary depending on the system examined and are influenced by such factors as the species studied, availability of T cell help, the antigens used for immunization, and the tissue in which the response occurs (Liu et al., 1991b; Manser, 2004; Pape et al., 2003; Shinall et al., 2000; Wang and Carter, 2005). Despite these variables, however, some common principles of GC formation have emerged. It is thought that the GC is seeded within a few days after the initiation of an immune response by activated B cells specific for foreign antigen. These activated B cells first move to the center of B cell follicles in secondary lymphoid organs and proliferate within the FDC network, as was established by careful kinetic studies in the rat (Liu et al., 1991b) and recently confirmed in the mouse (Wang and Carter, 2005). FDCs are radiation-resistant stromal cells that form a network of processes in B cell follicles with the ability to capture large amounts of antigen in the form of immune complexes in highly ordered units termed iccosomes (Cyster et al., 2000; Kosco-Vilbois, 2003; Szakal and Tew, 1991).
As the GC matures, two main compartments become evident, which were first described in 1930 through the careful study of cross-sections of cat lymph nodes (Röhlich, 1930). These compartments were termed dark and light zones based on their histological appearance, in which much of the light zone is occupied by FDC processes, whereas few processes extend into the dark zone where lymphocytes are closely packed (Figure 1). The FDCs of the light zone acquire features that distinguish them from FDCs in the primary follicles, including upregulation of VCAM-1 and FcγRIIB (Balogh et al., 2002; Cyster et al., 2000; Qin et al., 2000). It has been suggested that as the GC develops, some proliferating B cells move away from the FDC network to establish the dark zone (Liu et al., 1991b; Wang and Carter, 2005).
Figure 1. Schematic representation of GC compartments and cellular dynamics.
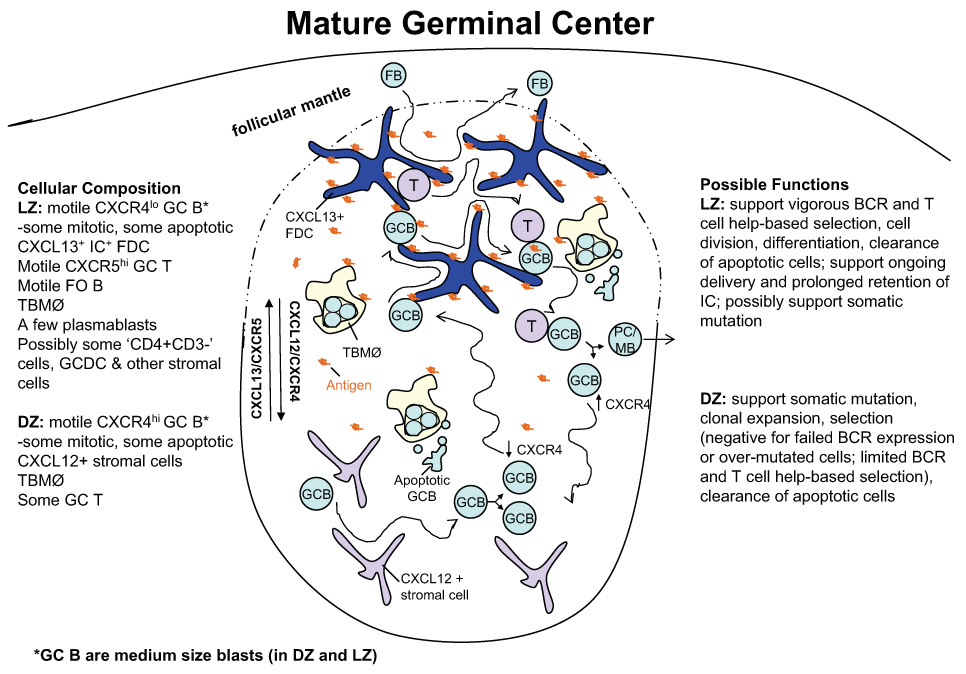
The cellular composition of the compartments is summarized on the left and possible functions of the compartments are described on the right. The diagram depicts the structure of an acute GC but chronic GCs, such as those typical of tonsils, may have additional levels of compartmentalization. GC B cells are medium sized blasts expressing low surface Ig and exhibiting a dendritic morphology, in both the dark and light zones. In humans, dark zone cells also express somewhat higher amounts of CD77 (Hardie et al., 1993) whereas CD44 may be higher on light zone cells (Feuillard et al., 1995). Additional cell types may sometimes be present including dendritic cells (Grouard et al., 1996), and CD4+CD3− cells (Kim et al., 2003). GC B cells migrate extensively within their respective compartments and move between compartments, most likely after modulating CXCR4 protein levels. Blebs of apoptotic GC B cells can associate with T cells, possibly limiting the availability of T cell help. Antigen is displayed as immune complexes (ICs) on FDCs but also possibly on tingible body macrophages (TBMØs) and motile B cells. Soluble antigen may also be present. Note the greater cellular heterogeneity and thus lower GC B cell density in the light zone than in the dark zone. See text for further details.
The light zone appears to be strategically positioned in secondary lymphoid organs toward the source of foreign antigens (Millikin, 1966). In the spleen, the light zone pole of the GC is proximal to the marginal sinus where blood-borne antigens enter the tissue. In lymph nodes, the light zone is positioned close to the subcapsular sinus, which receives afferent lymphatic drainage from the skin, mucosa, or viscera. In the Peyer’s patches, tonsils, and appendix, the light zone is oriented towards the mucosal surface. This orientation of the GC is concordant with evidence that antigen, often in the form of immune complexes, can be transported rapidly to GC light zones (Allen et al., 2007; Szakal et al., 1989).
The chemokine receptor, CXCR4, is needed for GC B cell positioning in the dark zone and its ligand, SDF-1 (CXCL12) is more abundant in this zone than in the light zone (Allen et al., 2004). SDF-1 appears to be produced locally by stromal cells (Allen et al., 2004) (Figure 1). Stromal cells have been described in the dark zone, although these apparently do not form a dense network (Opstelten et al., 1982; Rademakers, 1992). CXCL13 is more abundant in the light zone where it accumulates on the processes of FDCs and the CXCL13/CXCR5 chemokine/receptor pair is needed for GC B cells to accumulate normally in the light zone (Allen et al., 2004; Cyster et al., 2000) (Figure 1). Immunohistochemical staining shows that CXCR4 is more abundant in the dark zone than in the light zone and combined flow cytometric and BrdU incorporation analysis (discussed further below) reveals a profile of BrdU labeling in CXCR4hi and CXCR4lo GC B cells that corresponds to the kinetics of labeling in dark and light zones, respectively (Allen et al., 2004; Allen et al., 2007). In contrast with the differences in CXCR4 expression in dark and light zones, the GC B cell population as a whole has rather uniform CXCR5 expression. Although these data were largely collected in the mouse, staining of human tonsil also suggests CXCR4 is higher on dark zone cells (Forster et al., 1998). Taken together, these findings suggest that CXCR4 protein expression is tightly controlled on GC B cells and is a major factor controlling cell positioning in the dark zone versus the light zone.
Classical Model of GC Function in Affinity Maturation
Bringing together a century of observations from fixed tissue analysis and in vitro studies, MacLennan (1994) presented a model 13 years ago for the mechanism of GC organization and function. GC B cells in dark and light zones were historically defined as centroblasts and centrocytes, respectively. Centroblasts were named based on the observation of large, mitotically active cells in the dark zone that lacked surface immunoglobulin (Ig). These cells were proposed to undergo a rapid process of proliferation and somatic hypermutation of their antibody variable region genes. Centroblasts were then suggested to exit the cell cycle, reexpress surface Ig, and become smaller centrocytes that traveled to the light zone. This process was proposed to result in the generation of centrocytes expressing surface antibodies with a wide range of affinities for a given antigen. Centrocytes were then proposed to compete for binding to antigen in the form of immune complexes displayed on the surface of FDCs in the light zone. The presence of “tingible body” macrophages that have phagocytosed large numbers of GC B cells that were recently in cell cycle, suggests that most B cells die during this selection process. Selected centrocytes could then present antigen to helper T cells in the light zone, which could enhance survival or promote differentiation into antibody-secreting plasma cells or memory B cells.
This now classical model provided an important framework for further experimentation, and several features of the model have been well validated. However, some studies, including recent real-time imaging experiments, have led to findings that are not fully consistent with the original version of the model. In the sections below we discuss these findings and in the summary paragraph (and Figure 1 and Figure 3) we present a revised version of the model that attempts to incorporate this new information.
Figure 3. Two models of GC B cell selection within the GC.
