

The structure of recombinant T. brucei UDP-galactose-4′-epimerase cocrystallized with NAD+ and the substrate analogue UDP-4-deoxy-4-fluoro-α-d-galactose has been determined at medium resolution. Comparisons with structures of human and E. coli UDP-galactose-4′-epimerase–ligand complexes reveal that the hexose moieties are able to adopt different orientations in the active site.
Keywords: short-chain dehydrogenase/reductases, Trypanosoma brucei, UDP-galactose-4′-epimerase, UDP-4-deoxy-4-fluoro-α-d-galactose
Abstract
The structure of the NAD-dependent oxidoreductase UDP-galactose-4′-epimerase from Trypanosoma brucei in complex with cofactor and the substrate analogue UDP-4-deoxy-4-fluoro-α-d-galactose has been determined using diffraction data to 2.7 Å resolution. Despite the high level of sequence and structure conservation between the trypanosomatid enzyme and those from humans, yeast and bacteria, the binding of the 4-fluoro-α-d-galactose moiety is distinct from previously reported structures. Of particular note is the observation that when bound to the T. brucei enzyme, the galactose moiety of this fluoro-derivative is rotated approximately 180° with respect to the orientation of the hexose component of UDP-glucose when in complex with the human enzyme. The architecture of the catalytic centre is designed to effectively bind different orientations of the hexose, a finding that is consistent with a mechanism that requires the sugar to maintain a degree of flexibility within the active site.
1. Introduction
The tsetse-transmitted protozoan parasite Trypanosoma brucei causes African sleeping sickness in humans and nagana, a disease of cattle, in sub-Saharan Africa. The disease-causing bloodstream form of T. brucei is rich in galactose-containing glycoproteins, including the protective variant surface glycoproteins (VSGs) that, depending on the variant, contain galactose (Gal) in glycosylphosphatidylinositol (GPI) anchor side chains and/or N-linked oligosaccharides (Mehlert et al., 1998 ▶). In addition, the parasite’s transferrin receptor, which is critical for the acquisition of iron from the host, and various invariant surface glycoproteins also contain Gal in the form of poly N-acetyllactosamine, i.e. sugar chains containing Galβ1-4GlcNAc repeats (Nolan et al., 1999 ▶). Recently, ricin lectin affinity chromatography was used to isolate glycoproteins bearing terminal non-reducing βGal residues and these were found to contain a variety of Gal-containing N-linked oligosaccharides, including a family of novel giant structures that contain on average 54 N-acetyllactosamine repeats. These ricin-binding glycoproteins are localized in the flagellar pocket and throughout the endosomal/lysosomal system of the parasite (Atrih et al., 2005 ▶). The insect-dwelling procyclic form of the parasite also expresses Gal-containing glycoconjugates, notably the surface procyclin glycoproteins (Treumann et al., 1997 ▶) and free GPI structures (Vassella et al., 2003 ▶; Lillico et al., 2003 ▶; Nagamune et al., 2004 ▶). Importantly, neither life-cycle stage can transport Gal across the plasma membrane (Tetaud et al., 1997 ▶) and for galactose metabolism both are dependent on the NADH-dependent oxidoreductase UDP-glucose-4′-epimerase (EC 5.1.3.2; GalE) encoded by the TbGalE gene that interconverts UDP-Glc and UDP-Gal (Fig. 1 ▶; Roper et al., 2002 ▶, 2005 ▶). The same appears to be true of the related parasite T. cruzi, the causal agent of Chagas’ disease in South and Central America (MacRae et al., 2006 ▶). The African trypanosome requires UDP-glucose-4′-epimerase activity for growth and survival in vitro, providing genetic validation for this enzyme as a potential drug target against African trypanosomiasis (Roper et al., 2002 ▶).
Figure 1.

(a) The epimerization catalyzed by TbGalE, interconverting UDP-Glc and UDP-Gal. The blue arrow indicates the Pβ—O anomeric bond about which rotation occurs during catalysis. (b) The chemical structure of UDP-FGal.
