

Glucose-1-phosphate uridylyltransferase (UgpG) from S. elodea ATCC 31461 has been expressed, purified and crystallized. Seven crystal forms were obtained and characterized to a maximum resolution of 2.65 Å.
Keywords: GalU, UgpG, RmlA, pyrophosphorylase, sugar activation
Abstract
The cloning, expression, purification, crystallization and preliminary crystallographic analysis of glucose-1-phosphate uridylyltransferase (UgpG) from Sphingomonas elodea ATCC 31461 bound to glucose-1-phosphate are reported. Diffraction data sets were obtained from seven crystal forms in five different space groups, with highest resolutions ranging from 4.20 to 2.65 Å. The phase problem was solved for a P21 crystal form using multiple isomorphous replacement with anomalous scattering from an osmium derivative and a SeMet derivative. The best native crystal in space group P21 has unit-cell parameters a = 105.5, b = 85.7, c = 151.8 Å, β = 105.2°. Model building and refinement are currently under way.
1. Introduction
Glucose-1-phosphate uridylyltransferases (EC 2.7.7.9) are enzymes that mediate the reversible conversion of glucose-1-phosphate (G1P) and UTP into UDP-glucose and pyrophosphate. These proteins are also known as pyrophosphorylases, based on their ability to catalyse the reverse reaction. This enzymatic activity is present in all living organisms, although prokaryotic enzymes diverge significantly from those of eukaryotic origin (Daran et al., 1995 ▶; Eimert et al., 1996 ▶). The glucose-1-phosphate uridylyltransferases of prokaryotic organisms are essential to the synthesis of several polysaccharides, many of them involved in the pathogenesis of those organisms. In fact, glucose-1-phosphate uridylyltransferases are required for the synthesis of capsular polysaccharide (CPS) in Streptococcus pneumoniae, which is considered to be the virulence factor in this species (Bonofiglio et al., 2005 ▶), and have been suggested to be required for corneal infection by Pseudomonas aeruginosa and to be necessary for efficient systemic spread following lung infection (Priebe et al., 2004 ▶). These findings have prompted an increasing interest in prokaryotic glucose-1-phosphate uridylyltransferases enzymes as potential drug targets (Bonofiglio et al., 2005 ▶).
The UDP-glucose produced by glucose-1-phosphate uridylyltransferase can be interconverted into other sugar nucleotides or can be used as the glucosyl donor in the biosynthesis of various cellular carbohydrates, such as exopolysaccharides (EPS), lipopolysaccharides (LPS), capsular polysacharides (CPS) and storage compounds such as trehalose. Glucose-1-phosphate uridylyltransferases are also involved in the catabolism of galactose into glucose in the Leloir pathway (Frey, 1996 ▶).
In this work, we report the crystallization of a glucose-1-phosphate uridylyltransferase (commonly referred to as UgpG) from Sphingomonas elodea ATCC 31461 (Marques et al., 2003 ▶). This bacterium produces EPS gellan gum in high yield. Gellan gum produced by this method has been approved for use as a gelling and suspending agent in the USA and EU. In its native form, gellan is a linear anionic heteropolysaccharide based on a tetrasaccharide repeat unit composed of two molecules of d-glucose, one of d-glucuronic acid and one of l-rhamnose. The native gellan is partially esterified with acyl substituents: 1 mol of glycerate and 0.5 mol of acetate per repeat unit (Jay et al., 1988 ▶). Gellan biosynthesis starts with the intracellular synthesis of the nucleotide-sugar precursors UDP-glucose, UDP-glucuronic acid and dTDP-l-rhamnose. The repeat unit is then constructed by sequential transfer of the sugar donors to an activated lipid carrier by committed glycosyltransferases, followed by gellan polymerization and export (Sá-Correia et al., 2002 ▶).
In fact, UgpG recognizes both uridine (UTP) and deoxythymidine (dTTP) nucleotides substrates in vitro (Silva et al., 2005 ▶). However, in vivo a different enzyme, glucose-1-phosphate thymidylyltransferase (EC 2.7.7.24), is responsible for the formation of dTDP-glucose.