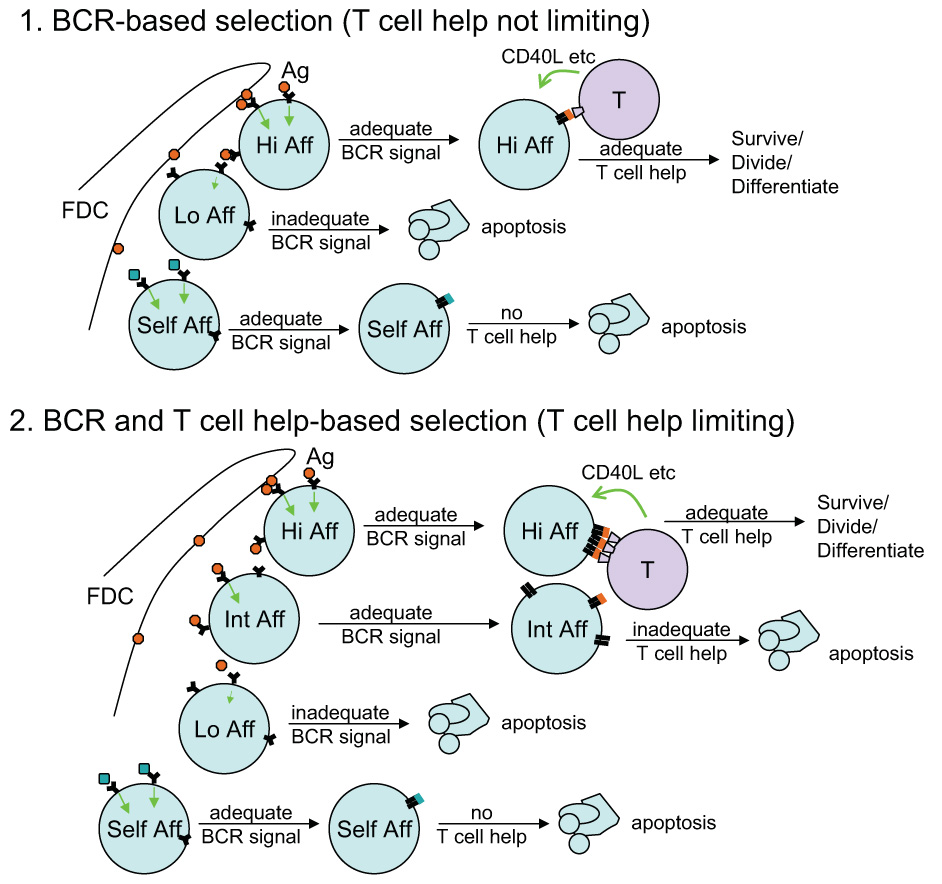
In the first model, discrimination between cells of different affinity for the foreign antigen occurs solely at the level of different strengths of BCR signaling. In the second model, sufficient BCR engagement is still necessary but further selection occurs due to competition for T cell help. B cells that have captured, processed, and presented more antigen as MHC-peptide complexes go on to receive T cell help at the expense of cells that have captured less antigen. See text for details.
Migration Dynamics of GC Cells
A striking feature that emerged in the real-time imaging studies was that GC B cells were highly motile and exhibited a dendritic morphology as they moved (Allen et al., 2007; Hauser et al., 2007; Schwickert et al., 2007). The overall motility rate was partially dependent on the chemokine CXCL13 (Allen et al., 2007). Unexpectedly, the morphology and velocities of cells in light and dark zones were similar. Moreover, real-time imaging showed that cell division and cell death occur in both compartments and not just in one zone (Allen et al., 2007). The latter findings will be discussed further in later sections but an important implication of these combined observations is that dark and light zone cells are more similar than the terms centroblast and centrocyte might suggest, a theme that we will continue to develop throughout this review.
An important feature of the classical model is that GC B cells migrate from the dark zone to the light zone (MacLennan, 1994). Evidence for this movement was originally obtained at the cell population level in studies where cells were labeled in S phase of the cell cycle with thymidine analogs, such as ³H-thymidine or bromodeoxyuridine (BrdU), and then their distribution was measured at various time points (Hanna, 1964; Koburg, 1966; Liu et al., 1991b). Enrichment of labeled cells in the dark zone after several hours was followed by the appearance of labeled cells in the light zone, suggesting that labeled cells had moved from the dark zone to the light zone. Indirect experimental results and theoretical modeling also suggested that some centrocytes might return to the dark zone to complete an additional round of mutation and selection (Kelsoe, 1996; Kepler and Perelson, 1993).
The migration of cells between dark and light zones has now been directly observed by real-time imaging (Allen et al., 2007; Hauser et al., 2007; Schwickert et al., 2007). In these studies, the light zone FDC network was visualized by the injection of fluorescently-labeled antibodies or through the deposition of fluorescent immune complexes. The studies differed in the criteria used to define cells that crossed between dark and light zones. Two groups focused on GCs that were of a size and orientation that allowed the dark and light zones to be seen in the xy plane, and then drew a line dividing the two zones and reported the frequency of cells that crossed this line (Hauser et al., 2007; Schwickert et al., 2007). The other study imaged a larger z-stack that permitted visualization of dark and light zones in 3-D and reported the frequency of cells that unidirectionally crossed a thick 20 µm boundary between the zones (Allen et al., 2007) (Figure 2). A common finding among all groups was that cells transited between dark and light zones in both directions, providing the first direct evidence that exchange occurs between these compartments. Although the rates of transit between dark and light zones differed in the three studies, it is important to emphasize that these rates should be viewed as imprecise estimates due to the relatively short observation periods and other technical limitations of two-photon microscopy. All of the groups used automated analysis software in which the cells were treated as spheres and their tracks were plotted over time. Two special problems arise, however, in this analysis: 1) when cells come too close together to determine the overall paths of the cells, the tracks are divided into multiple independent segments such that there might be several tracks generated for a single cell; and 2) cells frequently enter and exit the imaging volume and can only be tracked for a fraction of the time-lapse recording. Therefore, the density of cells in a recording and the overall volume imaged can have substantial effects on the ability to identify cells that traffic between zones. These issues also make it quite difficult to make precise statements about frequencies; for example, one cannot even precisely determine how many cells there were in the imaging volume in a given hour, when many of the cell tracks are shorter than 20 minutes. We therefore caution against interpreting these exchange rates to fit one model of selection versus another (Hauser et al., 2007), and instead emphasize the general finding that cells transit between dark and light zones.
Figure 2. GC B cell movement within and between GC dark and light zones.
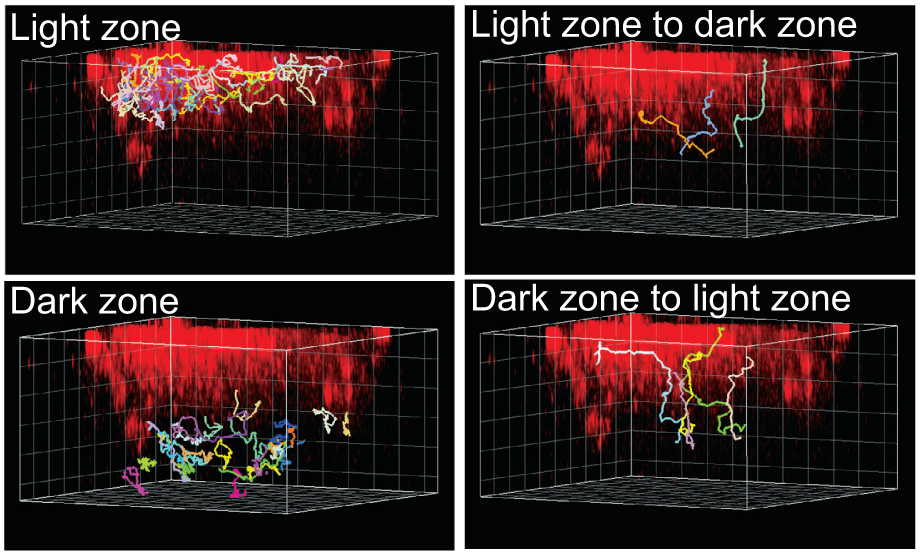
The light zone FDCs are identified by in vivo deposited PE immune complexes (red). Colored lines show tracks of GC B cells that moved within the light or dark zones or that crossed between zones, as indicated, during a 30 minute imaging session. The gridlines are separated by 20µm. Cell tracks were manually classified as being in the light or dark zones if the entire track stayed within the PE+ or PE− region, respectively, during the imaging session. Cell tracks were manually classified as traveling between zones if the tracks originated in one zone, crossed an approximately 20 µm border between the zones, then entered at least 10 µm into the other zone, and stayed in that zone for the duration of the analysis. Some tracks could not be definitively classified by these criteria, for example tracks that were too close to the border between the light and dark zones. From Allen et al., Science 315: 528 (2007).
The mechanism promoting cell movement from the dark zone to the light zone seems likely to be downregulation of surface CXCR4 expression (Allen et al., 2004). Movement from the light zone to the dark zone would reciprocally be promoted by CXCR4 upregulation. The ability of small changes in chemokine receptor expression to cause cells to migrate in a directed fashion across large distances to a new zone has been established for antigen-engaged B cells early in the antibody response (Okada et al., 2005; Reif et al., 2002). CXCR4 surface expression on GC B cells may perhaps be regulated directly by the cell cycle or in response to additional cues such as BCR triggering or ongoing SDF-1 exposure.
Proliferation Dynamics of GC Cells
The classical model of GC compartmentalization into dark and light zones defined the dark zone as the site of proliferation (MacLennan, 1994). However, recent work (Allen et al., 2007; Hauser et al., 2007; Rahman et al., 2003; Wang and Carter, 2005) has demonstrated that cells progress through the cell cycle in both dark and light zones. Cell division was observed in both zones during real-time imaging of GCs, although these events were difficult to quantify as they occurred infrequently in recordings that typically lasted less than one hour (Allen et al., 2007). When proliferation rates were quantified in both zones by flow cytometric cell cycle analysis, in which dark zone cells were identified by their higher CXCR4 expression, a similar proportion of cells were in S phase in both zones, with a somewhat higher frequency of cells in G2/M phase in the dark zone (Allen et al., 2007). While it is not possible to precisely delineate dark and light zone cells in flow cytometry by differences in CXCR4 expression, the validity of this procedure is supported by the corresponding immunohistochemical differences in CXCR4 expression between zones and by the genetic evidence that this receptor is needed for cell positioning in the dark zone (Allen et al., 2004).
It has been suggested that proliferation occurs in both dark and light zones in mice, but only in the dark zone in humans (Camacho et al., 1998) This suggestion was primarily based on staining for the cell cycle antigen Ki67 that failed to show dark and light zone polarity in mouse GCs (Camacho et al., 1998; Rahman et al., 2003; Wang and Carter, 2005), whereas this polarity is apparent in human GCs (Hardie et al., 1993; Liu et al., 1992). However, even in human GCs, it is evident that some Ki67+ cells are present in the light zone as well, calling into question the strength of this argument (Hardie et al., 1993; van Galen et al., 2004). It is also difficult to compare existing data on mouse and human GCs because most mouse studies were performed on acute GCs in the spleen or lymph nodes that were induced by immunization, whereas most human studies were performed on chronic GCs in the tonsils, which are continually exposed to foreign antigens. Substantial differences were seen in the degree of GC compartmentalization between human lymph node GCs and human tonsil GCs (Brachtel et al., 1996). For example, human tonsil GCs contain an outer zone rich in GC T cells, perhaps functioning to segregate GC B cell-T cell interactions in the outer zone from GC B cell-FDC interactions in the light zone. Recent studies also point to unique signaling requirements for chronic mouse GCs in mucosal lymphoid tissues (Casola and Rajewsky, 2006). Further studies will be needed to understand how the additional levels of organization described for chronic GCs may contribute to the function of these structures in the immune responses induced by chronic foreign antigen exposure.