GalE is a short-chain dehydrogenase/reductase (SDR) (Holden et al., 2003 ▶). Despite displaying an enormous spread of substrate specificities that regulate diverse biological processes, SDRs possess conserved motifs that are important for aspects of the enzyme structure, the recognition, binding and orientation of cofactor (NADH or NADPH) and substrates together with catalysis (Oppermann et al., 2003 ▶; Filling et al., 2002 ▶; Shi & Lin, 2004 ▶). Three amino acids are particularly important with respect to catalysis and two occur in a Tyr-XXX-Lys motif (Holm et al., 1994 ▶). The tyrosine is the catalytic base in the enzyme mechanism and the lysine contributes to binding the cofactor nicotinamide ribose (Gourley et al., 2001 ▶). In addition, a serine or threonine is often associated with the catalytic tyrosine or with the substrate. In TbGalE the relevant residues are Ser142, Tyr173 and Lys177.
A mechanism for the TbGalE-catalyzed conversion of an equatorial hydroxyl substituent at C4 of glucose to an axial position in galactose can be described in distinct stages (Shaw et al., 2003 ▶). UDP-Glc first binds to the binary complex TbGalE–NAD+. The nicotinamide abstracts a hydride from the glucose C4 as Tyr173 acquires a proton from the O4′ hydroxyl to produce a 4-keto intermediate. For inversion to occur, hydride transfer from the reduced cofactor must be to the opposite side of the hexose, a feat only possible after a rotation of the 4-keto intermediate within the active site. NADH then transfers the hydride back to C4 with concomitant reprotonation of the O4 hydroxyl group by Tyr173 to produce UDP-Gal. Ser142 OG accepts a hydrogen bond from the main-chain amide of Ala144 and acts as a hydrogen-bond donor to the O4′ hydroxyl of substrate. This is an important contribution to enzyme reactivity since it ensures that the O4′ group on the substrate is committed to be a hydrogen-bond donor with the phenolic Tyr173 OH and facilitates the H-atom transfers from and to O4′ that occur.
A complete understanding of the specificity and reactivity of TbGalE is sought to support the search for new enzyme inhibitors of TbGalE. Here, we describe the structure of this essential enzyme in ternary complex with NAD+ and the substrate analogue UDP-4-deoxy-4-fluoro-α-d-galactose (UDP-FGal; Fig. 1 ▶ b). The fluorine substitutes for the 4′-hydroxyl group from which a proton is abstracted in the first step of the proposed mechanism. A detailed analysis of the active site and comparisons with the human enzyme (HsGalE) highlights differences between substrate/product binding and provides insight into the mechanism of the enzyme together with clues for inhibitor design.
2. Materials and methods
2.1. Sample preparation
TbGalE, previously cloned into pET15b (Novagen), was heat-shock transformed in to Escherichia coli strain BL21(DE3)pLysS (Shaw et al., 2003 ▶). Cells were grown in Luria Broth supplemented with ampicillin (100 µg l−1) and chloramphenicol (100 µg l−1). At mid-log phase, the culture was cooled to 294 K, gene expression was induced with 0.4 mM isopropyl-β-d-thiogalactopyranoside and cell growth was continued overnight. Cells were harvested by centrifugation (2500g) at 277 K, resuspended in binding buffer (25 mM Tris–HCl, 50 mM NaCl pH 7.5) and lysed using a OneShot cell disrupter (Constant Systems). Insoluble debris was separated by centrifugation (40 000g) at 277 K for 20 min and the supernatant was filtered through a 0.45 µm syringe filter and then applied onto an Ni2+-resin column (GE Healthcare, 5 ml) pre-equilibrated with binding buffer using an ÄKTA Explorer (GE Healthcare). The resin was washed with binding buffer and protein and then eluted with an increasing imidazole gradient. Fractions were analyzed by SDS–PAGE and those containing TbGalE were pooled and dialysed overnight in binding buffer. The resulting mixture was filtered (0.45 µm) and applied onto a ResourceQ anion-exchange column (GE Healthcare, 6 ml). TbGalE did not bind to the column and was thus separated from contaminants that did. Fractions containing TbGalE were pooled, dialyzed overnight in 25 mM Tris–HCl, 1 mM dithiothreitol pH 7.5 at 277 K and then concentrated to approximately 20 mg ml−1 for crystallization. Previous work identified that crystals could be obtained without proteolytic removal of the histidine tag. The enzyme was judged to be greater than 95% pure as assessed by SDS–PAGE. The synthesis of UDP-FGal followed published methods (Burton et al., 1997 ▶).
