

Two methyltransferases from flaviviruses (Meaban and Yokose viruses) have been overexpressed and crystallized. Diffraction data and characterization of the two crystal forms are presented, together with a preliminary molecular-replacement solution for both enzymes.
Keywords: (nucleoside-2′-O-)-methyltransferases, flaviviruses, Meaban virus, Yokose virus
Abstract
Viral methyltranferases (MTase) are involved in the third step of the mRNA-capping process, transferring a methyl group from S-adenosyl-l-methionine (SAM) to the capped mRNA. MTases are classified into two groups: (guanine-N7)-methyltransferases (N7MTases), which add a methyl group onto the N7 atom of guanine, and (nucleoside-2′-O-)-methyltransferases (2′OMTases), which add a methyl group to a ribose hydroxyl. The MTases of two flaviviruses, Meaban and Yokose viruses, have been overexpressed, purified and crystallized in complex with SAM. Characterization of the crystals together with details of preliminary X-ray diffraction data collection (at 2.8 and 2.7 Å resolution, respectively) are reported here. The sequence homology relative to Dengue virus 2′OMTase and the structural conservation of specific residues in the putative active sites suggest that both enzymes belong to the 2′OMTase subgroup.
1. Introduction
The first pre-mRNA maturation steps in the RNA-transcription machinery of eukaryotes concern modifications at the terminal ends of the RNA molecule. Among such modifications, capping plays an essential role in the life cycle of mRNA, being required for efficient pre-mRNA splicing, export, stability and translation. Moreover, capping protects mRNA from enzymatic degradation (Parker & Song, 2004 ▶) and improves binding stability onto the ribosome (Shuman, 2001 ▶). Viruses are a particularly attractive system for the study of the molecular basis of RNA capping. However, some viruses replicate in the cytoplasm of eukaryotic cells, whose mRNA-capping machinery is located in the nucleus. Accordingly, some of these viruses have developed a strategy that does not require RNA capping, while others, such as flaviviruses, encode their own RNA-capping enzymes. The viral capping process originates at the 5′ end of the triphosphate mRNA, which is converted to a diphosphate by an RNA triphosphatase. A GTP unit is then added in a 5′-5′ phosphodiester bond by a guanylyltransferase to yield the ‘cap’ together with the first two nucleosides of the chain. The cap is further decorated by addition of methyl groups to the N7 position of the guanine (cap0) and to a hydroxyl group of the cap nucleotides (cap1). These steps are catalyzed by (guanine-N7)-methyltransferase (N7MTase) and (nucleoside-2′-O-)-methyltransferase (2′OMTase), respectively, which transfer the methyl group from S-adenosyl-l-methionine (SAM), the MTase cofactor. In some cases, methylation of the guanine base occurs before guanylyl transfer (Ahola & Kaariainen, 1995 ▶). Although RNA-cap structures originating from viral or cellular enzymes are often identical, the physical organization of genes, subunit composition, structure and the catalytic mechanisms of enzymes of the capping machinery differ significantly in fungi, metazoans, protozoa and viruses (Shuman, 2001 ▶). Such diversity makes RNA capping an attractive target for the design of antiviral drugs.
Meaban and Yokose viruses belong to the genus Flavivirus (family Flaviviridae). This genus of viruses includes important human pathogens such as West Nile virus (WNV), dengue virus (DENV) and yellow fever virus. Their single-stranded RNA genome is of positive polarity, displaying a cap1 structure (Chambers et al., 1990 ▶). Meaban virus has been isolated from seabird ticks in France, while Yokose virus has been isolated from Japanese bats. Little is known about the components of the flavivirus RNA-capping machinery. RNA triphosphatase activity has been described only for WNV and mapped to the C-terminus of the non-structural protein 3 (NS3) (Wengler, 1993 ▶). The guanylyltransferase enzyme has not yet been identified. The crystal structure of DENV NS5 2′OMTase (DVMTase) in complex with the methylation reaction product S-adenosyl-l-homocysteine has been reported (Egloff et al., 2002 ▶), but the exact GTP recognition and reactivity details in the N-terminal region of the MTase domain are still only marginally characterized.
