

Mutation of Tyr94 of E. coli thymidylate synthase to phenylalanine leads to a protein with k cat reduced by a factor of 400. The Y94F structure is essentially identical to the wild-type structure, which is consistent with a catalytic role for the phenolic OH.
Keywords: tyrosine mutant, TLS, catalytic mechanism
Abstract
Tyr94 of Escherichia coli thymidylate synthase is thought to be involved, either directly or by activation of a water molecule, in the abstraction of a proton from C5 of the 2′-deoxyuridine 5′-monophosphate (dUMP) substrate. Mutation of Tyr94 leads to a 400-fold loss in catalytic activity. The structure of the Y94F mutant has been determined in the native state and as a ternary complex with thymidine 5′-monophosphate (dTMP) and 10-propargyl 5,8-dideazafolate (PDDF). There are no structural changes ascribable to the mutation other than loss of a water molecule hydrogen bonded to the tyrosine OH, which is consistent with a catalytic role for the phenolic OH.
1. Introduction
Thymidylate synthase (TS) catalyzes the formation of dTMP from dUMP by a reductive methylation reaction involving sequential methylene and hydride transfers from 5,10-methylenetetrahydrofolate (CH2THF; Fig. 1 ▶). The reaction proceeds through a covalently bound ternary intermediate. The breakdown of this intermediate requires proton abstraction from C5 of the nucleotide by a base proposed to be Tyr94 (Hyatt et al., 1997 ▶). Mutation of Tyr94 in both Escherichia coli (Saxl et al., 2003 ▶) and Lactobacillus casei (Liu et al., 1999 ▶) thymidylate synthases led to proteins with substantially reduced catalytic abilities and the build-up of the covalently bound intermediate, supporting the hypothesis that Tyr94 is involved in C5 proton abstraction. Additional evidence supporting a role for Tyr94 as a catalytic base comes from kinetic studies on mutant proteins containing fluorinated tyrosine, where an inverse correlation between activity and acidity was observed (Liu et al., 1999 ▶). These results have led to efforts to design mechanism-based inhibitors of TS that target Tyr94 (Saxl et al., 2003 ▶; Liu et al., 1999 ▶).
Figure 1.

Mechanism of thymidylate synthase. Thymidylate synthase catalyzes the exchange of the dUMP C5 proton for a methyl group provided through methylene- and hydride-transfer steps. The reaction is initiated through Michael addition of Cys146 to C6 of dUMP (left), leading to a covalent ternary complex (center). The reaction proceeds with C5 hydrogen abstraction, involving Tyr94, hydride transfer and product formation (right). The methylene and hydride groups are italicized in the figure.
Structurally, the Tyr94 side chain lies in a hydrophobic pocket on the C5–C6 side of the nucleotide with the aromatic ring parallel to and partially stacked with the ring of His147 (Montfort et al., 1990 ▶; Hardy et al., 1987 ▶). Fig. 2 ▶ shows the position of Tyr94 with respect to the nucleotide and a cofactor analog in the thymidylate synthase ternary complex. In a variety of such ternary complex structures (reviewed in Stroud & Finer-Moore, 2003 ▶), the side-chain hydroxyl group of Tyr94 lies 3.8–4.0 Å from C5 of dUMP, suggesting that with only a modest change in conformation Tyr94 could directly abstract the C5 proton. In support of this, in the covalent ternary complex with 5-fluoro-dUMP (FdUMP), Tyr94 OH lies only 3.2 Å from C5—F, which has a geometry that is very similar to that of C5—H. Alternatively, there is a water molecule hydrogen bonded to Tyr94 OH that is ∼4 Å from C5 and in a position to participate in the reaction step. Tyr94 OH also forms hydrogen bonds directly with Cys146 N, His147 N and, through the hydrogen-bonded water molecule, to Ala144 O. Thus, Tyr94 OH provides three hydrogen bonds to residues lining the active-site cavity. In addition to the probable role Tyr94 plays in the reaction mechanism, these hydrogen bonds may be needed to maintain the geometry of the ligand-binding pocket. An anomaly of this residue is that in structures of unliganded E. coli TS, the main-chain ϕ/ψ angles of Tyr94 are in a relatively unfavorable conformation, with ϕ ≃ 0 and ψ ≃ −80°.