2.2. Crystallization and data collection
TbGalE was incubated with 2 mM β-NAD+ (Sigma–Aldrich) and 2 mM UDP-FGal at room temperature for 1 h and then used to assemble hanging drops consisting of 1 µl protein–ligand mixture and 1 µl reservoir solution (8% PEG 8000, 200 mM KCl, 100 mM Na2B4O7, 10% glycerol pH 8.5). Orthorhombic crystals grew over a period of 2 d and one (0.3 × 0.1 × 0.05 mm) was cryoprotected in 15% 2-methyl-2,4-pentanediol and then flash-cooled in a stream of nitrogen at 103 K for data collection. A data set of 238 images, each of 0.5° oscillation, was collected on a Rigaku R-AXIS IV++ image-plate detector coupled to a MicroMax-007HF rotating-anode X-ray generator (Cu Kα, λ = 1.5418 Å) operating at 40 kV and 20 mA. Data extending to 2.7 Å resolution were processed and scaled with DENZO and SCALEPACK, respectively (Otwinowski & Minor, 1997 ▶). Statistics are presented in Table 1 ▶. Although the outer shell of data (2.8–2.7 Å resolution) is less than 80% complete and weak, with an R merge of nearly 50%, we were content to include these diffraction terms, given that the 〈I/σ(I)〉 is 2.4, and to trust the benefits of maximum-likelihood weighting (Murshudov et al., 1997 ▶). The approach appears to have been successful given that the statistics are acceptable.
Table 1. Crystallographic statistics.
Values in parentheses are for the highest resolution bin (approximate width 0.07 Å).
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 101.72, b = 111.70, c = 160.87 |
| Resolution range (Å) | 92.0–2.7 |
| No. of observed/unique reflections | 222257/49501 |
| Wilson B factor (Å2) | 64.7 |
| Mosaic spread (°) | 0.9 |
| Completeness (%) | 96.8 (78.9) |
| Multiplicity | 4.5 (3.3) |
| Rmerge (%) | 9.0 (47.1) |
| 〈I/σ(I)〉 | 15.2 (2.4) |
| R factor | 19.9 (33.4) |
| Rfree (%) | 28.1 (39.8) |
| R.m.s.d. from ideal values, bond lengths (Å) | 0.012 |
| R.m.s.d. from ideal values, bond angles (°) | 1.464 |
| Average B values (Å2) | |
| Overall | 48.6 |
| Main chain | 48.3 |
| Side chain | 48.9 |
| Waters | 44.8 |
| NAD+ | 39.4 |
| UDP-FGal | 43.4 |
| Ramachandran plot analysis (%) | |
| Residues in most favourable regions | 86.3 |
| Residues in allowed regions | 13.4 |
| Residues in disallowed regions | 0.3 |
| Cruickshank’s DPI† (Å) based on Rfree | 0.4 |
Diffraction-component precision index (Cruickshank, 1999 ▶).