We have expressed, purified and crystallized the Meaban virus (MEAV) N-terminal NS5 MTase domain (MMTase; 265 residues) and the Yokose virus (YOKV) N-terminal NS5 MTase domain (YMTase; 293 residues) in complex with SAM. We report here the characterization of both crystals together with data on the preliminary X-ray diffraction experiments. The high sequence homology of both MTases relative to DVMTase and the conservation of characteristic residues in the putative active-site regions suggest that both viral enzymes act as 2′OMTases.
2. Materials and methods
2.1. Expression and purification
The MTase genes of Meaban and Yokose viruses were independently amplified from genomic RNA extracted from infected SW13 human cells (cultured as monolayers at 310 K under 5% CO2 in Eagle’s minimum essential medium with 10% calf serum and penicillin G, kanamycin and streptomycin at 100 IU ml−1, 100 µg ml−1 and 100 µg ml−1, respectively) using a long-range RT-PCR kit (cMaster RTplus PCR System, Eppendorf) and specific primers deduced from the genomic sequence. For each protein, this product was re-amplified using Gateway modified primers (Invitrogen) to encode N-terminal His6-tagged recombinant proteins (MMTase, amino acids 1–265, and YMTase, amino acids 1–293, of the respective NS5 genes). They were expressed in a pDEST 14 HN expression vector (Invitrogen). Cultures of Escherichia coli Rosetta (DE3) pRos transformed with the expression plasmid were grown in 2 l SB medium containing 100 µg ml−1 ampicillin and 34 µg ml−1 chloramphenicol at 310 K until the OD600 reached 0.8. After cooling the cultures to 298 K, expression of the recombinant protein was induced by the addition of 0.5 mM isopropyl β-d-thiogalactopyranoside (IPTG) and the culture was incubated overnight. The cells were harvested by centrifugation and resuspended in 80 ml lysis buffer [300 mM NaCl, 50 mM Tris–HCl pH 8, 25 mM imidazole, 0.25 mg ml−1 lysozyme, Complete EDTA-free protease inhibitor cocktail (Roche)] and stored at 193 K for 30 minutes. The cells were thawed and, after the addition of 10 µg ml−1 DNAse and 20 mM MgSO4, were lysed by sonication and then centrifuged at 23 000g for 1 h at 277 K. The supernatant was then injected onto a HisTrap HP column (all column materials were purchased from GE Healthcare) and eluted with 250 mM imidazole. In parallel procedures, the proteins were concentrated in an Amicon Ultra centrifugal filter device with a 10 kDa cutoff and then loaded onto a HiLoad 16/60 Superdex 200 column. MMTase was eluted in 800 mM NaCl, 1 mM DTT, 10% glycerol, 50 mM Bicine pH 7.5 and concentrated to 17.4 mg ml−1 for crystallization trials using an Amicon Ultra centrifugal filter. YMTase was eluted in 10 mM CHES, 500 mM NaCl, 1 mM DTT pH 9.0 and concentrated to 13 mg ml−1 for crystallization trials using an Amicon Ultra centrifugal filter. Both protein constructs retained their N-terminal His6 tags.
2.2. Dynamic light scattering
The purified proteins were analyzed using dynamic light scattering to assess their suitability for crystallization, as described by Ferre-D’Amare & Burley (1994 ▶) and Zulauf & D’Arcy (1992 ▶), using a DynaPro instrument (Protein Solutions, Charlottesville, USA). The proteins were centrifuged at 13 000g for 10 min prior to analysis. DLS measurements carried out at 283 and 293 K showed that at both temperatures the proteins were sufficiently monodisperse (polydispersity 12–15%) to justify crystallization trials.
