

Two S. agalactiae proteins, the inorganic pyrophosphatase and the serine/threonine phosphatase, were crystallized and diffraction data were collected and processed from these crystals. The data from the two protein crystals extended to 2.80 and 2.65 Å, respectively.
Keywords: Streptococcus agalactiae, inorganic pyrophosphatase, serine/threonine phosphatase
Abstract
Streptococcus agalactiae, which infects human neonates and causes sepsis and meningitis, has recently been shown to possess a eukaryotic-like serine/threonine protein phosphorylation signalling cascade. Through their target proteins, the S. agalactiae Ser/Thr kinase and Ser/Thr phosphatase together control the growth as well as the morphology and virulence of this organism. One of the targets is the S. agalactiae family II inorganic pyrophosphatase. The inorganic pyrophosphatase and the serine/threonine phosphatase have therefore been purified and crystallized and diffraction data have been collected from their crystals. The data were processed using XDS. The inorganic pyrosphosphatase crystals diffracted to 2.80 Å and the Ser/Thr phosphatase crystals to 2.65 Å. Initial structure-solution experiments indicate that structure solution will be successful in both cases. Solving the structure of the proteins involved in this cascade is the first step towards understanding this phenomenon in atomic detail.
1. Introduction
Streptococci are Gram-positive bacteria which, when cultured on blood agar plates, cause either partial (α) or complete (β) haemolysis. Those streptococci that cause complete haemolysis are classified into different groups based on antigenic differences in the polysaccharides that are located in the bacterial cell wall. One of the group B streptococci (GBS), characterized by the presence of the group B Lancefield antigen, is Streptococcus agalactiae, which asymptomatically colonizes the vaginal and rectal areas of 10–30% of pregnant women and may, after birth, cause pneumonia, sepsis and meningitis in human newborns. It has also recently been recognized as a pathogen of certain human adults including immunocompromized individuals, diabetics and the elderly. GBS infections in adults are varied and can range from skin, soft tissue and urinary tract infections to arthritis and endocarditis. Prevention and cure of S. agalactiae infections is thus medically important and one interesting strategy towards this would be the development of drugs to disrupt the signalling systems of S. agalactiae.
Protein phosphorylation is one type of signalling that causes reversible changes in the activities of target proteins, resulting in changes in the phenotypes of organisms (Hunter, 1995 ▶). It was previously thought to occur solely in eukaryotes; prokaryotic signalling was thought to be based only on the two-component system (Alex & Simon, 1994 ▶). In recent years, however, serine/threonine phosphorylation-based signal cascades have been found in many prokaryotes, including Bacillus subtilis (Yang et al., 1996 ▶; Adler et al., 1997 ▶), Yersinia pseudotuberculosis (Hakansson et al., 1996 ▶) and Mycobacterium tuberculosis (Boitel et al., 2003 ▶). Such a signalling cascade has also recently been discovered in S. agalactiae; changes in the phosphorylation status of S. agalactiae affect its growth, morphology and virulence (Rajagopal et al., 2003 ▶). This was shown by making mutations in the S. agalactiae Ser/Thr kinase (SaSTK) and Ser/Thr phosphatase (SaSTP) genes. Two proteins that are phosphorylated and dephosphorylated are adenylosuccinate synthetase (Rajagopal et al., 2005 ▶) and soluble inorganic pyrophosphatase (Rajagopal et al., 2003 ▶) (see below).
Soluble inorganic pyrophosphatases (EC 3.6.1.1) are essential metal-dependent enzymes (Chen et al., 1990 ▶) and can be classified into two families unrelated by divergent evolution, family I and the more recent family II (Shintani et al., 1998 ▶; Young et al., 1998 ▶), based on differences in the identities of the bound metal ions and in metal-coordinating ligands, in the sequences and in the overall structures. The S. agalactiae inorganic pyrophosphatase (SaPPase) can be classified as a family II enzyme, a number of highly related structures of which have already been solved (PDB codes 1i74, 1k20, 1k23, 1wpm and 1wpp; Merckel et al., 2001 ▶; Ahn et al., 2001 ▶; Fabrichniy et al., 2004 ▶). However, there is no evidence to date of phosphorylation affecting the catalytic activity of other inorganic pyrophosphatases, whether family I or family II, and so we decided to study the structure of SaPPase.