2.3. Structure determination and model refinement
The crystal is isomorphous with that previously studied (PDB code 1gy8; Shaw et al., 2003 ▶) and analysis was initiated by rigid-body refinement (REFMAC5; Collaborative Computational Project Number 4, 1994 ▶; Murshudov et al., 1997 ▶) using as a starting model the protein atoms of the four subunits that constitute the asymmetric unit. Rounds of restrained refinement interspersed with model building, inspection of electron-density and difference electron-density maps, ligand and water placement using Coot (Emsley & Cowtan, 2004 ▶) completed the analysis. Non-crystallographic restraints between the subunits were employed in the early stages of refinement and were released once waters and ligands were being identified. The resulting model is composed of four subunits forming two physiological dimers (subunits AB and CD). Subunit A comprises residues −1–150, 157–237 and 249–381, subunit B residues −1–150, 158–235 and 249–381, subunit C residues −1–152, 158–237 and 248–381 and subunit D residues −1–150, 157–235 and 249–381. The −1 refers to a serine residue which precedes the initiating methionine and is an artifact of the expression plasmid that generates an N-terminal extension. There are several missing residues which belong to flexible surface loops. Each active site is occupied by well ordered NAD+ and UDP-FGal and an example of the electron density for the latter is presented in Fig. 2 ▶. The geometry of the TbGalE model was acceptable, with 1232 residues in the most favorable (86.3%) or allowed (13.4%) regions of the Ramachandran plot (Table 1 ▶) as assessed with PROCHECK (Laskowski et al., 1993 ▶).
Figure 2.
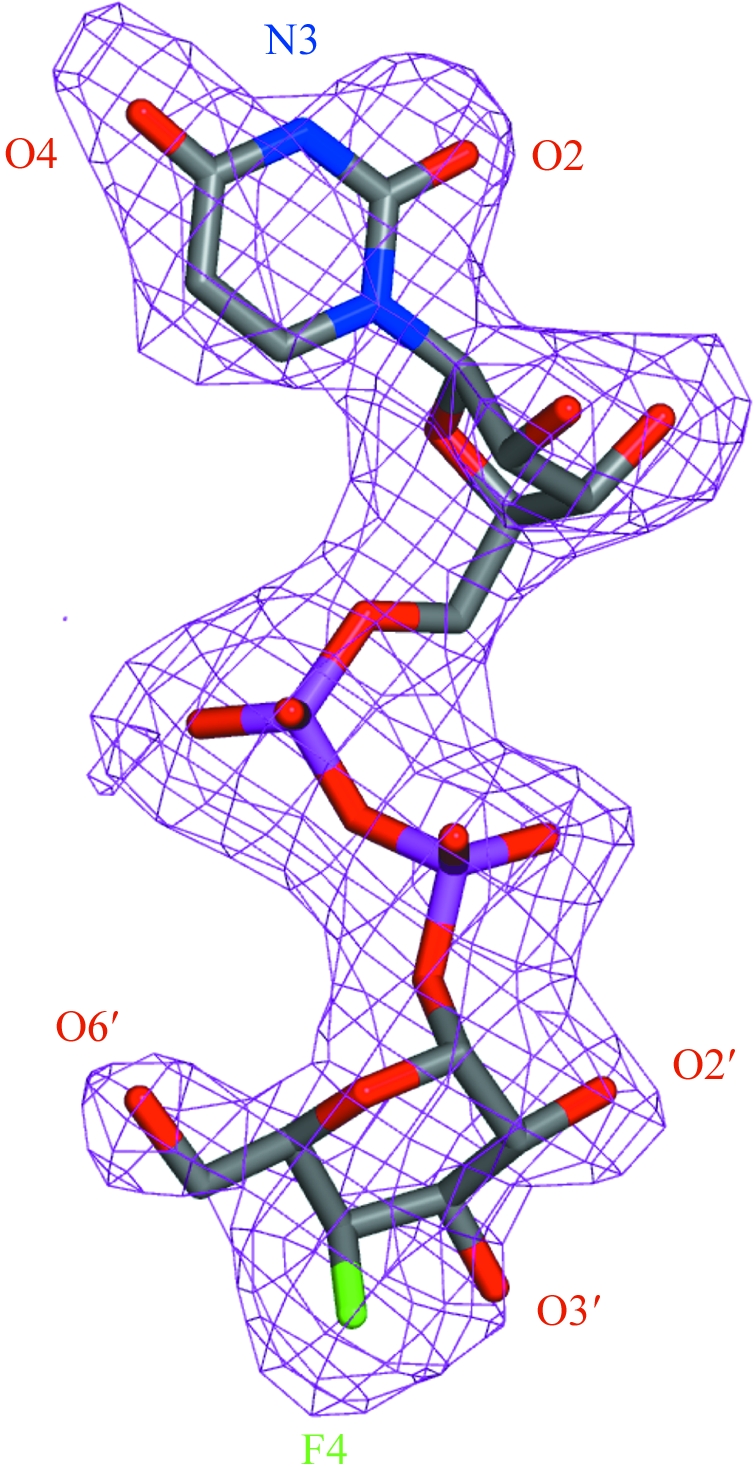
(a) The omit F o − F c difference density observed for UDP-FGal binding in the subunit A active site of TbGalE. The map, depicted as a purple mesh, is contoured at 2σ. The ligand is shown as a stick model with atomic positions coloured according to type: C, grey; N, blue; O, red; P, purple; F, green. This is the ligand orientation that will be used in all figures. Figs. 2–5 were prepared with PyMOL (DeLano, 2002 ▶).