Figure 2.

Ternary complex structure of E. coli wild-type TS showing the location of Tyr94. One TS monomer is shown as a ribbon drawing (the protein is an obligate dimer), with residues Cys146 and Tyr94 shown as sticks. dUMP and PDDF ligands are also shown. Cys146 is covalently bound to C6 of the dUMP substrate in the complex; the folate analog PDDF binds with the ring stacked with that of dUMP. The OH of Tyr94 points at dUMP and lies ∼3.8 Å from C5 of the pyrimidine ring.
The Y94F mutant differs from the wild-type protein only by the loss of the phenolic OH, resulting in a 400-fold drop in k cat (Liu et al., 1999 ▶). The Y94F protein is also less stable to trypsin digestion and subunit exchange than the wild-type protein, raising the possibility that the loss of hydrogen bonds to the tyrosine OH may cause the Y94F mutant protein to have a different conformation, undergo larger conformational fluctuations or display a greater tendency for dimer dissociation (Saxl et al., 2006 ▶). In order to assess whether differences in enzyme activity are a consequence of the loss of this OH or of unforeseen structural changes, we have determined two crystal structures of the TS-Y94F mutant protein, one of the protein alone and the second in a ternary complex structure containing the product dTMP and 10-propargyl-5,8-dideazafolate (PDDF), a transition-state analog.
2. Materials and methods
2.1. Protein production and crystallization
The procedure for wild-type and mutant recombinant protein preparation and purification has been reported elsewhere (Changchien et al., 2000 ▶; Saxl et al., 2003 ▶). Briefly, the full-length protein without purification tags was expressed in E. coli to a high level (∼50% of total protein) and purified to homogeneity using DE-52 anion-exchange and phenyl Sepharose column chromatography. The enzyme pools were concentrated in an Amicon ultrafiltration apparatus (W. R. Grace & Co., Beverly, MA, USA) with a 10 kDa cutoff filter. Proteins were stored as ammonium sulfate pellets at 203 K. There is a carbamate modification at the N-terminus that is required for catalysis and apparently forms through spontaneous addition of CO2 (Fauman et al., 1994 ▶).
Crystals were grown as previously reported using the hanging-drop vapor-diffusion method at room temperature (Montfort et al., 1990 ▶). Both crystal forms are obtained under essentially the same conditions, the difference being the presence of ligand in the crystallization drop. To obtain Y94F mutant protein crystals, the well buffer contained 1 ml 2.1–2.6 M (NH4)2SO4, 20 mM KH2PO4 pH 7.5 and 4 mM DTT. The 5 µl hanging drop was prepared by mixing equal volumes of the well buffer with a solution containing 10–20 mg ml−1 protein, 20 mM KH2PO4 pH 7.5 and 4 mM DTT. The same conditions were used to crystallize the ternary complex, except that 4 mM dTMP and 4 mM PDDF were added to the protein solution.
2.2. X-ray data measurement and refinement
2.2.1. Y94F–dTMP–PDDF ternary complex
Data were measured at room temperature using an Enraf–Nonius FAST detector mounted on an FR571 rotating-anode X-ray source using Cu Kα radiation (1.54 Å). Data were collected and scaled using MADNES (Messerschmidt & Pflugrath, 1987 ▶) and PROCOR (Kabsch, 1988 ▶) (Table 1 ▶). The structure 1tsn (Hyatt et al., 1997 ▶) was used as the starting model and initial refinement used GPRLSA (Furey et al., 1982 ▶). Refinement was completed using REFMAC5 (Murshudov et al., 1997 ▶; Winn et al., 2003 ▶). Dictionaries of restraints for ligand refinement were those distributed by CCP4 (Collaborative Computational Project, Number 4, 1994 ▶).
Table 1. Data collection and refinement.