As mentioned above, the SaSTP is responsible for dephosphorylation of phosphorylated SaPPase and adenylosuccinate synthetase. STPs (EC 3.1.3.16) can be classified into PPP and PPM subclasses (Barford et al., 1998 ▶), with SaSTP belonging to the metal-dependent PPM/PP2C subclass based on its amino-acid sequence. There are just two PPM/PP2C STP structures in the PDB, those of human STP (1a6q; Das et al., 1996 ▶) and of that from Mycobacterium tuberculosis (PDB code 1txo; Pullen et al., 2004 ▶). The sequence identity of the former to SaSTP is just 21% and that of the latter is only 33%, making structural studies of SaSTP worthwhile. In addition, the M. tuberculosis protein was solved as part of a structural genomics project and so there is no information about its target proteins and modes of interaction, unlike in S. agalactiae (Rajagopal et al., 2003 ▶, 2005 ▶). We have therefore focused our structural studies on the serine/threonine signalling cascade enzymes from S. agalactiae, including SaPPase, SaSTP and SaSTK. Here, we report on our progress on the first two enzymes.
2. Materials and methods
2.1. Cloning, expression and purification of the enzymes
The serine/threonine phosphatase and the inorganic pyrophosphatase genes from the A909 strain of S. agalactiae (NCBI RefSeq accession Nos. YP_330038 and YP_329040; Wheeler et al., 2004 ▶) had already been cloned into the pGEX4T3 (accession No. U13855) Schistosoma japonicum glutathione S-transferase fusion protein expression vector. The SaSTP gene was cloned between the BamHI and XhoI sites and the SaPPase gene between the BamHI and SalI sites, thus fusing both in frame with the C-terminus of the glutathione S-transferase gene (Rajagopal et al., 2003 ▶). The constructs contained the β-lactamase gene, which conferred resistance to the antibiotic ampicillin and allowed selection, and a thrombin cleavage site, which allowed cleavage of the purified proteins from their fusion partner. The cloning strategy did, however, result in constructs which introduced new residues to the N-termini of SaSTP and SaPPase, two from the C-terminal side of the thrombin recognition sequence and three (SaSTP) or four (SaPPase) from the linker region. The exact N-terminal sequence additions were 1GSRKKY for SaSTP and 1GSGKN for SaPPase. Consequently, the length of the genetically engineered SaSTP increased from 245 to 251 residues and the length of the genetically engineered SaPPase from 311 to 316 residues.
Expression and purification were performed as described in Rajagopal et al. (2003 ▶). Induction times were optimized for both time and yield and both proteins were purified with the same protocol. Briefly, expression was performed using Luria Broth medium [10 g l−1 tryptone, 5 g l−1 yeast extract (Fluka Biochemica) and 5 g l−1 NaCl; J. T. Baker]. 50 µg ml−1 ampicillin (Sigma–Aldrich) was added to the culture medium. Escherichia coli BL21 (DE3) host cells (Novagen) were grown at 310 K to an OD600 of 0.5 and expression was then induced with 1 mM IPTG (isopropyl β-d-thiogalactopyranoside; Promega). Expression continued for 4 h at the lowered temperature of 303 K to minimize the formation of inclusion bodies. The cells were collected with centrifugation using a Beckman Coulter Avanti J-20 XP centrifuge and a JLA 8.1000 rotor (6240g at 277 K for 10 min).