3. Results and discussion
3.1. Overall structure
The structure of TbGalE in this orthorhombic crystal form has been published (Shaw et al., 2003 ▶) and so only a brief overall description is given here. Each subunit comprises two domains: an N-terminal nucleotide-binding motif and a smaller C-terminal substrate-binding domain. The N-terminal domain (residues 1–203 and 271–306) contains a Rossmann fold and comprises a seven-stranded parallel and twisted β-sheet flanked on each side by three α-helices. The C-terminal domain (residues 204–270 and 307–381) forms a two-stranded parallel β-sheet and an α-helix bundle. The N-terminal domain binds the cofactor and the C-terminal domain binds the UDP-sugar substrate and the catalytic centre is located in a cleft at the domain–domain interface (Fig. 3 ▶). Least-squares superposition of the Cα atoms of all four subunits on each other indicates close similarity, with r.m.s.d. values in the range 0.4–0.6 Å. Only minor differences occur in some surface loops (data not shown). The cofactor and UDP-FGal overlay well with the corresponding ligands in the other subunits, with side chains and water-molecule positions together with inferred interactions being virtually identical between all four subunits. For this reason it is only necessary to provide details of a single subunit and that of subunit A has been chosen arbitrarily.
Figure 3.

Ribbon diagram to show the subunit fold and secondary structure of TbGalE. α-Helices are coloured red and β-strands cyan; UDP-FGal and the cofactor are depicted as sticks and coloured magenta and green, respectively.
The structure of the ternary complex TbGalE–NAD+–UDP-FGal is similar to that of the TbGalE–NAD+–UDP complex published previously (Shaw et al., 2003 ▶). Superposition of the two structures gives an r.m.s.d. of 0.4 Å for 1461 Cα positions. The close similarity even extends to the orientation of side chains in and around the active site. Superposition of subunit A of TbGalE onto a subunit of the human enzyme (HsGalE; PDB code 1ek6) gives an r.m.s.d. of 1.2 Å for 314 Cα atoms. An overlay with 307 Cα atoms of the E. coli enzyme (EcGalE; PDB code 1udb) produced an r.m.s.d. of 1.3 Å. The core of the GalE subunit is highly conserved in all three structures, with differences confined to areas distant from the active site, namely surface loops and α2 of TbGalE, which adopts a slightly different orientation compared with HsGalE and EcGalE (not shown) (Thoden et al., 1996 ▶).