Mutation studies, sequence comparisons and the four available glucose-1-phosphate thymidylyltransferase structures from Escherichia coli (Blankenfeldt, Asuncion et al., 2000 ▶; Blankenfeldt, Giraud et al., 2000 ▶), P. aeruginosa (Sivaraman et al., 2002 ▶), Salmonella enterica (Barton et al., 2001 ▶, 2002 ▶) and Methanobacterium thermoautotrophicum (PDB code 1lvw) have identified the possible key residues for substrate (G1P, dTTP/UTP) and product (TDP-glucose/UDP-glucose) binding, but there is still no report of a glucose-1-phosphate uridylyltransferase structure.
2. Materials and methods
2.1. Cloning and expression of UgpG
The complete sequence of the ugpG gene from Sphingomonas elodea ATCC 31461 was amplified by PCR using the oligonucleotides PUGP19 (5′-AAAGGATCCATGACGATCAAGC-3′) and PUGP29 (5′-AAAAAGCTTTCAGCCGAGCGCCTT-3′) and cosmid pC22 (16) as the template DNA. The PCR product (888 bp) was digested with BamHI and HindIII (shown in bold in the primers used) and cloned into compatible sites of pWH844 (Schirmer et al., 1997 ▶), generating pUgpG2. This recombinant plasmid carries the ugpG gene preceded by a sequence coding for six histidine residues followed by two residues, a glycine and a serine, before the initiating methionine. The insert cloned in pUgpG2 was then sequenced to confirm the fidelity of DNA amplification.
To overexpress the ugpG gene, transformants of E. coli BL21 Gold carrying pUgpG2 were cultivated at 298 K in 100 ml LB medium until an OD640nm of 0.6 ± 0.1 was attained. The cells were then induced with 0.1 mM isopropyl β-d-thiogalactoside (IPTG) for 3 h, harvested by centrifugation (5000g, 10 min, 277 K) and stored at 253 K overnight.
For the production of the SeMet-UgpG derivative protein (Molecular Dimensions protocol), methionine-auxotrophic E. coli strain B843 (DE3) harbouring pUgpG2 was cultivated at 298 K in 100 ml LB until an OD640nm of 0.6 ± 0.1 was attained. Cells were then harvested by centrifugation (5000g, 10 min, 278 K) and gently resuspended in SelenoMet Medium Base (Molecular Dimensions, UK). This process was repeated three times to remove traces of LB containing methionine. Finally, 250× concentrated selenomethionine solution (Molecular Dimensions, UK) was added and the cells were induced for 5 h with 0.1 mM IPTG.
2.2. Purification of UgpG
Cells were resuspended in 50 mM Tris–HCl pH 8.0, 10 mM imidazole, 50 mM NaCl and disrupted in a French press. Crude cell extract was obtained by centrifugation at 17 000g for 25 min and the supernatant was applied onto a 5 ml HisTrap column (Amersham Pharmacia, UK) pre-equilibrated with 50 mM Tris–HCl pH 8.0, 10 mM imidazole, 50 mM NaCl. The column was washed five times with buffer A (50 mM Tris–HCl pH 8.0, 10 mM imidazole, 500 mM sodium chloride) to remove any unbound proteins and a gradient of buffer B (50 mM Tris–HCl pH 8.0, 500 mM imidazole, 500 mM sodium chloride) was applied to 100%. UgpG was eluted at approximately 300 mM imidazole in a symmetrical chromatography peak. The eluted fractions were immediately transferred into 75 mM Tris–HCl buffer pH 8.0 and 40 mM sodium chloride using a PD10 desalting column (Amersham Pharmacia, UK) and the protein was concentrated to 10 mg ml−1 before storage at 253 K.
The SeMet-UgpG protein was purified, concentrated and stored as described above. MALDI–TOF mass spectrometry confirmed 100% selenomethionine incorporation and a molecular weight of 32 kDa in the UgpG produced by this method.