The notion that proliferation occurs only in the dark zone was also based on older studies that determined the frequency of mitotic figures by histological examination of secondary lymphoid organs in various species (reviewed in MacLennan, 1994). These studies indicated that a higher frequency of mitotic figures was seen in the dark zone than in the light zone. However, it is important to note that in these studies, mitotic figures were present in the light zone (Kindred, 1938), a finding that was largely overlooked in subsequent descriptions. A recent study has confirmed the presence of mitotic figures in the light zones of mouse GCs (Wang and Carter, 2005). When considering the frequency of mitotic figures in dark and light zones, it is important to consider that the density of B cells is much higher in the dark zone than in the light zone, because much of the light zone space is occupied by the processes of FDCs (referred to in the older literature as reticulum cells) (Figure 1). Follicular helper T cells are also more abundant in the light zone than in the dark zone (Vinuesa et al., 2005), and some naive follicular B cells are seen to enter the GC at the light zone pole (Schwickert et al., 2007), perhaps accounting for the higher frequency of small lymphocytes seen in the light zone in older studies that were not undergoing mitosis. When the frequency of mitotic figures was considered only among medium sized lymphocytes, less than a two-fold difference was observed between dark and light zones (Kindred, 1938), consistent with a recent analysis (Allen et al., 2007). Therefore, these studies establish that mitosis occurs in light zones, but perhaps at a somewhat reduced rate compared with dark zones. These studies do not exclude the possibility that the mitoses in light and dark zones may have distinct functions, an idea that we will discuss further in our summary model below.
Further insight into the relationship between the cell cycle and GC B cell trafficking between dark and light zones has come from BrdU pulse-labeling studies (Allen et al., 2007; Hauser et al., 2007; Liu et al., 1991a). Due to a short in vivo half-life, a single dose of this thymidine analog pulse labels cells in S phase for less than 1–2 hours, after which the labeled cells can be tracked by immunohistochemistry. In one study the position of labeled cells was also assessed by flow cytometry, using CXCR4 surface expression as a proxy for dark and light zone cells (Allen et al., 2007). A combination of these techniques showed that cells were BrdU-labeled in S phase in both dark and light zones, but after completing mitosis, most of these labeled cells were cleared from the light zone and accumulated in the dark zone, which was evident five hours after BrdU injection. It remains unknown whether the BrdU-labeled cells in the light zone returned to the dark zone, exited the GC, or underwent apoptosis. After 12 hours, BrdU-labeled cells reemerged in the light zone but had not completed a second round of mitosis, suggesting that they migrated from the dark zone to the light zone, consistent with previous reports (Hanna, 1964; Koburg, 1966). Further supporting this interpretation, the frequency of dark zone cells that were BrdU-labeled decreased as the frequency of light zone cells that were BrdU-labeled increased. When taken together with the real-time imaging observations of cell transit between zones as described above, it appears likely that a substantial fraction of light zone cells are turned over within hours of time and replenished by cells from the dark zone that recently completed a cell cycle. These global analyses do not exclude the possibility, however, that a small fraction of GC B cells might stay resident in one zone or another for longer periods of time.
One further issue raised by recent studies relates to the cell cycle time of GC B cells. Previous work had indicated a range of possible cell cycle times, with an estimate of 5–7 hours becoming widely accepted over time (MacLennan, 1994). However, recent data obtained by sensitive flow cytometric techniques show that only a fraction of the originally BrdU-labeled cells had reentered S phase by 8–12 hours (Allen et al., 2007; Hauser et al., 2007). Although these studies differ somewhat in their experimental conditions and the rate at which cells first reenter S phase, both show that the cell cycles were not synchronous and that some cells would reenter S phase at times later than 12 hours. Indeed, such a conclusion was made by an older study that carefully analyzed the fraction of labeled mitoses in the GC after administration of ³H-thymidine (Zaitoun, 1980). Thus, these data can be interpreted to give the minimum cell cycle times, but not the average cell cycle times, which are longer and more difficult to determine. The basis for the asynchrony is not yet clear but seems consistent with the need for a positive input, perhaps from the BCR or helper T cells, to reenter S phase. Due to the variable distribution of antigen and cells in the GC, the time needed to obtain this input would vary.
Differences Between Dark and Light Zones and Implications for Selection
The similar morphology and motility of dark and light zone cells, the presence of proliferating cells in both compartments, and the rapid rate at which the light zone compartment is replaced leads us to reassess other reported differences between centroblasts and centrocytes. A classical distinction was that centroblasts lacked surface Ig, whereas centrocytes reexpressed surface Ig (Liu et al., 1992; Manser, 2004). It appears that this notion might have arisen from immunohistochemical analyses of GCs, in which more Ig was seen in the light zone. These studies must be interpreted with caution, however, as the FDCs in the light zone are known to capture large amounts of immune complexes containing various Ig isotypes (Kosco-Vilbois, 2003; Nieuwenhuis and Opstelten, 1984; Szakal et al., 1989). In one study, cells expressing high amounts of Ig mRNA were found at the base of the light zone; however, these cells were relatively infrequent and expressed large amounts of cytoplasmic Ig protein, suggesting that they may have been antibody-secreting plasma cells (Close et al., 1990). Several other studies have identified rare cells with low expression of plasma cell markers in the light zone (Angelin-Duclos et al., 2000; Carbone et al., 2001; Falini et al., 2000). It is interesting to note that no study to date has demonstrated by the routine quantitative method of flow cytometry that centroblasts and centrocytes differ in the amount of surface Ig. In mouse studies nearly all GC B cells have been described to express surface Ig by flow cytometry (Shinall et al., 2000; Wang and Carter, 2005; Wolniak et al., 2006), typically in low amounts compared with naïve B cells (Rossbacher et al., 2006). Taken together with immunohistochemical observations that Ig levels appear low in the dark zone, where immune complexes do not deposit on FDCs, (Kosco-Vilbois, 2003; Liu et al., 1992; Opstelten et al., 1982), these data suggest that most GC B cells express reduced surface Ig compared with naïve cells, but that this feature does not distinguish centroblasts from centrocytes.
It has also been suggested that somatic hypermutation occurs in the dark zone but not in the light zone (Zhang et al., 1988). While this remains an attractive notion, it is currently supported by only limited direct evidence. Activation-induced cytidine deaminase (AID), a protein essential for somatic hypermutation and class switch recombination, was found to be expressed primarily in the dark zone in human tonsil GCs by immunohistochemistry (Cattoretti et al., 2006; Moldenhauer et al., 2006). A very small number of cells in the light zone were positive for AID, perhaps even fewer cells than were positive for Ki67, although no quantitative data were provided. Because of the low GC B cell density in the light zone compared with the dark zone, it is not clear what fraction of GC B cells in the light zone expressed AID. It is also not yet known whether such a polarized distribution occurs in GCs in other tissues or in mice.
Recently several groups have performed microarray studies of gene expression in human B cell subsets that included GC B cells separated on the basis of differences in CD77 expression, a marker suggested to be more abundant on centroblasts than on centrocytes (Klein et al., 2003; Nakayama et al., 2006; Shaffer et al., 2002). While these studies have shown a large number of genes differentially expressed between GC B cells and naïve B cells, relatively few differences were seen between centroblasts and centrocytes. The main differences observed were the expression of some plasma cell specific genes among centrocytes, suggesting that a fraction of centrocytes were differentiating into plasma cells. These microarray results are therefore also consistent with the view that centroblasts and centrocytes are much more similar than previously thought. A caveat to these studies is that CD77 shows a broad range of staining intensity on GC B cells and has been argued to be insufficient to discriminate between centroblasts and centrocytes (Feuillard et al., 1995). In addition, the microarray results suggest that most observed differences between dark and light zone cells may be regulated at the protein level. In this regard is it notable that although CXCR4 protein was more abundant in dark zone cells compared with light zone cells, similar amounts of CXCR4 mRNA were detected in these cells (Allen et al., 2004). Polycomb proteins and the nuclear GANP protein have also been described to be differentially expressed in dark and light zones in GCs, although it is unclear whether this expression is regulated at the mRNA or protein level (Kuwahara et al., 2000; van Galen et al., 2004). Further studies will be needed to define other differences in the proteins expressed by dark zone cells and light zone cells and their functional importance.
In the classical model of GC organization and function, selection depends on competition among centrocytes for firm adhesion to light zone FDCs based on the affinity of their BCRs for antigen held on the FDC processes in the form of immune complexes (MacLennan, 1994). However, several experimental results present challenges to this classical model. In mice lacking serum Ig and therefore deficient in immune complexes, GC size was not affected (Hannum et al., 2000). Some evidence for selection was obtained, although the affinity maturation process could not be fully assessed in these studies due to the use of transgenic mice expressing multiple copies of a single Ig heavy chain (Hannum et al., 2000). A complexity to the interpretation of these studies is that the reduced ability to form immune complexes may itself lead to a non-physiological elevation in free antigen due to impaired antigen clearance. When FDC lacked the expression of complement receptors 1 and 2 and showed a marked decrease in the trapping of immune complexes, GC formation still occurred although sustained IgG responses were affected (Fang et al., 1998). Perhaps the strongest evidence to date against the view that selection depends on competition for firm adhesion to FDCs is the data from real-time imaging studies (Allen et al., 2007; Hauser et al., 2007; Schwickert et al., 2007). Most GC B cells migrate rapidly and continuously within the network of FDC and tingible body macrophages. Although rare cases of prolonged contacts were observed in all the studies, these contacts have not been carefully quantified and their significance is not yet clear. A limitation in the studies to date has been that antigen distribution in the GC and capture by GC B cells have not been tracked, a challenge that must be addressed in future studies. However, it seems reasonable to suggest from the information available that GC B cells capture antigen by rapidly moving over the FDC network - as well as tingible body macrophages and each other - in a scavenging-type behavior rather than by making prolonged synaptic contacts with antigen-bearing cells. The dendritic morphology of the migrating cells seems likely to facilitate antigen detection by increasing their surface area. Whether cell movement speeds averaging 6–7 µm/min permit cell processes to capture antigen through local membrane spreading and contraction events (Fleire et al., 2006) needs further assessment. The high expression of integrin ligands VCAM-1 and ICAM-1 on FDCs (Freedman et al., 1990; Koopman et al., 1991) suggests that they will be used by migrating GC B cells, perhaps to enhance the efficiency of signaling in response to antigen engagement (Carrasco and Batista, 2006), though a direct role for these adhesion molecules in affinity maturation remains to be established.