3.2. Active site: cofactor and UDP-FGal binding
The cofactor-binding site, as in all SDR family members, is located at the C-terminal ends of the seven-stranded β-sheet. NAD+ binds with the adenine in an anti conformation and the nicotinamide syn with respect to their associated ribose groups. The interactions between the cofactor and protein are conserved in the TbGalE–NAD+–UDP-FGal and TbGalE–NAD+–UDP complexes. There are 14 hydrogen bonds formed between the cofactor and the protein and a further five between cofactor and water molecules in the active site. Several water molecules also mediate interactions with the protein (not shown). The majority of residues which hydrogen bond with NAD+ (Tyr11, Ile12, Asp32, Asp75, Met98, Asn117, Tyr173 and Lys177) are conserved between species (Shaw et al., 2003 ▶). Hydrophobic interactions, several of which involve highly conserved residues (Gly7, Ser140 and Tyr200), have also been noted (Shaw et al., 2003 ▶).
Key residues in the active site of TbGalE that interact with UDP-FGal together with selected hydrogen-bonding interactions are depicted in Fig. 4 ▶. The published structure of TbGalE contained a fragment of the substrate, UDP, in the active site (Shaw et al., 2003 ▶) and, in a similar fashion to the observations made above regarding cofactor binding, we note a consistency in the association of common structural components of UDP and UDP-FGal in the two complexes. Uracil binds in a hydrophobic pocket, sandwiched between Leu222 and Phe255, forming hydrogen-bonding interactions with the main-chain atoms of Pro253 and Phe255; a pair of residues located on β8. O4 interacts with a water molecule and potentially forms a C—H⋯O hydrogen bond with Pro253. The ribose hydroxyl groups form hydrogen bonds with the side chains of Cys266 (not shown) and Asp338. The pyrophosphate moiety interacts with His221 (another potential C—H⋯O hydrogen bond), Leu222, Arg268, Arg335 and nearby water molecules. Although weak (Duax et al., 2003 ▶; Leonard et al., 1995 ▶ and references cited therein), these C—H⋯O hydrogen-bond interactions serve to assist the association of protein with ligand because they can alleviate the destabilizing interactions that would arise owing to the presence of an unsatisfied hydrogen-bond acceptor. C—H⋯O associations are commonly observed in high-resolution SDR structures, in particular contributing to cofactor placement (Duax et al., 2003 ▶).
Figure 4.

A stereo representation of ligand binding in the active site showing selected residues. Atomic positions are coloured according to type. C-atom positions of UDP-FGal, NAD+ and TbGalE are magenta, grey and wheat, respectively; all O atoms are red, all N atoms blue and P atoms orange; F is green. Two water molecules are shown as red spheres. Selected potential hydrogen bonds are depicted as yellow dashed lines and C—H⋯O interactions as black dashed lines.
The new complex now allows us to describe how a galactose derivative interacts with TbGalE. The nicotinamide creates the floor of the hexose-binding site and is in close proximity (approximately 3.0 Å) to the galactose and so is positioned to participate in hydride transfers. All functional groups on the galactose participate in at least one hydrogen bond with surrounding residues or waters (Fig. 4 ▶). The hydroxyl O2′ forms a hydrogen bond with Asn202 ND2, while O3′ interacts with two main-chain carbonyl groups provided by Tyr200 and Phe201 and Ser142 OG. O6′ interacts with the carbonyl of Leu102, His221 NE2 and a well ordered water molecule that in turn associates with O5′, α- and β-phosphate O atoms. The fluorine, F4, is 2.7 Å distant from Ser142 OG and 2.8 Å from Tyr173 OH. The location of F4 is equivalent to the hydroxyl group position if a substrate (e.g. UDP-Gal; Fig. 1 ▶ b) were bound and, as discussed, Tyr173 is the catalytic base that extracts the 4′-hydroxyl hydrogen to produce the ketose intermediate.