The purity of both native and Se-labelled proteins was confirmed by SDS–PAGE. Protein concentrations were determined by the method of Bradford (1976 ▶) using bovine serum albumin fraction V (Sigma, France) as the standard.
Gel-filtration chromatography and dynamic light scattering confirmed that the protein was stable and monodisperse under the working conditions. These techniques show UgpG to have a molecular weight between 130 and 140 kDa in solution.
2.3. Crystallization of UgpG protein
Crystallization screens with native protein were performed with a Tecan Genesis robot using the vapour-diffusion method. Drops consisting of 1 µl native protein solution (75 mM Tris–HCl pH 8.0, 40 mM sodium chloride) at 5 or 10 mg ml−1 and 1 µl precipitant solution were equilibrated against 576 conditions based on the crystallization screens Crystal Screen, Crystal Screen 2, Crystal Screen Lite, PEG/Ion Screen, Quick Screen, Index, Crystal Screen Ammonium Sulfate, Screen Malonate, Screen Formate, PEG 6K, PEG LiCl, MPD and MME 5000 from Hampton Research (Aliso Viejo, USA). Crystalline material was found in several of these screens. In Crystal Screen Lite, five drops contained crystals that were visible through a microscope. From those, solution No. 9 was the most promising in terms of diffraction and was subsequently refined (crystallization condition A1; see Table 1 ▶). In Crystal Screen I, solution No. 40 was optimized by the use of macroseeding and temperature screens between 277 and 308 K, which led to larger needles (condition A2, Table 1 ▶).
Table 1. Crystallization conditions for UgpG.
| Crystallization conditions | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Crystal | Salt/alcohol | Buffer | Precipitant | Cocrystallization | Drop size/ratio, protein + precipitant (µl) | Crystal dimensions (mm) | Growth time (d) | Temperature (K) | Cryoprotectant | Observations |
| A1 | 0.2 M NH4OAc | 0.1 M sodium citrate pH 4.6 | 33% PEG 4K | — | 1 + 1 | 0.02 × 0.02 × 0.08 | 15 | 281 | 25%(v/v) glycerol/mother liquor | — |
| A2 | 20% 2-propanol | 0.1 M sodium citrate pH 6.0 | 20% PEG 6K | — | 2 + 2 | 0.03 × 0.03 × 0.10 | 15 | 291 | 25%(v/v) glycerol/mother liquor | Macroseeding |
| B | 0.1 M NH4OAc | 0.1 M sodium citrate pH 5.0 | 31% PEG 4K | — | 3 + 3 | 0.05 × 0.05 × 0.10 | 21 | 277 | 25%(v/v) glycerol/mother liquor | SeMet protein |
| C | 1.6 M (NH4)2SO4 | 0.1 M Bicine pH 9.0 | — | — | 3 + 2 | 0.06 × 0.03 × 0.03 | 4 | 293 | 35%(w/v) sucrose/mother liquor | — |
| D | 1.6 M (NH4)2SO4 | 0.1 M Bicine pH 9.0 | — | — | 3 + 2 | 0.08 × 0.04 × 0.04 | 7 | 293 | 35%(w/v) sucrose/mother liquor | — |
| E | — | 0.1 M sodium citrate pH 5.6 | 10% PEG MME | +0.5 µl dioxane/drop | 5 + 4.5 | 0.12 × 0.08 × 0.03 | 2 | 293 | 35%(w/v) sucrose/mother liquor | 0.1% LM agarose gel, SeMet protein |
| F | — | 0.1 M sodium citrate pH 6.0 | 15% PEG 4K | 10 mM UDP-Glc | 2 + 2 | 0.02 × 0.03 × 0.05 | 15 | 293 | 35%(w/v) sucrose/mother liquor | — |
| G1 | 0.1 M NH4OAc | 0.1 M sodium citrate pH 4.6 | 15% PEG MME | 5 mM G1P | 5 + 2 | 0.20 × 0.05 × 0.05 | 15 | 293 | Not needed | 30 min soak in 10 mM K2OsO4 |
| G2 | 0.1 M NH4OAc | 0.1 M sodium citrate pH 4.6 | 13% PEG MME | 5 mM G1P | 5 + 2 | 0.10 × 0.04 × 0.04 | 15 | 293 | Not needed | SeMet protein |
| G3 | 0.1 M NH4OAc | 0.1 M sodium citrate pH 4.6 | 15% PEG MME | 5 mM G1P | 5 + 2 | 0.20 × 0.05 × 0.05 | 15 | 293 | Not needed | — |