Values in parentheses are for the highest resolution shell.
| Y94F | Y94F–dTMP–PDDF | |
|---|---|---|
| PDB code | 2ftn | 2fto |
| Space group | I213 | P3121 |
| Unit-cell parameters (Å) | a = 131.17 | a = 72.08, c = 115.21 |
| Z (molecules per ASU) | 1 | 1 |
| Temperature (K) | 100 | 293 |
| Wavelength (Å) | 0.9 | 1.54 |
| Resolution (Å) | 1.6 | 2.0 |
| Total/unique reflections | 504632/48596 | 70659/21450 |
| Completeness (%) | 99 (100) | 89† |
| Mean I/σ(I) | 18.9/3.1 | † |
| Reflections with I > 3σ(I) (%) | 67/32 | |
| Rsym‡ | 0.08 (0.37) | 0.07 (0.23) |
| Rcryst/Rfree§ | 0.18 (0.20) | 0.16 (0.18) |
| R.m.s.d. bonds (Å) | 0.014 | 0.015 |
| R.m.s.d. angles (°) | 1.5 | 1.5 |
| Residues in most favorable region of Ramachandran plot (%) | 94 | 93 |
| Average B (Å2) | 18.8 | 24.8 |
I/σ(I) and completeness in the highest resolution shell are not reported by PROCOR.
R
sym = 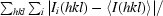
 , where 〈I(hkl)〉 is the mean intensity of all symmetry-related reflections I
i(hkl).
, where 〈I(hkl)〉 is the mean intensity of all symmetry-related reflections I
i(hkl).
R
cryst = 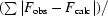
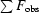 . R
free is as for R
cryst, but using a random subset of the data (5%) not included in the refinement.
. R
free is as for R
cryst, but using a random subset of the data (5%) not included in the refinement.
2.2.2. Y94F protein
Data were collected at 100 K at beamline 14-BMC of the Advanced Photon Source using a Quantum-4 CCD detector and were reduced using CrystalClear (d*TREK; Pflugrath, 1999 ▶) (Table 1 ▶). A previously refined structure based on PDB entry 1tms of the unliganded wild-type protein was used as a starting model and refined using REFMAC5. Other crystallographic calculations were performed using the CCP4 package (Collaborative Computational Project, Number 4, 1994 ▶). Rebuilding was performed using O (Jones et al., 1991 ▶) and Coot (Emsley & Cowtan, 2004 ▶). Figures were prepared with PyMOL (DeLano, 2002 ▶).
2.2.3. TLS refinement
The refined structure of Y94F (unliganded) was submitted to the TLS Motion Determination server (http://skuld.bmsc.washington.edu/~tlsmd/index.html) to determine an initial partition into TLS groups (Painter & Merritt, 2006 ▶). After initial refinement with ten TLS groups, the crystallographic and free R factors dropped by 0.015 and 0.018, respectively. Only four of the TLS groups had appreciable anisotropic motion and each of these four contained residues known to participate in a conformational change upon ligand binding. Further TLS refinement was carried out with five TLS groups, one containing the stationary core of the protein as previously determined and four corresponding to segments known to move upon ligand binding (residues 10–24, 51–119, 123–149 and 260–264; Montfort et al., 1990 ▶). Upon refinement with five TLS groups, the drop in crystallographic and free R was essentially identical to that obtained using ten TLS groups (0.014 and 0.017). In contrast, a drop in R cryst and R free of only 0.002 was seen for the Y94F ternary complex structure. Refined TLS parameters are included in the deposited PDB file for each entry.
3. Results and discussion
The unliganded Y94F mutant protein and the Y94F–dTMP–PDDF ternary complex crystallize in space groups with Z = 1. Thus, in both cases the second subunit of the physiological dimer is generated by a crystallographic twofold axis and is therefore identical to the first subunit.