In the HsGalE–NAD+–UDP-Glc complex (PDB code 1ek6; Thoden et al., 2000 ▶), each functional group associated with the hexose also participates in hydrogen-bonding interactions with the protein. In the TbGalE–NAD+–UDP-FGal structure there are seven residues which bind the galactose group. These are Leu102, Ser142, Tyr173, Tyr200, Phe201, Asn202 and His221. Five of these are strictly conserved in HsGalE (Ser132, Tyr157, Tyr185, Phe186 and Asn187). Leu102 corresponds to Lys92, but since the interaction with the ligand involves a main-chain group the identity of the amino acid is less important. His221 in TbGalE is altered to Asn207 in HsGalE, a conservative substitution since both residue types present an N—H hydrogen-bond donor group directed towards the hexose-binding site. This position is also occupied by an asparagine in EcGalE (Asn199) and in the yeast enzyme (Asn214). However, although conserved residues are involved in binding the glucose moiety, the hexose has rotated/flipped through approximately 180° (Fig. 5 ▶), with the O6′ hydroxyl groups changing position by 7.5 Å as a consequence of the different orientations. This alteration to the glucose group results in different hydrogen-bonding details to those shown in Fig. 4 ▶ for the galactose. In HsGalE–NAD+–UDP-Glc, the O2′ and O6′ groups of the glucose interact with Asn207 and Asn187, respectively. Asn187 also forms a hydrogen bond to the β-phosphate of the pyrophosphate moiety. O3′ interacts with Tyr157 OH and the carbonyl group of Lys92, whilst O4′ interacts with Ser132 OG and Tyr157 OH. Since the relevant side-chain functional groups are conserved in TbGalE, the same interactions are predicted to occur when UDP-Glc is bound to the parasite enzyme.
Figure 5.

Overlay of two UDP-FGal molecules observed in the complex with TbGalE (this work) and EcGalE (PDB code 1uda; Thoden et al., 1997 ▶) together with UDP-Glc from the complex with human GalE (PDB code 1ek6; Thoden et al., 2000 ▶). The UDP-FGal bound to TbGalE is depicted as in Fig. 2 ▶. The ligand from the bacterial enzyme complex is shown as black sticks with green to mark the F position; UDP-Glc is drawn as cyan sticks.
The structure of UDP-FGal bound to TbGalE also shows the galactose in a different orientation compared with the same ligand when in complex with EcGalE (Fig. 5 ▶; Thoden et al., 1997 ▶). In the bacterial enzyme complex the galactose adopts a conformation intermediate between that of the T. brucei and human enzyme structures (Fig. 5 ▶). These three complexes clearly indicate that the GalE active site has enough space to allow conformational freedom of the hexose rings with respect to UDP and the correct placement of functional groups to accommodate or stabilize different orientations of the sugars. The design of novel inhibitors targeting TbGalE will need to take into consideration the variety of hydrogen-bonding partners that create this open hydrophilic hexose-binding site.
Supplementary Material
PDB reference: UDP-galactose-4′-epimerase complex, 2cnb, r2cnbsf
Acknowledgments
We thank Terry Smith for useful discussions, The Wellcome Trust (programme grant 071463 to MAJF and Senior Fellowship to WNH) and BBSRC (Structural Proteomics of Rational Targets) for financial support.
References
- Atrih, A., Richardson, J. M., Prescott, A. R. & Ferguson, M. A. J. (2005). J. Biol. Chem.280, 865–871. [DOI] [PubMed] [Google Scholar]
- Burton, A., Wyatt, P. & Boons, G. J. (1997). J. Chem. Soc., Perkin Trans. I, 16, 2375–2382.
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50 760–763. [Google Scholar]
- Cruickshank, D. W. J. (1999). Acta Cryst. D55, 583–601. [DOI] [PubMed] [Google Scholar]
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA.