Hauser et al. (2007) conclude that the frequency of GC B cell recirculation between dark and light zones is insufficient to account for a selection mechanism that occurs only in the light zone, and therefore propose an intrazonal recirculation process in which most cells stay resident in dark or light zones and selection in the dark and light zones operates independently. Another group recently suggested that proliferation and selection occur independently in dark and light zones based on the study of CD19 signaling mutants (Wang and Carter, 2005). These studies raise the important issue that there is little if any direct evidence that selection occurs in the light zone but not in the dark zone. Although large amounts of immune complexes are deposited in the light zone, these observations have not excluded that antigen may be available to cells in the dark zone below the limits of detection by standard methods such as immunohistochemistry. Arguing against a purely intrazonal recirculation and selection mechanism, however, the real-time imaging and BrdU pulse-labeling studies described above indicate that cells do transit between zones at a rate that would result in substantial exchange within twelve hours, which is less than the average cell cycle time of GC B cells. In addition, as discussed further below, CD4 T cells are enriched in the light zone and there is good evidence that T cells are required for selection of GC B cells. Hauser et al. (2007) suggest that 100% exchange must occur in each cell cycle for selection to occur only in the light zone, and while some results are not inconsistent with this possibility (Allen et al., 2007; Schwickert et al., 2007), we also caution that this statement is based on theoretical modeling which cannot yet fully account for the complex cellular milieu and in vivo dynamics of the GC.
As expected, based on the long recognized segregation of the GC structure from the surrounding follicular mantle, the imaging analysis showed that most GC B cells and follicular mantle B cells turned at the interface between the compartments and did not cross from one region into the other (Allen et al., 2007). Some cells clearly crossed this boundary however although their ultimate fate could not be ascertained. Schwickert et al. (2007) proposed that the entry of follicular mantle B cells into the GC light zone might allow new clones to participate in the GC response, and demonstrated that this can occur by the recruitment of adoptively transferred high affinity B cells into preexisting GCs. The extent to which this process operates in normal immune responses remains to be determined. A recent modeling study has argued that migration of cells between GCs may play a general role in achieving maximal affinity maturation (Or-Guil et al., 2007). Of possible relevance in this regard, more sensitive techniques have demonstrated that antigen-specific GCs may be present for many weeks of time (Gatto et al., 2007; Takahashi et al., 2001), in contrast to the 2–3 week duration suggested by earlier studies (Liu et al., 1991b; Takahashi et al., 1998). In the immune response to the hapten nitrophenyl, selection occurs in GCs for a mutation of Trp33 to Leu in VH186.2 within the first 2–3 weeks giving an approximately 10-fold increase in affinity, but after 6 weeks a novel VH186.2 VDJ rearrangement emerges with Trp33 and Gly99 that exhibits even higher affinity (Furukawa et al., 1999). It is unclear whether this novel rearrangement emerges because of the entry of additional cells into existing GCs or because of competition between cells that emerged from distinct GCs.
T Cells in the GC
The GC response has a well characterized dependence on helper T cells and CD40 (Gaspal et al., 2006; Grewal and Flavell, 1998; MacLennan, 1994; Vinuesa et al., 2005). The T cell costimulatory molecules CD28 and ICOS are also important and the cytokines IL-4 and IL-21 contribute to the response (Cunningham et al., 2004; Ozaki et al., 2002; Reiter and Pfeffer, 2002; Sharpe and Freeman, 2002; Vajdy et al., 1995). GCs that are formed in response to T-independent antigens collapse shortly after compartmentalization into dark and light zones, suggesting that signals from T cells are essential for maintaining the response (Manser, 2004; Vinuesa et al., 2000). GC T cells belong to the follicular T helper cell subset and are characterized by high expression of CXCR5 and ICOS (Vinuesa et al., 2005). T cells constitute approximately 5–20% of GC cells and are enriched within the light zone, though low numbers can also be found in the dark zone (Arnold et al., 2007; Brachtel et al., 1996; Haynes et al., 2007). Positioning in the light zone is CXCR5 dependent and in mice where T cells lack CXCR5, the GC response is reduced in magnitude, consistent with the model that T-dependent selection events preferentially occur in the light zone (Arnold et al., 2007; Haynes et al., 2007). However, the alternative possibility that the reduced GC response was due to reduced overall recruitment of T cells into the B cell follicle has not been ruled out.
GC B cells express elevated amounts of MHC class II compared to follicular B cells (Fallas et al., 2007; Glazier et al., 2002; Shinall et al., 2000). By contrast, they have reduced amounts of HLA-DO (H2-O in the mouse), a molecule that is thought to modify the peptide exchange activity of HLA-DM (Fallas et al., 2007). In addition, GC B cells endocytose antigen into MHC class II antigen processing compartments more rapidly than naïve B cells (Chalouni et al., 2003). These findings suggest that there is a reprogramming of the antigen uptake, processing and presentation pathway in GC B cells, consistent with the possibility that these cells have specialized antigen-presentation requirements.
Important early insights into the kinetics of B–T interactions in the first phases of the antibody response were obtained by fixed tissue analysis, in which it was possible to observe colocalized antigen-specific B and T cells surrounded by numerous non-specific cells (Garside et al., 1998; MacLennan et al., 1997). This approach was less informative for the GC because the majority of B cells that surround each T cell recognize the same antigen, making it unclear whether specific interactions are taking place. Dynamic imaging analysis has overcome this limitation since it allows cell conjugates to be tracked over time. This analysis revealed that most GC B cell - T cell contacts were of short (less than 5 minute) duration and most likely did not engage the T cell in a productive conjugate (Allen et al., 2007). In addition, although GC B cells have been estimated to be 5–20 times more abundant in the GC than GC T cells, less than a third of the T cells were moving at the slow B cell velocities that have so far typified cognate B–T conjugates (Allen et al., 2007; Okada et al., 2005). These findings suggest that GC B cell interactions with GC T cells are tightly regulated, and that GC B cells compete for cognate interactions with GC T cells. They favor a model, elaborated upon further below, in which one layer of competition between GC B cells is for cognate T cell help (Figure 3). Competition for T cell help within the GC was also proposed in a recent theoretical study as possibly the most effective mechanism for achieving selection of high affinity clones (Meyer-Hermann et al., 2006). Consistent with few GC B cells receiving cognate T cell help at any one time, analysis of GC cells for nuclear cRel as an indicator of CD40 signaling identified only small numbers of positive cells in the light zone (Basso et al., 2004). In another study, CD45-deficient B cells that showed defective BCR signaling were found to enter GC responses and undergo affinity maturation, though the response largely collapsed after seven days (Huntington et al., 2006). To account for the observed affinity maturation in this system, it was suggested that selection occurred by BCR-mediated internalization and antigen presentation to T cells rather than by differential BCR signaling (Huntington et al., 2006).
A variety of factors are likely to regulate the formation of stable conjugates between GC B and T cells. Based on findings for DC-T cell interactions, the propensity for long contacts is strongly associated with the amounts of MHC-peptide complexes displayed on the antigen presenting cell (Garcia et al., 2007). Within the GC, some T cells were found to be engaged in stable interactions with blebs from apoptotic GC B cells, a process that may contribute to the competition for T cell help (Allen et al., 2007). MHC class II expressing microvesicles have been observed inside GCs by immuno-electron microscopy, though they were suggested to be exosomes, which are microvesicles that are smaller than those observed by two-photon microscopy and that can be released from viable cells (Denzer et al., 2000). It will be of interest in future studies to determine whether GC T cells interact with B cell-derived exosomes. Heterogeneity in the GC T cell compartment, as suggested for example by differences in CD57 expression (Bowen et al., 1991; Kim et al., 2001), may further limit the number of T cells available as helpers. Many GC T cells have high expression of the negative costimulatory molecule, PD-1 (Cai et al., 2004; Haynes et al., 2007; Iwai et al., 2002). PD-1 is upregulated on chronically stimulated (‘exhausted’) T cells (Barber et al., 2006; Day et al., 2006) and it seems possible that the high level of PD-1 on GC T cells is an indication of chronic encounter with antigen-presenting GC B cells. CTLA-4 is abundantly expressed on GC T cells (Castan et al., 1997; Marinova et al., 2007) and the ligands CD80 and CD86 are upregulated on GC B cells (Glazier et al., 2002). In an intriguing series of experiments, Lane and coworkers obtained evidence that CD28 engagement is not required within established GCs and that instead engagement of CTLA-4 is important in limiting T cell help since interrupting CTLA-4 function increased GC size several fold (Walker et al., 2003). CTLA-4 can function in T cells to limit the activity of the positive costimulatory ICOS molecule (Riley et al., 2001). PD-1 and CTLA-4 may be components of a program acting to increase the threshold of MHC-peptide recognition that is needed to overcome negative signaling and permit a helper response. FoxP3+ regulatory T cells may further influence the availability of T cell help, though only limited data are available on their presence inside GC (Fields et al., 2005). A recent in vitro study has suggested that CD57+ GC T cells may have some regulatory capabilities without expressing FoxP3 (Marinova et al., 2007). It seems likely that many of the same mechanisms that limit T cell help to the highest affinity clones also function to ensure that help is not provided to any newly emerging autoreactive clones. Competition for T cell help may achieve selection against high affinity clones that cross-react with self-antigens, since these clones would internalize a mixture of foreign- and self-antigens and as a result would likely dilute the amount of foreign peptide-MHC complexes that they display compared to non-cross-reactive high affinity clones. Autoreactive clones may also be selectively recognized and controlled or eliminated by regulatory T cells (Fields et al., 2005; Lim et al., 2005; Zhao et al., 2006).