Since the initial screens showed amourphous precipitate in numerous conditions, lower buffer and salt in the protein solution (50 mM Tris–HCl pH 7.6, 30 mM sodium chloride) were used. A further set of crystallization screens, this time for both native and selenated proteins, were performed using the vapour-diffusion method as implemented on the EMBL Cartesian HTX platform, which led to over 30 conditions producing crystals. Manual reproduction of the condition giving crystals with promising diffraction (again solution No. 9 from Crystal Screen Lite) and protein/precipitant composition and ratio optimization led to the first good-sized needle-like crystals of the SeMet protein (condition B in Table 1 ▶). Optimization of crystals from solution No. B6 of Crystal Screen Ammonium Sulfate led to rod-like crystals of the native protein (conditions C and D in Table 1 ▶). SeMet-UgpG crystals growing in condition No. C2 of Screen MME 500 grew very fast, reaching 0.1 × 0.08 × 0.03 mm in less than 2 h, but unfortunately did not deliver diffraction higher than 4.5 Å resolution. Temperature, precipitant composition and cocrystallization with glycerol and other additives were not successful in slowing nucleation. The use of 0.1% LM agarose (Hampton Research) in the drops retarded nucleation by 24 h before the appearance of the first crystals. Optimization of this condition led to condition E in Table 1 ▶. A similar condition with the native protein was also found to be successful (condition F in Table 1 ▶).
Since crystal quality had not been sufficient to enable structure determination, another set of 576 crystallization trials for the native and selenated UgpG were performed using the EMBL Cartesian HTX platform, this time also attempting cocrystallization with 1 and 5 mM G1P, UDP-Glc or UTP. Again, the best conditions were found to be similar to those already obtained before and optimization led to condition G3 (Fig. 1 ▶). Crystal soaks and cocrystallization trials with salts of Hg, Pt, Sm, W and Os were screened in order to search for heavy-atom derivatives. Soaking times ranged from 10 min to 3 h and soaking concentrations varied from 2 to 50 mM. The crystals soaked with potassium osmate(VI) darkened with increasing incubation time (see supplementary material1), suggesting metal incorporation (crystals G1, G2 and G3). Only W and Os produced diffracting crystals.
Figure 1.

Crystal of native UgpG crystallized from 5 µl protein solution at a concentration of 9 mg ml−1 plus 2 µl buffer solution (condition G3, Table 1 ▶).
2.4. X-ray diffraction analysis and structure determination
All diffraction data were collected at the European Synchrotron Radiation Facility (ESRF). Conditions A1, A2, B, C, D, E and F gave crystals that diffracted to a maximum of 2.9 Å resolution when cryocooled (Table 2 ▶). Cryoprotection was always achieved by soaking the crystals in cryoprotectant for the shortest possible time; longer soaks did not result in improved diffraction. Flash-cooling took place by rapidly placing crystal loops in a 100 K nitrogen stream. Heavy-atom screens were conducted with salts of Hg, Pt, Sm, W and Os, but either suitable derivatives were not obtained or molecular replacement (using homologous Rm1A structures and a number of software packages) was not successful in structure determination. Other attempts to use the homologous Rm1A structures as search models for glucose-1-phosphate uridylyltransferase molecular replacement have also been reported to fail (Kim et al., 2004 ▶). The best crystals grew from type G conditions, from which were obtained an osmium and a selenomethionine derivative and the highest resolution native data sets, G1, G2 and G3, respectively.
Table 2. Data-collection statistics for the various crystal types.