- Duax, W. L., Pletnev, V., Addlagatta, A., Bruenn, J. & Weeks, C. M. (2003). Proteins, 53, 931–943. [DOI] [PubMed] [Google Scholar]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Filling, C., Berndt, K. D., Benach, J., Knapp, S., Prozorovski, T., Nordling, E., Ladenstein, R., Jornvall, H. & Oppermann, U. (2002). J. Biol. Chem.277, 25677–25684. [DOI] [PubMed] [Google Scholar]
- Gourley, D. G., Schüttelkopf, A. W., Leonard, G. A., Luba, J., Hardy, L. W., Beverley, S. M. & Hunter, W. N. (2001). Nature Struct. Biol.8, 521–525. [DOI] [PubMed] [Google Scholar]
- Holden, H. M., Rayment, I. & Thoden, J. B. (2003). J. Biol. Chem.278, 43885–43888. [DOI] [PubMed] [Google Scholar]
- Holm, L., Sander, C. & Murzin, A. (1994). Nature Struct. Biol.1, 146–147. [DOI] [PubMed] [Google Scholar]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291. [Google Scholar]
- Leonard, G. A., McAuley-Hecht, K., Brown, T. & Hunter, W. N. (1995). Acta Cryst. D51, 136–139. [DOI] [PubMed] [Google Scholar]
- Lillico, S., Field, M. C., Blundell, P., Coombs, G. H. & Mottram, J. C. (2003). Mol. Biol. Cell, 14, 1182–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacRae, J. I., Obado, S. O., Roper, J. R., Kierans, M., Kelly, J. M. & Ferguson, M. A. J. (2006). Mol. Biochem. Parasitol.147, 126–136. [DOI] [PubMed] [Google Scholar]
- Mehlert, A., Zitzmann, N., Richardson, J. M., Treumann, A. & Ferguson, M. A. J. (1998). Mol. Biochem. Parasitol.91, 145–152. [DOI] [PubMed] [Google Scholar]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Nagamune, K., Acosta-Serrano, A., Uemura, H., Brun, R., Kunz-Renggli, C., Maeda, Y., Ferguson, M. A. & Kinoshita, T. (2004). J. Exp. Med.199, 1445–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan, D. P., Geuskens, M. & Pays, E. (1999). Curr. Biol.9, 1169–1172. [DOI] [PubMed] [Google Scholar]
- Oppermann, U., Filling, C., Hult, M., Shafqat, N., Wu, X., Lindh, M., Shafqat, J., Nordling, E., Kallberg, Y., Persson, B. & Jornvall, H. (2003). Chem.-Biol. Interact.144, 247–253. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Roper, J. R., Güther, M. L., MacRae, J. I., Prescott, A. R., Hallyburton, I., Acosta-Serrano, A. & Ferguson, M. A. (2005). J. Biol. Chem., 280, 19728–19736. [DOI] [PubMed] [Google Scholar]
- Roper, J. R., Güther, M. L., Milne, K. G. & Ferguson, M. A. (2002). Proc. Natl. Acad. Sci. USA, 99, 5884–5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, M. P., Bond, C. S., Roper, J. R., Gourley, D. G., Ferguson, M. A. & Hunter, W. N. (2003). Mol. Biochem. Parasitol.126, 173–180. [DOI] [PubMed] [Google Scholar]
- Shi, R. & Lin, S.-X. (2004). J. Biol. Chem.279, 16778–16785. [DOI] [PubMed] [Google Scholar]
- Tetaud, E., Barrett, M. P., Bringaud, F. & Baltz, T. (1997). Biochem. J.325, 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoden, J. B., Frey, P. A. & Holden, H. M. (1996). Biochemistry, 35, 5137–5144. [DOI] [PubMed] [Google Scholar]
- Thoden, J. B., Hegeman, A. D., Wesenberg, G., Chapeau, M. C., Frey, P. A. & Holden, H. M. (1997). Biochemistry, 36, 6294–6304. [DOI] [PubMed] [Google Scholar]
- Thoden, J. B., Wohlers, T. M., Fridovich-Keil, J. L. & Holden, H. M. (2000). Biochemistry, 39, 5691–5701. [DOI] [PubMed] [Google Scholar]
- Treumann, A., Zitzmann, N., Hülsmeier, A., Prescott, A. R., Almond, A., Sheehan, J. & Ferguson, M. A. J. (1997). J. Mol. Biol.269, 529–547. [DOI] [PubMed] [Google Scholar]
- Vassella, E., Butikofer, P., Engstler, M., Jelk, J. & Roditi, I. (2003). Mol. Biol. Cell, 14, 1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: UDP-galactose-4′-epimerase complex, 2cnb, r2cnbsf