GC B Cell Apoptosis
Many of the B cells generated within the GC die in situ. The nuclei of dead B cells are identified as tingible bodies clustered inside macrophages (MacLennan, 1994) (Figure 1). Tingible body macrophages are present in both dark and light zones of GC, with some bias toward the junctional region (Liu et al., 1992), and imaging analysis revealed examples of GC B cells that underwent apoptosis in both zones (Allen et al., 2007). These findings provide evidence that selection occurs in both zones, though they do not shed light on the type of selection. In addition to undergoing negative selection due to inadequate BCR engagement or T cell help, cells may die because of acquiring mutations that inactivate BCR expression causing loss of BCR-mediated pro-survival signals, or because they have accumulated too many mutations and activated a DNA damage checkpoint. Cells may also die as part of the homeostatic mechanism that controls GC size and limits the spread of these rapidly-dividing cells. As noted above, dying GC B cells were observed to release small and large blebs that were often taken up by nearby macrophages, but the small blebs could also be picked up by motile GC T cells (Allen et al., 2007). Release of apoptotic blebs was only reported in one of the three imaging studies (Allen et al., 2007), perhaps because of differences in the intensity of GFP reporter expression and instrument sensitivity, but clearly indicating the need for further analysis in multiple systems. Consistent with the imaging data, human GC B cells have been observed to release spherical membranous vesicles during cell death in vitro (Segundo et al., 1999). Studies in several cell types showed that caspases can cleave and activate the Rho kinase ROCK-I, in turn causing myosin light chain kinase activation and inducing the events necessary for plasma membrane blebbing (Coleman et al., 2001; Sebbagh et al., 2001; Shiratsuchi et al., 2002). Although blebbing can occur prior to macrophage uptake, DNA fragmentation may not occur until after engulfment (Nakamura et al., 1996). In two-photon microscopy it was observed that despite the frequent release of blebs from dying cells, a large cell fragment always remained and was sometimes seen to be engulfed whole by a nearby macrophage (Allen et al., 2007), consistent with blebbing being restricted to cytoplasmic contents. An absence of nuclear breakdown outside macrophages is likely to be important for avoiding autoimmunity since defective clearance of apoptotic cells in GCs has been correlated with induction of anti-nuclear antibodies (Hanayama et al., 2004).
Summary Model
Based on the studies summarized above, we suggest the following revised model for GC organization and function in selection (see also Figure 1 & Figure 3). GC B cells are constantly in cycle and accumulate in the dark zone after undergoing mitosis. Cellular growth during the early G1 phase of the cell cycle occurs preferentially in the dark zone, possibly to leave open space in the light zone for cells undergoing selection. Somatic mutation may also occur preferentially during this period. Several hours after entering the G1 phase of the cell cycle, CXCR4 is downmodulated on some of the cells. These cells are then no longer retained by SDF-1 in the dark zone and can migrate to the light zone in response to CXCL13. During this period, the cells most likely have substantially turned over their low amounts of surface Ig, replacing the original BCRs with newly mutated versions. Within the light zone, the GC B cells rapidly move about the FDC network where they may pick up antigen, receiving pro-survival BCR signals while also internalizing, processing and presenting the antigen as peptides on MHC class II molecules. Higher affinity cells may be able to capture more antigen than lower affinity cells during rapid movement past the FDC processes, such as by achieving greater shear force. In addition, as the GC B cells are in close proximity, higher affinity cells may be able to pick up antigen from the surface of lower affinity cells in direct cell-cell competition prior to antigen internalization. The GC B cells then compete with other GC B cells and dead B cell blebs for T cell help in the light zone. As T cells must polarize in a fashion that only allows interaction with one antigen presenting cell at a time, a single GC T cell is suggested to only form a stable interaction with the GC B cell that presents the highest number of antigen peptide-MHC class II complexes compared with neighboring cells. The threshold for helper T cell activation may be continuously adjusted in relation to the average amount of MHC-peptide exposure in a given GC. The time window for selection in the light zone is tightly regulated, such that all GC B cells must undergo apoptosis, exit the GC, or return to the dark zone within a few hours of entering the light zone. Cells exiting the GC rapidly differentiate into long-lived plasma cells or memory B cells.
The logic for cell proliferation in the light zone as well as in the dark zone is not yet clear but we suggest two non-mutually exclusive possibilities. First, the zones may not represent fully segregated states and there may therefore be further diversification of surface Ig by somatic mutation and cell division in the light zone. Second, positive selection by antigen-recognition and T cell help may be necessarily followed by an asymmetric cell division event to generate distinct daughter cells, such as a plasmablast or memory cell precursor and another GC B cell. Evidence has recently been obtained for asymmetric cell division playing a role in the generation of effector and memory CD8 T cells (Chang et al., 2007).
We emphasize that despite the many recent advances, any model of GC organization and function continues to be very much a working model due to the large number of unknowns about this complex structure. Certainly we can anticipate that the ongoing application of real-time imaging analysis combined with more sophisticated cell and antigen labeling methods and an array of further perturbations will lead to more revelations. Already, though, one has the emerging sense that the system supporting antibody affinity maturation is an example of a robust biological system (Kitano, 2004) where numerous, seemingly redundant processes operate to ensure that the system functions stably even in the face of extensive variability in inputs or perturbations. For example, selection may well frequently occur in both zones but may be more effective in the light zone for certain antigens; in some cases, antigen abundance and complexity may be such that BCR based selection is sufficient to achieve affinity maturation whereas in other cases competition for T cell help may be an essential requirement. Thus, it may be wise in future studies to avoid assumptions of absoluteness regarding requirements for selection in GCs and to instead begin with the view that there will be a range of mechanisms that operate around a common framework.
Acknowledgments
We thank Ian C.M. MacLennan, Tri G. Phan, and Jesse A. Green for helpful comments on the manuscript. C.D.C.A. is a predoctoral fellow and J.G.C. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen CD, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, Cyster JG. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. 2004;5:943–952. doi: 10.1038/ni1100. [DOI] [PubMed] [Google Scholar]
- Allen CD, Okada T, Tang HL, Cyster JG. Imaging of germinal center selection events during affinity maturation. Science. 2007;315:528–531. doi: 10.1126/science.1136736. [DOI] [PubMed] [Google Scholar]
- Angelin-Duclos C, Cattoretti G, Lin KI, Calame K. Commitment of B lymphocytes to a plasma cell fate is associated with Blimp-1 expression in vivo. J Immunol. 2000;165:5462–5471. doi: 10.4049/jimmunol.165.10.5462. [DOI] [PubMed] [Google Scholar]
- Arnold CN, Campbell DJ, Lipp M, Butcher EC. The germinal center response is impaired in the absence of T cell-expressed CXCR5. Eur J Immunol. 2007;37:100–109. doi: 10.1002/eji.200636486. [DOI] [PubMed] [Google Scholar]
- Balogh P, Aydar Y, Tew JG, Szakal AK. Appearance and phenotype of murine follicular dendritic cells expressing VCAM-1. Anat Rec. 2002;268:160–168. doi: 10.1002/ar.10148. [DOI] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- Basso K, Klein U, Niu H, Stolovitzky GA, Tu Y, Califano A, Cattoretti G, Dalla-Favera R. Tracking CD40 signaling during germinal center development. Blood. 2004;104:4088–4096. doi: 10.1182/blood-2003-12-4291. [DOI] [PubMed] [Google Scholar]
- Benson MJ, Erickson LD, Glesson MW, Noelle RJ. Affinity of antigen encounter and other early B-cell signals determine B-cell fate. Curr Opin Immunol. 2007;19:275–280. doi: 10.1016/j.coi.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berek C, Berger A, Apel M. Maturation of the immune response in germinal centers. Cell. 1991;67:1121–1129. doi: 10.1016/0092-8674(91)90289-b. [DOI] [PubMed] [Google Scholar]