All data reduction was performed with MOSFLM (Leslie, 1992 ▶), SCALA and TRUNCATE (Collaborative Computational Project, Number 4, 1994 ▶). Estimation of the number of molecules in the asymmetric unit (ASU) was based on normalized probabilities for the occurrence of multimerization states of the Matthews coefficient (V M) and solvent content (V s) (Kantardjieff & Rupp, 2003 ▶; Matthews, 1968 ▶). Values in parentheses are for the outer shell.
| Crystal | A1 | A2 | B | C | D | E | F | G1 | G2 | G3 |
|---|---|---|---|---|---|---|---|---|---|---|
| ESRF beamline | ID14-1 | ID14-2 | ID14-2 | ID14-4 | ID14-4 | ID14-4 | ID14-2 | ID29 | ID14-2 | ID29 |
| Wavelength (Å) | 0.934 | 0.934 | 0.933 | 0.933 | 0.933 | 0.979 | 0.933 | 1.141 | 0.933 | 0.976 |
| Special observations | — | — | SeMet | — | — | SeMet | UDP-Glc | Os, G1P | SeMet, G1P | G1P |
| Resolution (Å) | 24.50–3.50 (3.65–3.50) | 81.60–3.10 (3.22–3.10) | 77.40–4.20 (4.34–4.22) | 76.40–3.00 (3.10–3.00) | 76.20–2.90 (3.04–2.90) | 76.40–3.00 (3.17–3.02) | 83.60–4.00 (4.28–4.00) | 74.70–3.50 (3.67–3.50) | 75.20–3.50 (3.59–3.50) | 102.0–2.65 (2.67–2.65) |
| Space group | P3121 | P3121 | P312 | P1 | P1 | P212121 | P3121 | P21 | P21 | P21 |
| Unit-cell parameters | ||||||||||
| a (Å) | 92.6 | 93.0 | 122.6 | 92.3 | 89.7 | 76.6 | 92.1 | 105.3 | 106.6 | 105.5 |
| b (Å) | 92.6 | 93.0 | 122.6 | 92.7 | 124.3 | 102.2 | 92.1 | 85.7 | 85.9 | 85.7 |
| c (Å) | 257.0 | 256.6 | 159.7 | 124.8 | 167.7 | 152.9 | 245.6 | 152.3 | 152.3 | 151.8 |
| α (°) | 90.0 | 90.0 | 90.0 | 96.6 | 86.7 | 90 | 90.0 | 90.0 | 90.0 | 90.0 |
| β (°) | 90.0 | 90.0 | 90.0 | 90.1 | 85.9 | 90 | 90.0 | 104.8 | 105.4 | 105.2 |
| γ (°) | 120.0 | 120.0 | 120.0 | 115.6 | 83.6 | 90 | 120.0 | 90.0 | 90.0 | 90.0 |
| No. of reflections | 177577 | 296577 | 72806 | 144310 | 536358 | 162303 | 98321 | 192656 | 440196 | 490748 |
| No. of unique reflections | 16645 | 27833 | 10841 | 82535 | 140323 | 24180 | 18944 | 33222 | 33342 | 85566 |
| Redundancy | 10.7 | 10.6 | 6.7 | 1.7 | 3.8 | 6.7 | 5.2 | 5.8 | 13.2 | 5.7 |
| 〈I/σ(I)〉 | 15.4 (3.0) | 20.3 (4.0) | 18.4 (5.5) | 9.0 (2.1) | 10.5 (2.3) | 13.8 (3.1) | 13.8 (3.8) | 12.0 (2.4) | 18.1 (4.5) | 14.1 (2.0) |
| Rmerge† | 0.11 (0.60) | 0.08 (0.51) | 0.11 (0.44) | 0.10 (0.25) | 0.11 (0.50) | 0.13 (0.48) | 0.11 (0.50) | 0.13 (0.64) | 0.15 (0.65) | 0.10 (0.65) |
| Completeness (%) | 99.5 (98.9) | 99.9 (99.8) | 100.0 (100.0) | 90.6 (90.1) | 90.9 (90.2) | 100 (100) | 98.4 (98.1) | 99.6 (98.5) | 99.2 (99.0) | 100 (100) |
| Mosaicity (°) | 0.4 | 0.4 | 1.0 | 0.6 | 1.5 | 0.7 | 1 | 0.5 | 0.5 | 0.5 |
| VM (Å3 Da−1) | 2.48 | 2.50 | 2.71 | 2.49 | 2.89, 2.41 | 2.34 | 2.35 | 2.60 | 2.62 | 2.59 |
| Solvent content (%) | 50.5 | 50.7 | 54.6) | 50.5 | 57.5, 49.0 | 47.4 | 47.7 | 52.6 | 53.0 | 52.4 |
| Estimated No. of molecules in ASU | 4 | 4 | 4 | 12 | 20, 24 | 4 | 4 | 8 | 8 | 8 |
| Isomorphous phasing power | — | — | — | — | — | — | — | 0.73 | 0.53 | — |
| Figure of merit (centric/acentric reflections) | — | — | — | — | — | — | — | 0.25/0.27 | 0.19/0.10 | — |
R
merge = 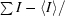
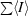 , where I is the observed intensity.