The bacterial pellet from a 1 l growth was resuspended in 20 ml PBS (phosphate-buffered saline; 0.2 g l−1 KCl, 0.2 g l−1 KH2PO4, 8.0 g l−1 NaCl, 1.1 g l−1 Na2HPO4) buffer (reagents from J. T. Baker) and the solution was sonicated for 30 min using a Braun Labsonic sonicator at low power output with a 0.6 s duty cycle. After clarification by centrifugation using an Eppendorf 5810 R centrifuge and an F34-6-38 rotor (15 600g at 277 K for 20 min), the supernatant was applied onto a 10 ml Glutathione Sepharose 4B column (Amersham Biosciences), which was subsequently washed with several column volumes of PBS buffer. The inorganic pyrophosphatase was distinctly yellow, indicating bound metal ions, whereas the serine/threonine phosphatase was colourless. After binding, the protein on the column was treated overnight with thrombin protease (Amersham Biosciences) at room temperature using 100 units of protease per 10 mg of protein. The eluted protein concentrations were determined by Bradford assay (Bradford, 1976 ▶) and the purities and molecular weights of the enzymes were determined by SDS–PAGE (Laemmli, 1970 ▶). The gels were stained using Coomassie Brilliant Blue (Sigma Aldrich). The proteins were precipitated with ammonium sulfate (J. T. Baker) using 0.5 g ammonium sulfate per millilitre of protein solution and stored at 203 K.
2.2. Crystallization experiments
The proteins were buffer-exchanged to 20 mM Tris buffer (ICN Biomedicals Inc.) pH 7.0 or 8.0 (pyrophosphatase and phosphatase, respectively) using 6000–8000 kDa molecular-weight cutoff dialysis membrane (Membrane Filtration Products Inc.). They were then concentrated to 5–20 mg ml−1 using 10 kDa cutoff Microcon spin concentrators (Millipore). For initial crystallization experiments, Hampton Research Crystal Screens I and II were used (Jancarik & Kim, 1991 ▶). Hanging-drop vapour-diffusion experiments were set up by pipetting drops constisting of 1 µl protein solution and 1 µl well solution onto cover slips. The cover slips were laid over greased plastic plates and the experiments were allowed to equilibrate at room temperature for one to two weeks. For flash-freezing, the crystals were harvested with Hampton Research CryoLoops and transferred to 50 µl drops consisting of mother liquor to which 15% glycerol had been added. The crystals were soaked for 1 min, reharvested into CryoLoops and frozen in liquid nitrogen. Crystallization reagents were from Sigma–Aldrich (monobasic ammonium phosphate, magnesium acetate and PEG 8000) and from Fluka Biochemica (sodium cacodylate). Glycerol was from J. T. Baker.
2.3. Data collection and processing
Crystals from both proteins diffracted to medium resolution. We collected data from SaPPase crystals using Cu Kα radiation generated by a Rigaku RU3000 rotating-anode generator operated at 50 kV and 20 mA and monochromated with confocal mirrors. An R-AXIS IV++ image-plate detector was used to collect the data. Data collection was performed at 100 K and the crystal diffracted to 2.8 Å resolution. Using 0.5° frames with 12 min exposure times, a total of 100° of data were collected with a crystal-to-detector distance of 250 mm. The data were processed with XDS (Kabsch, 1993 ▶) in space group R32 and the R merged-F was 8.5% overall (Table 1 ▶).
Table 1. Data-collection statistics for the inorganic pyrophosphatase.
Values in parentheses are for the highest resolution shell.
| Space group | R32 |
| Wavelength (Å) | 1.5418 |
| Unit-cell parameters (Å) | a = b = 182.0, c = 132.6 |
| Resolution (Å) | 20–2.8 (2.9–2.8) |
| Reflections measured | 126779 (8798) |
| Unique reflections | 20708 (1993) |
| Completeness (%) | 99.2 (96.4) |
| Redundancy | 6.1 (4.4) |
| I/σ(I) | 12.3 (6.23) |
| Rmerged-F† (%) | 8.5 (14) |
R
merged-F = 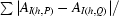
 , where AI = I
1/2 if I ≥ 0, A
I = −I
1/2 if I < 0 and P and Q are two subsets of data (Diederichs & Karplus, 1997 ▶).
, where AI = I
1/2 if I ≥ 0, A
I = −I
1/2 if I < 0 and P and Q are two subsets of data (Diederichs & Karplus, 1997 ▶).