- Blink EJ, Light A, Kallies A, Nutt SL, Hodgkin PD, Tarlinton DM. Early appearance of germinal center-derived memory B cells and plasma cells in blood after primary immunization. J Exp Med. 2005;201:545–554. doi: 10.1084/jem.20042060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen MB, Butch AW, Parvin CA, Levine A, Nahm MH. Germinal center T cells are distinct helper-inducer T cells. Hum Immunol. 1991;31:67–75. doi: 10.1016/0198-8859(91)90050-j. [DOI] [PubMed] [Google Scholar]
- Brachtel EF, Washiyama M, Johnson GD, Tenner-Racz K, Racz P, MacLennan IC. Differences in the germinal centres of palatine tonsils and lymph nodes. Scand J Immunol. 1996;43:239–247. [PubMed] [Google Scholar]
- Cai G, Karni A, Oliveira EM, Weiner HL, Hafler DA, Freeman GJ. PD-1 ligands, negative regulators for activation of naive, memory, and recently activated human CD4+ T cells. Cell Immunol. 2004;230:89–98. doi: 10.1016/j.cellimm.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Camacho SA, Kosco-Vilbois MH, Berek C. The dynamic structure of the germinal center. Immunol Today. 1998;19:511–514. doi: 10.1016/s0167-5699(98)01327-9. [DOI] [PubMed] [Google Scholar]
- Carbone A, Gloghini A, Larocca LM, Capello D, Pierconti F, Canzonieri V, Tirelli U, Dalla-Favera R, Gaidano G. Expression profile of MUM1/IRF4, BCL-6, and CD138/syndecan-1 defines novel histogenetic subsets of human immunodeficiency virus-related lymphomas. Blood. 2001;97:744–751. doi: 10.1182/blood.v97.3.744. [DOI] [PubMed] [Google Scholar]
- Carrasco YR, Batista FD. B-cell activation by membrane-bound antigens is facilitated by the interaction of VLA-4 with VCAM-1. Embo J. 2006;25:889–899. doi: 10.1038/sj.emboj.7600944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casola S, Rajewsky K. B cell recruitment and selection in mouse GALT germinal centers. Curr Top Microbiol Immunol. 2006;308:155–171. doi: 10.1007/3-540-30657-9_7. [DOI] [PubMed] [Google Scholar]
- Castan J, Tenner-Racz K, Racz P, Fleischer B, Broker BM. Accumulation of CTLA-4 expressing T lymphocytes in the germinal centres of human lymphoid tissues. Immunology. 1997;90:265–271. doi: 10.1046/j.1365-2567.1997.00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattoretti G, Buttner M, Shaknovich R, Kremmer E, Alobeid B, Niedobitek G. Nuclear and cytoplasmic AID in extrafollicular and germinal center B cells. Blood. 2006;107:3967–3975. doi: 10.1182/blood-2005-10-4170. [DOI] [PubMed] [Google Scholar]
- Chalouni C, Banchereau J, Vogt AB, Pascual V, Davoust J. Human germinal center B cells differ from naive and memory B cells by their aggregated MHC class II-rich compartments lacking HLA-DO. Int Immunol. 2003;15:457–466. doi: 10.1093/intimm/dxg037. [DOI] [PubMed] [Google Scholar]
- Chang JT, Palanivel VR, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, Longworth SA, Vinup KE, Mrass P, Oliaro J, et al. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science. 2007;315:1687–1691. doi: 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]
- Close PM, Pringle JH, Ruprai AK, West KP, Lauder I. Zonal distribution of immunoglobulin-synthesizing cells within the germinal centre: an in situ hybridization and immunohistochemical study. J Pathol. 1990;162:209–216. doi: 10.1002/path.1711620306. [DOI] [PubMed] [Google Scholar]
- Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3:339–345. doi: 10.1038/35070009. [DOI] [PubMed] [Google Scholar]
- Cunningham AF, Serre K, Toellner KM, Khan M, Alexander J, Brombacher F, MacLennan IC. Pinpointing IL-4-independent acquisition and IL-4-influenced maintenance of Th2 activity by CD4 T cells. Eur J Immunol. 2004;34:686–694. doi: 10.1002/eji.200324510. [DOI] [PubMed] [Google Scholar]
- Cyster JG, Ansel KM, Reif K, Ekland EH, Hyman PL, Tang HL, Luther SA, Ngo VN. Follicular stromal cells and lymphocyte homing to follicles. Immunol Rev. 2000;176:181–193. doi: 10.1034/j.1600-065x.2000.00618.x. [DOI] [PubMed] [Google Scholar]
- Dal Porto JM, Haberman AM, Kelsoe G, Shlomchik MJ. Very low affinity B cells form germinal centers, become memory B cells, and participate in secondary immune responses when higher affinity competition is reduced. J Exp Med. 2002;195:1215–1221. doi: 10.1084/jem.20011550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Porto JM, Haberman AM, Shlomchik MJ, Kelsoe G. Antigen drives very low affinity B cells to become plasmacytes and enter germinal centers. J Immunol. 1998;161:5373–5381. [PubMed] [Google Scholar]
- Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- Denzer K, van Eijk M, Kleijmeer MJ, Jakobson E, de Groot C, Geuze HJ. Follicular dendritic cells carry MHC class II-expressing microvesicles at their surface. J Immunol. 2000;165:1259–1265. doi: 10.4049/jimmunol.165.3.1259. [DOI] [PubMed] [Google Scholar]
- Eisen HN, Siskind GW. Variations in Affinities of Antibodies During the Immune Response. Biochemistry. 1964;3:996–1008. doi: 10.1021/bi00895a027. [DOI] [PubMed] [Google Scholar]
- Falini B, Fizzotti M, Pucciarini A, Bigerna B, Marafioti T, Gambacorta M, Pacini R, Alunni C, Natali-Tanci L, Ugolini B, et al. A monoclonal antibody (MUM1p) detects expression of the MUM1/IRF4 protein in a subset of germinal center B cells, plasma cells, and activated T cells. Blood. 2000;95:2084–2092. [PubMed] [Google Scholar]
- Fallas JL, Yi W, Draghi NA, O'Rourke HM, Denzin LK. Expression patterns of H2-O in mouse B cells and dendritic cells correlate with cell function. J Immunol. 2007;178:1488–1497. doi: 10.4049/jimmunol.178.3.1488. [DOI] [PubMed] [Google Scholar]
- Fang Y, Xu C, Fu YX, Holers VM, Molina H. Expression of complement receptors 1 and 2 on follicular dendritic cells is necessary for the generation of a strong antigen-specific IgG response. J Immunol. 1998;160:5273–5279. [PubMed] [Google Scholar]
- Feuillard J, Taylor D, Casamayor-Palleja M, Johnson GD, MacLennan IC. Isolation and characteristics of tonsil centroblasts with reference to Ig class switching. Int Immunol. 1995;7:121–130. doi: 10.1093/intimm/7.1.121. [DOI] [PubMed] [Google Scholar]
- Fields ML, Hondowicz BD, Metzgar MH, Nish SA, Wharton GN, Picca CC, Caton AJ, Erikson J. CD4+ CD25+ regulatory T cells inhibit the maturation but not the initiation of an autoantibody response. J Immunol. 2005;175:4255–4264. doi: 10.4049/jimmunol.175.7.4255. [DOI] [PubMed] [Google Scholar]
- Fleire SJ, Goldman JP, Carrasco YR, Weber M, Bray D, Batista FD. B cell ligand discrimination through a spreading and contraction response. Science. 2006;312:738–741. doi: 10.1126/science.1123940. [DOI] [PubMed] [Google Scholar]
- Forster R, Kremmer E, Schubel A, Breitfeld D, Kleinschmidt A, Nerl C, Bernhardt G, Lipp M. Intracellular and surface expression of the HIV-1 coreceptor CXCR4/fusin on various leukocyte subsets: rapid internalization and recycling upon activation. J Immunol. 1998;160:1522–1531. [PubMed] [Google Scholar]
- Freedman AS, Munro JM, Rice GE, Bevilacqua MP, Morimoto C, McIntyre BW, Rhynhart K, Pober JS, Nadler LM. Adhesion of human B cells to germinal centers in vitro involves VLA-4 and INCAM-110. Science. 1990;249:1030–1033. doi: 10.1126/science.1697696. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Akasako-Furukawa A, Shirai H, Nakamura H, Azuma T. Junctional amino acids determine the maturation pathway of an antibody. Immunity. 1999;11:329–338. doi: 10.1016/s1074-7613(00)80108-9. [DOI] [PubMed] [Google Scholar]
- Futterer A, Mink K, Luz A, Kosco-Vilbois MH, Pfeffer K. The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity. 1998;9:59–70. doi: 10.1016/s1074-7613(00)80588-9. [DOI] [PubMed] [Google Scholar]
- Garcia Z, Pradelli E, Celli S, Beuneu H, Simon A, Bousso P. Competition for antigen determines the stability of T cell-dendritic cell interactions during clonal expansion. Proc Natl Acad Sci U S A. 2007;104:4553–4558. doi: 10.1073/pnas.0610019104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garside P, Ingulli E, Merica RR, Johnson JG, Noelle RJ, Jenkins MK. Visualization of specific B and T lymphocyte interactions in the lymph node. Science. 1998;281:96–99. doi: 10.1126/science.281.5373.96. [DOI] [PubMed] [Google Scholar]
- Gaspal FM, McConnell FM, Kim MY, Gray D, Kosco-Vilbois MH, Raykundalia CR, Botto M, Lane PJ. The generation of thymus-independent germinal centers depends on CD40 but not on CD154, the T cell-derived CD40-ligand. Eur J Immunol. 2006;36:1665–1673. doi: 10.1002/eji.200535339. [DOI] [PubMed] [Google Scholar]
- Gatto D, Martin SW, Bessa J, Pellicioli E, Saudan P, Hinton HJ, Bachmann MF. Regulation of memory antibody levels: the role of persisting antigen versus plasma cell life span. J Immunol. 2007;178:67–76. doi: 10.4049/jimmunol.178.1.67. [DOI] [PubMed] [Google Scholar]
- Glazier KS, Hake SB, Tobin HM, Chadburn A, Schattner EJ, Denzin LK. Germinal center B cells regulate their capability to present antigen by modulation of HLA-DO. J Exp Med. 2002;195:1063–1069. doi: 10.1084/jem.20012059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16:111–135. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- Grouard G, Durand I, Filgueira L, Banchereau J, Liu YJ. Dendritic cells capable of stimulating T cells in germinal centres. Nature. 1996;384:364–367. doi: 10.1038/384364a0. [DOI] [PubMed] [Google Scholar]
- Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, Nagata S. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304:1147–1150. doi: 10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- Hanna MG., Jr. An Autoradiographic Study of the Germinal Center in Spleen White Pulp During Early Intervals of the Immune Response. Lab Invest. 1964;13:95–104. [PubMed] [Google Scholar]
- Hannum LG, Haberman AM, Anderson SM, Shlomchik MJ. Germinal center initiation, variable gene region hypermutation, and mutant B cell selection without detectable immune complexes on follicular dendritic cells. J Exp Med. 2000;192:931–942. doi: 10.1084/jem.192.7.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DL, Johnson GD, Khan M, MacLennan IC. Quantitative analysis of molecules which distinguish functional compartments within germinal centers. Eur J Immunol. 1993;23:997–1004. doi: 10.1002/eji.1830230502. [DOI] [PubMed] [Google Scholar]
- Hauser AE, Junt T, Mempel TR, Sneddon MW, Kleinstein SH, Henrickson SE, von Andrian UH, Shlomchik MJ, Haberman AM. Definition of germinal-center B cell migration in vivo reveals predominant intrazonal circulation patterns. Immunity. 2007;26:655–667. doi: 10.1016/j.immuni.2007.04.008. [DOI] [PubMed] [Google Scholar]
- Haynes NM, Allen CDC, Lesley R, Ansel KM, Killeen N, Cyster JG. Role of CXCR5 and CCR7 in follicular T helper cell positioning and appearance of a PD-1hi germinal center associated population. 2007 doi: 10.4049/jimmunol.179.8.5099. submitted. [DOI] [PubMed] [Google Scholar]
- Huntington ND, Xu Y, Puthalakath H, Light A, Willis SN, Strasser A, Tarlinton DM. CD45 links the B cell receptor with cell survival and is required for the persistence of germinal centers. Nat Immunol. 2006;7:190–198. doi: 10.1038/ni1292. [DOI] [PubMed] [Google Scholar]
- Iwai Y, Okazaki T, Nishimura H, Kawasaki A, Yagita H, Honjo T. Microanatomical localization of PD-1 in human tonsils. Immunol Lett. 2002;83:215–220. doi: 10.1016/s0165-2478(02)00088-3. [DOI] [PubMed] [Google Scholar]
- Jacob J, Kassir R, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. I. The architecture and dynamics of responding cell populations. J Exp Med. 1991a;173:1165–1175. doi: 10.1084/jem.173.5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centres. Nature. 1991b;354:389–392. doi: 10.1038/354389a0. [DOI] [PubMed] [Google Scholar]
- Kelsoe G. Life and death in germinal centers (redux) Immunity. 1996;4:107–111. doi: 10.1016/s1074-7613(00)80675-5. [DOI] [PubMed] [Google Scholar]
- Kepler TB, Perelson AS. Cyclic re-entry of germinal center B cells and the efficiency of affinity maturation. Immunol Today. 1993;14:412–415. doi: 10.1016/0167-5699(93)90145-B. [DOI] [PubMed] [Google Scholar]
- Kim CH, Rott LS, Clark-Lewis I, Campbell DJ, Wu L, Butcher EC. Subspecialization of CXCR5+ T cells: B helper activity is focused in a germinal center-localized subset of CXCR5+ T cells. J Exp Med. 2001;193:1373–1381. doi: 10.1084/jem.193.12.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Gaspal FM, Wiggett HE, McConnell FM, Gulbranson-Judge A, Raykundalia C, Walker LS, Goodall MD, Lane PJ. CD4(+)CD3(−) accessory cells costimulate primed CD4 T cells through OX40 and CD30 at sites where T cells collaborate with B cells. Immunity. 2003;18:643–654. doi: 10.1016/s1074-7613(03)00110-9. [DOI] [PubMed] [Google Scholar]
- Kindred JE. A quantitative study of the lymphoid organs of the albino rat. American Journal of Anatomy. 1938;62:453–473. [Google Scholar]
- Kitano H. Biological robustness. Nat Rev Genet. 2004;5:826–837. doi: 10.1038/nrg1471. [DOI] [PubMed] [Google Scholar]
- Klein U, Tu Y, Stolovitzky GA, Keller JL, Haddad J, Jr, Miljkovic V, Cattoretti G, Califano A, Dalla-Favera R. Transcriptional analysis of the B cell germinal center reaction. Proc Natl Acad Sci U S A. 2003;100:2639–2644. doi: 10.1073/pnas.0437996100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koburg E. Cell Production and Cell Migration in the Tonsil; Germinal Centers in Immune Responses; University of Bern, Switzerland. New York Inc.: Springer-Verlag; 1966. [Google Scholar]
- Koni PA, Flavell RA. Lymph node germinal centers form in the absence of follicular dendritic cell networks. J Exp Med. 1999;189:855–864. doi: 10.1084/jem.189.5.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman G, Parmentier HK, Schuurman HJ, Newman W, Meijer CJ, Pals ST. Adhesion of human B cells to follicular dendritic cells involves both the lymphocyte function-associated antigen 1/intercellular adhesion molecule 1 and very late antigen 4/vascular cell adhesion molecule 1 pathways. J Exp Med. 1991;173:1297–1304. doi: 10.1084/jem.173.6.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosco-Vilbois MH. Are follicular dendritic cells really good for nothing? Nat Rev Immunol. 2003;3:764–769. doi: 10.1038/nri1179. [DOI] [PubMed] [Google Scholar]
- Kuwahara K, Yoshida M, Kondo M, Sakata A, Watanabe Y, Abe E, Kouno Y, Tomiyasu S, Fujimura S, Tokuhisa T, et al. A novel nuclear phosphoprotein, GANP, is up-regulated in centrocytes of the germinal center and associated with MCM3, a protein essential for DNA replication. Blood. 2000;95:2321–2328. [PubMed] [Google Scholar]
- Lim HW, Hillsamer P, Banham AH, Kim CH. Cutting edge: direct suppression of B cells by CD4+ CD25+ regulatory T cells. J Immunol. 2005;175:4180–4183. doi: 10.4049/jimmunol.175.7.4180. [DOI] [PubMed] [Google Scholar]
- Liu YJ, Cairns JA, Holder MJ, Abbot SD, Jansen KU, Bonnefoy JY, Gordon J, MacLennan IC. Recombinant 25-kDa CD23 and interleukin 1 alpha promote the survival of germinal center B cells: evidence for bifurcation in the development of centrocytes rescued from apoptosis. Eur J Immunol. 1991a;21:1107–1114. doi: 10.1002/eji.1830210504. [DOI] [PubMed] [Google Scholar]
- Liu YJ, Johnson GD, Gordon J, MacLennan IC. Germinal centres in T-celldependent antibody responses. Immunol Today. 1992;13:17–21. doi: 10.1016/0167-5699(92)90199-H. [DOI] [PubMed] [Google Scholar]
- Liu YJ, Zhang J, Lane PJ, Chan EY, MacLennan IC. Sites of specific B cell activation in primary and secondary responses to T cell-dependent and T cell-independent antigens. Eur J Immunol. 1991b;21:2951–2962. doi: 10.1002/eji.1830211209. [DOI] [PubMed] [Google Scholar]
- MacLennan IC. Germinal centers. Annu Rev Immunol. 1994;12:117–139. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- MacLennan IC, Gulbranson-Judge A, Toellner KM, Casamayor-Palleja M, Chan E, Sze DM, Luther SA, Orbea HA. The changing preference of T and B cells for partners as T-dependent antibody responses develop. Immunol Rev. 1997;156:53–66. doi: 10.1111/j.1600-065x.1997.tb00958.x. [DOI] [PubMed] [Google Scholar]
- Manser T. Textbook germinal centers? J Immunol. 2004;172:3369–3375. doi: 10.4049/jimmunol.172.6.3369. [DOI] [PubMed] [Google Scholar]
- Marinova E, Han S, Zheng B. Germinal center helper T cells are dual functional regulatory cells with suppressive activity to conventional CD4+ T cells. J Immunol. 2007;178:5010–5017. doi: 10.4049/jimmunol.178.8.5010. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Lo SF, Carruthers CJ, Min J, Mariathasan S, Huang G, Plas DR, Martin SM, Geha RS, Nahm MH, Chaplin DD. Affinity maturation without germinal centres in lymphotoxin-alpha-deficient mice. Nature. 1996;382:462–466. doi: 10.1038/382462a0. [DOI] [PubMed] [Google Scholar]
- McHeyzer-Williams LJ, Malherbe LP, McHeyzer-Williams MG. Checkpoints in memory B-cell evolution. Immunol Rev. 2006;211:255–268. doi: 10.1111/j.0105-2896.2006.00397.x. [DOI] [PubMed] [Google Scholar]
- Meyer-Hermann ME, Maini PK, Iber D. An analysis of B cell selection mechanisms in germinal centers. Math Med Biol. 2006;23:255–277. doi: 10.1093/imammb/dql012. [DOI] [PubMed] [Google Scholar]
- Milikin PD. Anatomy of germinal centers in human lymphoid tissue. Arch Pathol. 1966;82:499–505. [PubMed] [Google Scholar]
- Moldenhauer G, Popov SW, Wotschke B, Bruderlein S, Riedl P, Fissolo N, Schirmbeck R, Ritz O, Moller P, Leithauser F. AID expression identifies interfollicular large B cells as putative precursors of mature B-cell malignancies. Blood. 2006;107:2470–2473. doi: 10.1182/blood-2005-06-2502. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Yagi H, Kayaba S, Ishii T, Gotoh T, Ohtsu S, Itoh T. Death of germinal center B cells without DNA fragmentation. Eur J Immunol. 1996;26:1211–1216. doi: 10.1002/eji.1830260604. [DOI] [PubMed] [Google Scholar]
- Nakayama Y, Stabach P, Maher SE, Mahajan MC, Masiar P, Liao C, Zhang X, Ye ZJ, Tuck D, Bothwell AL, et al. A limited number of genes are involved in the differentiation of germinal center B cells. J Cell Biochem. 2006;99:1308–1325. doi: 10.1002/jcb.20952. [DOI] [PubMed] [Google Scholar]
- Nieuwenhuis P, Opstelten D. Functional anatomy of germinal centers. Am J Anat. 1984;170:421–435. doi: 10.1002/aja.1001700315. [DOI] [PubMed] [Google Scholar]