, where I is the observed intensity.
Crystals cryocooled as grown (no cryoprotectant required) to 100 K from condition G1 diffracted to 3.5 Å resolution, from condition G2 to 3.5 Å resolution and from condition G3 to 2.65 Å resolution. Gel-filtration and dynamic light-scattering studies show that UgpG oligomerizes as a tetramer, 4 × 32 kDa, in solution, as reported by others (Chang et al., 1999 ▶). Taking this into consideration, the Matthews coefficient (V M = 2.6 Å3 Da−1) suggests two tetramers in the asymmetric unit with 53% solvent content. Diffraction data were collected at the Os L III absorption peak from a type G1 crystal soaked with 10 mM potassium osmate(VI) for 60 min. Using data from the G3 type native and osmium derivative, AutoSHARP (Vonrhein et al., 2006 ▶) was directed to search for osmium sites and successfully located 12 sites. To improve the electron-density maps, 80 selenium peaks were located from the phased anomalous difference map calculated using diffraction data collected at the peak of the selenium K absorption edge from a G2 type crystal and input into SHARP (de La Fortelle & Bricogne, 1997 ▶) together with the phase information from the osmium data (Fig. 2 ▶). Eight monomers were built by manual model building using Coot (Emsley & Cowtan, 2004 ▶) on eightfold-averaged maps calculated with DM (Cowtan, 1994 ▶).
Figure 2.

Section of the experimental electron-density map at 3.5 Å resolution produced by AutoSHARP and after density modification by SOLOMON (12 osmium sites and 83 selenium sites for phasing).
3. Results
Glucose-1-phosphate uridylyltransferase from S. elodea ATCC 31461 was cloned and expressed. Extensive crystallization screens were performed in order to address the phase problem. Diffracting crystals were obtained in five different space groups and seven crystal forms, from which diffraction data were collected and characterized. SeMet and heavy-atom derivatives were screened. The phase problem was finally solved by MIRAS using Os and SeMet derivatives and the best diffracting crystal form, a complex with one G1P bound molecule in a monoclinic system that diffracted to 2.65 Å resolution. In the crystal, with eight UgpG molecules per asymmetric unit and a solvent content of 52%(v/v), the protein oligomerizes as tetramers, matching that observed in solution by other biochemical techniques.
Supplementary Material
Supplementary material file. DOI: 10.1107/S1744309106030107/ll5075sup1.pdf
Acknowledgments
The authors thank the staff at IBS and EMBL Grenoble for the use of high-throughput crystallization robots and ESRF for the provision of synchrotron radiation. The authors also thank Ana Varela Coelho and the ITQB/IBET Mass Spectrometry Laboratory for mass-spectrometric analysis. DA acknowledges a grant from FCT, SFRH (BD/6480/2001). This work was partially funded by FEDER and Fundação para a Ciência e a Tecnologia (FCT), Portugal (grants POCTI/BME/44441/2002, POCTI/BIO/58401/2004).
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: LL5075).