The SaSTP crystals diffracted to 2.65 Å resolution. The data were collected at 100 K using radiation from the ESRF fixed-wavelength beamline ID14-1 equipped with a MAR detector. Using 1° frames with 2 s exposure times, a total of 132° of data were collected with a crystal-to-detector distance of 255 mm. The diffraction data, processed using XDS, had an overall R merged-F of 11.5% in space group P21212 (Table 2 ▶).
Table 2. Data-collection statistics for the serine/threonine phosphatase.
Values in parentheses are for the highest resolution shell.
| Space group | P21212 |
| Wavelength (Å) | 0.933 |
| Unit-cell parameters (Å) | a = 91.8, b = 139.0, c = 86.7 |
| Resolution (Å) | 20–2.65 (2.7–2.65) |
| Reflections measured | 169847 (7368) |
| Unique reflections | 32767 (1731) |
| Completeness (%) | 99.5 (97.5) |
| Redundancy | 5.2 (4.3) |
| I/σ(I) | 11.72 (3.69) |
| Rmerged-F† (%) | 11.5 (43.9) |
R
merged-F =
, where AI = I
1/2 if I ≥ 0, A
I = −I
1/2 if I < 0 and P and Q are two subsets of data (Diederichs & Karplus, 1997 ▶).
3. Results
3.1. Inorganic pyrophosphatase
The level of expression of the S. agalactiae inorganic pyrophosphatase was followed by taking aliquots at three time points and analyzing them on a Coomassie Brilliant Blue-stained SDS–PAGE gel (Fig. 1 ▶). The expression level increased to 50% of the total protein after 5.5 h induction (Fig. 1 ▶, lane 10). To save time, we decided to harvest after 4 h of induction, as this did not result in significant protein loss and we hoped it would lead to better quality protein. Purification was also followed by SDS–PAGE (not shown), which indicated that binding to the Glutathione Sepharose 4B column was 90% efficient, possibly because 10% of the protein was not folded properly. Cleavage was performed in the column with thrombin protease and was about 90% efficient. We obtained 40 mg of pure SaPPase per litre of culture with 80% total purification efficiency. This corresponds to an expression yield of 50 mg l−1 after a 4 h induction period.
Figure 1.

Expression of the serine/threonine phosphatase (STP) fusion protein, the inorganic pyrophosphatase (PPase) fusion protein and a pGEX4T3 control followed as a function of time. Samples were taken at 1.5, 2.5 and 5.5 h post-induction. Lane 1, molecular-weight markers (kDa) (Fermentas SMO431). Lanes 2, 5 and 8, expression of the empty vector control (pGEX4T3; C). Lanes 3, 6 and 9, expression of the SaSTP fusion protein (S). Lanes 4, 7 and 10, expression of the SaPPase fusion protein (P).
SaPPase crystallized readily from Crystal Screen I solution No. 3 (0.4 M monobasic ammonium phosphate). A pyrophosphate analogue, amidodiphosphonate [PNP; (HO)2POHNOP(OH)2], was used in the crystallization drop at 1 mM concentration and the crystals grew to 0.5 × 0.5 × 0.3 mm without optimization. Three different protein concentrations (10, 15 and 20 mg ml−1) yielded crystals. Crystals were cryoprotected using 15% glycerol (see §2) and flash-frozen in liquid nitrogen. The best crystal, taken from the drops with 20 mg ml−1 protein, diffracted to 2.8 Å resolution (Table 1 ▶). The data were processed in space group R32, with unit-cell parameters a = 182.0, c = 132.6 Å (hexagonal indexing). The Matthews coefficient V M (Matthews, 1968 ▶) is 3.1 Å3 Da−1 for two molecules per asymmetric unit or 2.1 Å3 Da−1 for three molecules per asymmetric unit. These correspond to 60.1 or 40.1% solvent content, respectively.
The sequence identity between our S. agalactiae PPase and the solved S. mutans PPase is 77% (PDB code 1i74; Merckel et al., 2001 ▶). Although this is not the structure with the highest sequence identity available (the highest being S. gordonii PPase, 83% identity; PDB code 1k20; Ahn et al., 2001 ▶), our previous experience has shown that molecular replacement in family II PPases is relatively straightforward.