- Okada T, Miller MJ, Parker I, Krummel MF, Neighbors M, Hartley SB, O'Garra A, Cahalan MD, Cyster JG. Antigen-engaged B cells undergo chemotaxis toward the T zone and form motile conjugates with helper T cells. PLoS Biol. 2005;3:e150. doi: 10.1371/journal.pbio.0030150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opstelten D, Stikker R, Deenen GJ, Nieuwenhuis P. Germinal centers and the B-cell system. VII. Complement receptors, antigen receptors, immunoglobulin and alkaline phosphatase in germinal centers of the rabbit appendix and popliteal lymph nodes. Cell Tissue Res. 1982;224:505–516. doi: 10.1007/BF00213748. [DOI] [PubMed] [Google Scholar]
- Or-Guil M, Wittenbrink N, Weiser AA, Schuchhardt J. Recirculation of germinal center B cells: a multilevel selection strategy for antibody maturation. Immunol Rev. 2007;216:130–141. doi: 10.1111/j.1600-065X.2007.00507.x. [DOI] [PubMed] [Google Scholar]
- Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, Sher A, Morse HC, 3rd, Liu C, Schwartzberg PL, Leonard WJ. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298:1630–1634. doi: 10.1126/science.1077002. [DOI] [PubMed] [Google Scholar]
- Pape KA, Kouskoff V, Nemazee D, Tang HL, Cyster JG, Tze LE, Hippen KL, Behrens TW, Jenkins MK. Visualization of the genesis and fate of isotype-switched B cells during a primary immune response. J Exp Med. 2003;197:1677–1687. doi: 10.1084/jem.20012065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paus D, Phan TG, Chan TD, Gardam S, Basten A, Brink R. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J Exp Med. 2006;203:1081–1091. doi: 10.1084/jem.20060087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin D, Wu J, Vora KA, Ravetch JV, Szakal AK, Manser T, Tew JG. Fc gamma receptor IIB on follicular dendritic cells regulates the B cell recall response. J Immunol. 2000;164:6268–6275. doi: 10.4049/jimmunol.164.12.6268. [DOI] [PubMed] [Google Scholar]
- Rademakers LH. Dark and light zones of germinal centres of the human tonsil: an ultrastructural study with emphasis on heterogeneity of follicular dendritic cells. Cell Tissue Res. 1992;269:359–368. doi: 10.1007/BF00319629. [DOI] [PubMed] [Google Scholar]
- Rahman ZS, Rao SP, Kalled SL, Manser T. Normal induction but attenuated progression of germinal center responses in BAFF and BAFF-R signaling-deficient mice. J Exp Med. 2003;198:1157–1169. doi: 10.1084/jem.20030495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reif K, Ekland EH, Ohl L, Nakano H, Lipp M, Forster R, Cyster JG. Balanced responsiveness to chemoattractants from adjacent zones determines B-cell position. Nature. 2002;416:94–99. doi: 10.1038/416094a. [DOI] [PubMed] [Google Scholar]
- Reiter R, Pfeffer K. Impaired germinal centre formation and humoral immune response in the absence of CD28 and interleukin-4. Immunology. 2002;106:222–228. doi: 10.1046/j.1365-2567.2002.01405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley JL, Blair PJ, Musser JT, Abe R, Tezuka K, Tsuji T, June CH. ICOS costimulation requires IL-2 and can be prevented by CTLA-4 engagement. J Immunol. 2001;166:4943–4948. doi: 10.4049/jimmunol.166.8.4943. [DOI] [PubMed] [Google Scholar]
- Röhlich K. Beitrag zur Cytologie der Keimzentren der Lymphknoten. Z Mikrosk Anat Forsch. 1930;20:287–297. [Google Scholar]
- Rossbacher J, Haberman AM, Neschen S, Khalil A, Shlomchik MJ. Antibody-independent B cell-intrinsic and -extrinsic roles for CD21/35. Eur J Immunol. 2006;36:2384–2393. doi: 10.1002/eji.200636172. [DOI] [PubMed] [Google Scholar]
- Schwickert TA, Lindquist TL, Shakhar G, Livshits G, Skokos D, Kosco-Vilbois MH, Dustin ML, Nusenzweig MC. In vivo imaging of germinal centres reveals a dynamic open structure. Nature. 2007;446:83–87. doi: 10.1038/nature05573. [DOI] [PubMed] [Google Scholar]
- Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J, Breard J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol. 2001;3:346–352. doi: 10.1038/35070019. [DOI] [PubMed] [Google Scholar]
- Segundo C, Medina F, Rodriguez C, Martinez-Palencia R, Leyva-Cobian F, Brieva JA. Surface molecule loss and bleb formation by human germinal center B cells undergoing apoptosis: role of apoptotic blebs in monocyte chemotaxis. Blood. 1999;94:1012–1020. [PubMed] [Google Scholar]
- Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, Giltnane JM, Yang L, Zhao H, Calame K, Staudt LM. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 2002;17:51–62. doi: 10.1016/s1074-7613(02)00335-7. [DOI] [PubMed] [Google Scholar]
- Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2:116–126. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- Shih TA, Meffre E, Roederer M, Nussenzweig MC. Role of BCR affinity in T cell dependent antibody responses in vivo. Nat Immunol. 2002;3:570–575. doi: 10.1038/ni803. [DOI] [PubMed] [Google Scholar]
- Shinall SM, Gonzalez-Fernandez M, Noelle RJ, Waldschmidt TJ. Identification of murine germinal center B cell subsets defined by the expression of surface isotypes and differentiation antigens. J Immunol. 2000;164:5729–5738. doi: 10.4049/jimmunol.164.11.5729. [DOI] [PubMed] [Google Scholar]
- Shiratsuchi A, Mori T, Nakanishi Y. Independence of plasma membrane blebbing from other biochemical and biological characteristics of apoptotic cells. J Biochem (Tokyo) 2002;132:381–386. doi: 10.1093/oxfordjournals.jbchem.a003233. [DOI] [PubMed] [Google Scholar]
- Szakal AK, Kosco MH, Tew JG. Microanatomy of lymphoid tissue during humoral immune responses: structure function relationships. Annu Rev Immunol. 1989;7:91–109. doi: 10.1146/annurev.iy.07.040189.000515. [DOI] [PubMed] [Google Scholar]
- Szakal AK, Tew JG. Significance of iccosomes in the germinal centre reaction. Res Immunol. 1991;142:261–263. doi: 10.1016/0923-2494(91)90072-q. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Dutta PR, Cerasoli DM, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. V. Affinity maturation develops in two stages of clonal selection. J Exp Med. 1998;187:885–895. doi: 10.1084/jem.187.6.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Ohta H, Takemori T. Fas is required for clonal selection in germinal centers and the subsequent establishment of the memory B cell repertoire. Immunity. 2001;14:181–192. doi: 10.1016/s1074-7613(01)00100-5. [DOI] [PubMed] [Google Scholar]
- Tarlinton DM, Smith KG. Dissecting affinity maturation: a model explaining selection of antibody-forming cells and memory B cells in the germinal centre. Immunol Today. 2000;21:436–441. doi: 10.1016/s0167-5699(00)01687-x. [DOI] [PubMed] [Google Scholar]
- Vajdy M, Kosco-Vilbois MH, Kopf M, Kohler G, Lycke N. Impaired mucosal immune responses in interleukin 4-targeted mice. J Exp Med. 1995;181:41–53. doi: 10.1084/jem.181.1.41. [DOI] [PubMed] [Google Scholar]
- van Galen JC, Dukers DF, Giroth C, Sewalt RG, Otte AP, Meijer CJ, Raaphorst FM. Distinct expression patterns of polycomb oncoproteins and their binding partners during the germinal center reaction. Eur J Immunol. 2004;34:1870–1881. doi: 10.1002/eji.200424985. [DOI] [PubMed] [Google Scholar]
- Vinuesa CG, Cook MC, Ball J, Drew M, Sunners Y, Cascalho M, Wabl M, Klaus GG, MacLennan IC. Germinal centers without T cells. J Exp Med. 2000;191:485–494. doi: 10.1084/jem.191.3.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody responses and autoimmunity. Nat Rev Immunol. 2005;5:853–865. doi: 10.1038/nri1714. [DOI] [PubMed] [Google Scholar]
- Walker LS, Wiggett HE, Gaspal FM, Raykundalia CR, Goodall MD, Toellner KM, Lane PJ. Established T cell-driven germinal center B cell proliferation is independent of CD28 signaling but is tightly regulated through CTLA-4. J Immunol. 2003;170:91–98. doi: 10.4049/jimmunol.170.1.91. [DOI] [PubMed] [Google Scholar]
- Wang Y, Carter RH. CD19 regulates B cell maturation, proliferation, and positive selection in the FDC zone of murine splenic germinal centers. Immunity. 2005;22:749–761. doi: 10.1016/j.immuni.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Wang Y, Huang G, Wang J, Molina H, Chaplin DD, Fu YX. Antigen persistence is required for somatic mutation and affinity maturation of immunoglobulin. Eur J Immunol. 2000;30:2226–2234. doi: 10.1002/1521-4141(2000)30:8<2226::AID-IMMU2226>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Wolniak KL, Noelle RJ, Waldschmidt TJ. Characterization of (4-hydroxy-3-nitrophenyl)acetyl (NP)-specific germinal center B cells and antigen-binding B220- cells after primary NP challenge in mice. J Immunol. 2006;177:2072–2079. doi: 10.4049/jimmunol.177.4.2072. [DOI] [PubMed] [Google Scholar]
- Zaitoun AM. Cell population kinetics of the germinal centres of lymph nodes of BALB/c mice. J Anat. 1980;130:131–137. [PMC free article] [PubMed] [Google Scholar]
- Zhang J, MacLennan IC, Liu YJ, Lane PJ. Is rapid proliferation in B centroblasts linked to somatic mutation in memory B cell clones? Immunol Lett. 1988;18:297–299. doi: 10.1016/0165-2478(88)90178-2. [DOI] [PubMed] [Google Scholar]
- Zhao DM, Thornton AM, DiPaolo RJ, Shevach EM. Activated CD4+CD25+ T cells selectively kill B lymphocytes. Blood. 2006;107:3925–3932. doi: 10.1182/blood-2005-11-4502. [DOI] [PMC free article] [PubMed] [Google Scholar]