References
- Barton, W. A., Biggins, J. B., Jiang, J., Thorson, J. S. & Nikolov, D. B. (2002). Proc. Natl Acad. Sci. USA, 99, 13397–13402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton, W. A., Lesniak, J., Biggins, J. B., Jeffrey, P. D., Jiang, J., Rajashankar, K. R., Thorson, J. S. & Nikolov, D. B. (2001). Nature Struct. Biol.8, 545–551. [DOI] [PubMed] [Google Scholar]
- Blankenfeldt, W., Asuncion, M., Lam, J. S. & Naismith, J. H. (2000). EMBO J.19, 6652–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenfeldt, W., Giraud, M. F., Leonard, G., Rahim, R., Creuzenet, C., Lam, J. S. & Naismith, J. H. (2000). Acta Cryst. D56, 1501–1504. [DOI] [PubMed] [Google Scholar]
- Bonofiglio, L., Garcia, E. & Mollerach, M. (2005). Curr. Microbiol.51, 217–221. [DOI] [PubMed] [Google Scholar]
- Bradford, M. M. (1976). Anal. Biochem.72, 248–254. [DOI] [PubMed] [Google Scholar]
- Chang, H. Y., Huang, H. C., Lee, J. H. & Peng, H. L. (1999). Proc. Natl Sci. Counc. Repub. China B, 23, 74–84. [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Cowtan, K. (1994). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.31, 34–38.
- Daran, J. M., Dallies, N., Thines-Sempoux, D., Paquet, V. & Francois, J. (1995). Eur. J. Biochem.233, 520–530. [DOI] [PubMed] [Google Scholar]
- Eimert, K., Villand, P., Kilian, A. & Kleczkowski, L. A. (1996). Gene, 170, 227–232. [DOI] [PubMed] [Google Scholar]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Frey, P. A. (1996). FASEB J.10, 461–470. [PubMed] [Google Scholar]
- Jay, A. J., Colquhoun, I. J., Ridout, M. J., Brownsey, G. J., Morris, V. J., Fialho, A. M., Leitão, J. H. & Sa-Correia, I. (1988). Carbohydr. Polym.35, 179–188.
- Kantardjieff, K. & Rupp, B. (2003). Protein Sci.12, 1865–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H., Wu, C. A., Kim, D. Y., Han, Y. H., Ha, S. C., Kim, C. S., Suh, S. W. & Kim, K. K. (2004). Acta Cryst. D60, 1447–1449. [DOI] [PubMed] [Google Scholar]
- La Fortelle, E. & de Bricogne, G. (1997). Methods Enzymol.276, 472–494. [DOI] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Marques, A. R., Ferreira, P. B., Sá-Correia, I. & Fialho, A. M. (2003). Mol. Genet. Genomics, 268, 816–824. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Priebe, G. P., Dean, C. R., Zaidi, T., Meluleni, G. J., Coleman, F. T., Coutinho, Y. S., Noto, M. J., Urban, T. A., Pier, G. B. & Goldberg, J. B. (2004). Infect. Immun.72, 4224–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sá-Correia, I., Fialho, A. M., Videira, P., Moreira, L. M., Marques, A. R. & Albano, H. (2002). J. Ind. Microbiol. Biotechnol.29, 170–176. [DOI] [PubMed] [Google Scholar]
- Schirmer, F., Ehrt, S. & Hillen, W. (1997). J. Bacteriol.179, 1329–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva, E., Marques, A. R., Fialho, A. M., Granja, A. T. & Sá-Correia, I. (2005). Appl. Environ. Microbiol.71, 4703–4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaraman, J., Sauve, V., Matte, A. & Cygler, M. (2002). J. Biol. Chem.277, 44214–44219. [DOI] [PubMed] [Google Scholar]
- Vonrhein, C., Blanc, E., Roversi, P. & Bricogne, G. (2006). Macromolecular Crystallography Protocols, Vol. 2, edited by S. Doublié. Totowa, NJ, USA: Humana Press.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material file. DOI: 10.1107/S1744309106030107/ll5075sup1.pdf