Our preliminary results show that molecular replacement using 1i74 as a model is successful. MOLREP (Vagin & Teplyakov, 1997 ▶) from the CCP4 package (Collaborative Computational Project, Number 4, 1994 ▶) finds two monomers in the asymmetric unit, resulting in an R factor of 43.2% and a correlation coefficient of 0.38. We are currently refining the structure, confirming the space group of the crystal as R32. Our progress in the structure-solution process also shows that the data set is not twinned or otherwise erroneous.
3.2. Serine/threonine phosphatase
Expression of the S. agalactiae serine/threonine phosphatase was also followed by taking aliquots and analyzing them on a Coomassie Brilliant Blue-stained SDS–PAGE gel (Fig. 1 ▶). For the SaSTP, the expression level also increased to 50% of the total protein after 5.5 h (Fig. 1 ▶, lane 9) and again the decision was made to harvest the cells after 4 h. As with SaPPase, purification was also followed by SDS–PAGE (data not shown) and binding to the affinity column was 90% efficient. Cleavage performed in the column with thrombin protease was 100% efficient and we obtained 40 mg of pure SaSTP per litre. With 90% total purification efficiency, this corresponds to an expression yield of 44 mg l−1 after a 4 h induction period.
SaSTP crystallized from Crystal Screen I solution No. 18 (0.1 M sodum cacodylate pH 6.5, 0.2 M magnesium acetate, 20% PEG 8000). Varying the buffer, the pH and the PEG concentration produced several different crystal forms. At pH 6.5, we obtained crystals in space group C2, with unit-cell parameters a = 160.2, b = 68.0, c = 85.1 Å, β = 104.4°. These crystals only diffracted to 4 Å and the data-collection statistics were poor, so we searched for other conditions.
We found another crystal form using 100 mM Tris buffer pH 8.5, 0.2 M magnesium acetate and 18% PEG 8000 as the well solution. The crystals typically grew to 0.5 × 0.1 × 0.05 mm with a protein concentration of 5 mg ml−1. A 1 min soak in a drop of mother liquor that contained 15% glycerol as a cryoprotectant, together with flash-freezing in liquid nitrogen, protected the crystal successfully. The best crystal diffracted to 2.65 Å (Table 2 ▶) and the space group was primitive orthorhombic. There were systematic absences along two of the three axes, indicating that there were two screw axes, and so we processed the data in P21212, with unit-cell parameters a = 91.8, b = 1139.0, c = 86.7 Å. With four monomers per asymmetric unit, the Matthews coefficient V M would be 2.7 Å3 Da−1. This corresponds to 53.8% solvent content.
Unlike for the SaPPase, there are few models in the PDB for SaSTP. The best serine/threonine phosphatase molecular-replacement model is from M. tuberculosis (PDB code 1txo; Pullen et al., 2004 ▶). The sequence identity between the two STPs is 33% with a number of large gaps. Consequently, molecular replacement was difficult and MOLREP did not find the correct solution using this protein as a model. The R factor was 58.2% and the correlation coefficient was 0.24 with a search for four monomers in the asymmetric unit and refinement of this solution failed. This suggested that either the data were somehow not good enough to solve the structure, the space group was wrong or the model was too inaccurate. However, we recently collected SAD (single-wavelength anomalous dispersion) data (not shown). Using this phase information, we were able to build an initial model which indicates that the difference between the M. tuberculosis STP and SaSTP was too great to allow molecular replacement to be successful. The structure is currently under refinement.
Acknowledgments
We acknowledge the European Synchrotron Radiation Facility MX beamlines for provision of synchrotron radiation, which was made available under the European Union Improving Human Potential Programme (Access to Research Infrastructures) under contract No. HPRI-CT-1999-00022. We would like to thank the beamline personnel for assistance in data collection. This work was supported by grants from the Sigrid Juselius Foundation and from the Academy of Finland (grant number 1105157) to AG, who is a member of the Biocentrum Helsinki research organization, and by funding from the National Institutes of Health, Grant No. RO1 AI056073 to CER. We thank Professor Arto Annila for support for MKR.