Fig. 3 ▶ displays a close-up view of the Tyr94 and Phe94 environments in the unliganded wild-type and mutant proteins. Both Tyr94 and Phe94 are well ordered in all structures. The phenyl ring of the Phe94 side chain lies in a hydrophobic pocket with the ring planar to that of His147 and occupies almost exactly the same position as that of Tyr94 (r.m.s.d. of side-chain atoms = 0.16 Å). One water molecule that is hydrogen bonded to Tyr94 and an alternate conformation of Cys146 SG in the wild-type unliganded structure is not present in the mutant structure. This water molecule also does not appear in ternary complex structures of the wild-type protein (see, for instance, PDB code 2g8o; Newby et al., 2006 ▶). A second water molecule in the vicinity of Tyr94 OH is retained and is part of a chain of water molecules connecting, through hydrogen bonds, the carbonyl oxygen of Cys146 and the main-chain N atom of Tyr/Phe94. Interestingly, the loss of the phenolic oxygen does not change the main-chain torsional angles: for the unliganded Y94F protein, the residue still lies in a relatively disfavored area of the Ramachandran plot (ϕ = −1, ψ = −81°), with the side chain apparently locked into this conformation because of the hydrophobic pocket it occupies. There are no significant changes in the structure of the protein chain. The r.m.s. difference in the positions of the main-chain atoms in the unliganded wild-type and Y94F proteins is 0.15 Å, with no differences over 0.35 Å except in poorly ordered regions of the protein (the loop near Arg21, loop 104–108 and residues 231–235 on the protein surface). Cys146 and His147 have been built in two positions in the cryocooled wild-type structure, but the alternate position of His147 was absent in the room-temperature Y94F structure. In ternary complex structures, O4 of the bound nucleotide extends into the position occupied by the alternate conformation of His147; Cys146 is restricted to the primary conformation through Michael addition to the nucleotide (Newby et al., 2006 ▶; Montfort et al., 1990 ▶). Consequently, we believe the alternate conformations of His147 and Cys146 in the absence of ligands are not relevant to the enzyme mechanism.
Figure 3.

Stereo close-up of residues surrounding Tyr94 and Phe94 in unliganded wild-type TS and mutant proteins. (a) Native TS (PDB code 2ftq), displaying two conformations for Cys146 and His147. Dotted lines show hydrogen bonds linking Tyr94 OH to the protein (Cys146 N, 2.8 Å; His147 N, 3.1 Å) and a solvent molecule (2.7 Å). Also shown are a hydrogen bond between Tyr94 N and solvent (2.9 Å), two hydrogen bonds between solvent molecules (2.7 and 2.8 Å) and a hydrogen bond between solvent and Cys146 N (3.2 Å). Not shown are hydrogen bonds between solvent and Ala144 O and solvent and Cys146 S in the alternate conformation. (b) Y94F, with electron density contoured at 1.2σ. Hydrogen bonds involving Tyr94 OH and the associated solvent molecule are missing, while the string of three solvent molecules remain, with hydrogen-bond distances similar to those in the wild-type protein. His146 displays a single conformation.
On ternary complex formation between Y94F, the product dTMP and the inhibitor PDDF, a conformational change occurs, as in the wild-type protein. One effect of this is that the torsional angles of Phe94 move to a more favorable region of the Ramachandran plot (ϕ = −27, ψ = −67°). Again, there are no significant differences in the positions of the Tyr94 and Phe94 side chains (r.m.s.d. for side-chain atoms is 0.25 Å).
While there are no significant positional differences between Y94F and wild-type TS, it is possible that differences in the disorder and dynamics of the region near residue 94 might exist and could be revealed through thermal motion analyses of the crystal structures. A comparison of isotropic displacement parameters shows that there are no regions where displacement parameters for the Y94F protein are significantly larger than for the same regions of the wild-type protein. Although the individual isotropic thermal parameters show no differences, it is still conceivable that regions of residues in the crystal move in concert. We undertook TLS refinement to search for changes in coordinate motions. If the loss of hydrogen bonds to Phe94 causes an increase in disorder or dynamics, one would expect to see larger TLS displacements. After TLS refinement, a drop of more than 1.5% was seen in the free R values for both wild-type and Y94F unliganded structures. No significant drop in free R was seen for the ternary complex structure. Thus, the unliganded structures contain evidence of a range of structures along the path of the known conformational change being present in the crystal (and by extension in solution), whereas the ternary complex appears to be in a single closed conformation and does not show disorder of this type.
Analysis of the TLS parameters shows that TLS displacements are similar for wild-type and Y94F proteins, albeit slightly smaller for the higher resolution mutant structure. Thus, it appears that the loss of the hydrogen bond between Tyr94 OH and Cys146 N does not result in increased thermal motion or disorder in this region or elsewhere in the protein, at least in the crystal lattice. Although one hydrogen bond is lost between the helix containing Tyr/Phe94 and the region containing the active-site Cys146, other hydrogen bonds, including Leu90 O–Ala142 N and Lys96 NZ– Glu137 OE1 serve to maintain the structural integrity of this region. Nonetheless, increased sensitivity toward proteolysis for the Y94F protein suggests there is a change in folding stability or dynamic behavior or dimer dissociation in solution (Saxl et al., 2006 ▶).