References
- Adler, E., Donella-Deana, A., Arigoni, F., Pinna, L. A. & Stragler, P. (1997). Mol. Microbiol.23, 57–62. [DOI] [PubMed] [Google Scholar]
- Ahn, S., Milner, A. J., Futterer, K., Konopka, M., Ilias, M., Young, T. W. & White, S. A. (2001). J. Mol. Biol.313, 797–811. [DOI] [PubMed] [Google Scholar]
- Alex, L. A. & Simon, M. I. (1994). Trends Genet.10, 133–138. [DOI] [PubMed] [Google Scholar]
- Barford, D., Das, A. K. & Egloff, M. P. (1998). Annu. Rev. Biophys. Biomol. Struct.27, 133–164. [DOI] [PubMed] [Google Scholar]
- Boitel, B., Ortiz-Lombardia, M., Duran, R., Pompeo, F., Cole, S. T., Cervenansky, C. & Alzari, P. M. (2003). Mol. Microbiol.49, 1493–1508. [DOI] [PubMed] [Google Scholar]
- Bradford, M. M. (1976). Anal. Biochem.72, 248–254. [DOI] [PubMed] [Google Scholar]
- Chen, J., Brevet, A., Fromant, M., Leveque, F., Schmitter, J. M., Blanquet, S. & Plateau, P. (1990). J. Bacteriol.172, 5686–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Das, A. K., Helps, N. R., Cohen, P. T. & Barford, D. (1996). EMBO J.15, 6798–6809. [PMC free article] [PubMed] [Google Scholar]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol.4, 269–275. [DOI] [PubMed] [Google Scholar]
- Fabrichniy, I. P., Lehtiö, L., Salminen, A., Zyryanov, A. B., Baykov, A. A., Lahti, R. & Goldman, A. (2004). Biochemistry, 43, 14403–14411. [DOI] [PubMed] [Google Scholar]
- Hakansson, S., Galyov, E. E., Rosqvist, R. & Wolf-Watz, H. (1996). Mol. Microbiol.20, 593–603. [DOI] [PubMed] [Google Scholar]
- Hunter, T. (1995). Cell, 80, 225–236. [DOI] [PubMed] [Google Scholar]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411. [Google Scholar]
- Kabsch, W. (1993). J. Appl. Cryst.26, 795–800. [Google Scholar]
- Laemmli, U. K. (1970). Nature (London), 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Merckel, M. C., Fabrichniy, I. P., Salminen, A., Kalkkinen, N., Baykov, A. A., Lahti, R. & Goldman, A. (2001). Structure, 9, 289–297. [DOI] [PubMed] [Google Scholar]
- Pullen, K. E., Ng, H. L., Sung, P. Y., Good, M. C., Smith, S. M. & Alber, T. (2004). Structure, 12, 1947–1954. [DOI] [PubMed] [Google Scholar]
- Rajagopal, L., Clancy, A. & Rubens, C. E. (2003). J. Biol. Chem.278, 14429–14441. [DOI] [PubMed] [Google Scholar]
- Rajagopal, L., Vo, A., Silvestroni, A. & Rubens, C. E. (2005). Mol. Microbiol.56, 1329–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani, T., Uchiumi, T., Yonezawa, T., Salminen, A., Baykov, A. A., Lahti, R. & Hachimori, A. (1998). FEBS Lett.439, 263–266. [DOI] [PubMed] [Google Scholar]
- Vagin, A. A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025. [Google Scholar]
- Wheeler, D. L., Church, D. M., Edgar, R., Federhen, S., Helmberg, W., Madden, T. L., Pontius, J. U., Schuler, G. D., Schriml, L. M., Sequeira, E., Suzek, T. O., Tatusova, T. A. & Wagner, L. (2004). Nucleic Acids Res.32, D35–D40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X., Kang, C. M., Brody, M. S. & Price, C. W. (1996). Genes Dev.10, 2265–2275. [DOI] [PubMed] [Google Scholar]
- Young, T. W., Kuhn, N. J., Wadeson, A., Ward, S., Burges, D. & Cooke, G. D. (1998). Microbiology, 144, 2563–2571. [DOI] [PubMed] [Google Scholar]