In summary, there are no structural differences caused by the Tyr94 to Phe94 mutation. Differences in kinetic behavior are likely to be a consequence of the loss of the tyrosine OH, further supporting the view that Tyr94 is the base responsible for proton abstraction in the wild-type protein. Residual activity in the mutant protein is likely to arise from other means of proton abstraction involving transient positional fluctuations in solvent or protein in the thymidylate synthase active site.
Supplementary Material
PDB reference: Y94F thymidylate synthase, 2ftn, r2ftnsf
PDB reference: ternary complex, 2fto, r2ftosf
Acknowledgments
Diffraction data were measured at BioCARS, Sector 14, Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science under Contract No. W-31-109-Eng-38. Use of the BioCARS Sector 14 was supported by the National Institutes of Health, National Center for Research Resources under grant No. RR07707. This work was supported in part by ACS Grant RPG-93-041-04-CDD (WRM), ADCRC Grant 1-208A (WRM) and the DOD Breast Cancer Research Program, DAMD 17-02-1-0405 (FM).
References
- Changchien, L. M., Garibian, A., Frasca, V., Lobo, A., Maley, G. F. & Maley, F. (2000). Protein Expr. Purif.19, 265–270. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- DeLano, W. L. (2002). The PyMOL Molecular Visualization System. http://www.pymol.org.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Fauman, E. B., Rutenber, E. E., Maley, G. F., Maley, F. & Stroud, R. M. (1994). Biochemistry, 33, 1502–1511. [DOI] [PubMed] [Google Scholar]
- Furey, W., Wang, B.-C. & Sax, M. (1982). J. Appl. Cryst.15, 160–166. [Google Scholar]
- Hardy, L. W., Finer-Moore, J. S., Montfort, W. R., Jones, M. O., Santi, D. V. & Stroud, R. M. (1987). Science, 235, 448–455. [DOI] [PubMed] [Google Scholar]
- Hyatt, D. C., Maley, F. & Montfort, W. R. (1997). Biochemistry, 36, 4585–4594. [DOI] [PubMed] [Google Scholar]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. (1988). J. Appl. Cryst.21, 916–934. [Google Scholar]
- Liu, Y., Barrett, J. E., Schultz, P. G. & Santi, D. V. (1999). Biochemistry, 38, 848–852. [DOI] [PubMed] [Google Scholar]
- Messerschmidt, A. & Pflugrath, J. W. (1987). J. Appl. Cryst.20, 306–315. [Google Scholar]
- Montfort, W. R., Perry, K. M., Fauman, E. B., Finer-Moore, J. S., Maley, G. F., Hardy, L., Maley, F. & Stroud, R. M. (1990). Biochemistry, 29, 6964–6977. [DOI] [PubMed] [Google Scholar]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Newby, Z., Lee, T. T., Morse, R. J., Liu, Y., Liu, L., Venkatraman, P., Santi, D. V., Finer-Moore, J. S. & Stroud, R. M. (2006). Biochemistry, 45, 7415–7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter, J. & Merritt, E. A. (2006). J. Appl. Cryst.39, 109–111. [Google Scholar]
- Pflugrath, J. W. (1999). Acta Cryst. D55, 1718–1725. [DOI] [PubMed] [Google Scholar]
- Saxl, R. L., Maley, G. F., Hauer, C. R., MacColl, R., Changchien, L. M. & Maley, F. (2006). Submitted.
- Saxl, R. L., Reston, J., Nie, Z., Kalman, T. I. & Maley, F. (2003). Biochemistry, 42, 4544–4551. [DOI] [PubMed] [Google Scholar]
- Stroud, R. M. & Finer-Moore, J. S. (2003). Biochemistry, 42, 239–247. [DOI] [PubMed] [Google Scholar]
- Winn, M. D., Murshudov, G. N. & Papiz, M. Z. (2003). Methods Enzymol.374, 300–321. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Y94F thymidylate synthase, 2ftn, r2ftnsf
PDB reference: ternary complex, 2fto, r2ftosf