2.3. Crystallization
Vapour-diffusion crystallization experiments were prepared using an Oryx-6 crystallization robot (Douglas Instruments, East Garston, UK). In a typical experiment, 0.4 µl screening solution was added to 0.4 µl protein solution in 96-well Corning Crystalex round plates (Hampton Research); the reservoir wells contained 100 µl screening solution. The screening solutions used for the experiments were those from Crystal Screens I and II, Index and SaltRx Screens from Hampton Research (Aliso Viejo, CA, USA). All crystallization trials were performed at 293 K. In the absence of crystalline precipitates after two months, all crystal screening trials were repeated as described above, but using protein solutions containing 10 mM SAM. Crystals of MMTase were obtained, with low reproducibility, after two months of vapour diffusion against 20% PEG MME 2000, 0.01 M NiCl2, 0.1 M Tris pH 8.5 (Fig. 1 ▶ a). In contrast, small isolated crystals of YMTase were obtained after 2 d of vapor diffusion against 1 M sodium formate pH 4.6. Optimization of such growth conditions (to 1.5 M sodium formate pH 4.0) yielded crystals of YMTase that were suitable for X-ray analysis (Fig. 1 ▶ b).
Figure 1.

Crystals of Meaban and of Yokose virus methyltransferases grown in the presence of 10 mM S-adenosyl-l-methionine. (a) MMTase crystal of about 120 × 35 × 35 µm in size. (b) YMTase crystals; the largest crystals are approximately 120 × 60 × 30 µm in size.
3. Results and discussion
3.1. MMTase X-ray data collection and characterization
Native X-ray diffraction data were collected from a flash-cooled crystal of MMTase suspended in the crystallization mother liquor supplemented with 20% glycerol for cryoprotection at the European Synchrotron Radiation Facility (ESRF-Grenoble, France) on beamline ID29; the native crystal diffracted to a maximum resolution of 2.8 Å. The diffraction data were processed with MOSFLM (Leslie, 1992 ▶) and intensities were merged with SCALA (Collaborative Computational Project, Number 4, 1994 ▶). The MMTase crystals belong to the tetragonal space group P43, with unit-cell parameters a = b = 83.0, c = 170.2 Å. The calculated crystal-packing coefficient (VM = 2.5 Å3 Da−1) suggests the presence of four MMTase molecules per asymmetric unit and a corresponding solvent content of 49.9% (Matthews, 1968 ▶). X-ray data-collection statistics are summarized in Table 1 ▶.
Table 1. MMTase X-ray data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Space group | P43 |
| Unit-cell parameters (Å) | a = b = 83.0, c = 170.2 |
| Resolution (Å) | 40.00–2.90 (3.06–2.90) |
| Mosaicity (°) | 0.67 |
| No. of unique reflections | 24302 (3582) |
| Completeness (%) | 95.4 (96.8) |
| Redundancy | 3.2 |
| Rmerge† (%) | 9.5 (52.6) |
| Average I/σ(I) | 12.4 (3.1) |
| R factor‡ (%) | 34.9 |
| Rfree§ (%) | 41.2 |
R
merge = 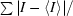
 × 100, where I is the intensity of a reflection and 〈I〉 is the average intensity.
× 100, where I is the intensity of a reflection and 〈I〉 is the average intensity.
R factor = 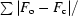
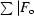 | × 100.
| × 100.
R free is calculated from a randomly selected 5% of data for cross-validation.
3.2. YMTase X-ray data collection and characterization
The YMTase crystals were cryoprotected by transfer to their mother liquor supplemented with 20% glycerol prior to flash-cooling in liquid nitrogen. A 2.7 Å X-ray diffraction data set was collected at ESRF beamline ID23-1. The data were integrated and scaled using the programs MOSFLM (Leslie, 1992 ▶) and SCALA (Collaborative Computational Project, Number 4, 1994 ▶) (see Table 2 ▶). YMTase crystals belong to the orthorhombic space group P21212, with unit-cell parameters a = 58.7, b = 126.6, c = 156.1 Å. Calculation of the Matthews crystal-packing coefficient (V M = 2.9 Å3 Da−1) suggests the presence of three YMTase molecules in the asymmetric unit, with a solvent content of 57.7% (Matthews, 1968 ▶). Data-collection statistics are summarized in Table 2 ▶.
Table 2. YMTase X-ray data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Space group | P21212 |
| Unit-cell parameters (Å) | a = 58.7, b = 126.6, c = 156.1 |
| Resolution (Å) | 40.00–2.80 (2.95–2.80) |
| Mosaicity (°) | 0.4 |
| No. of unique reflections | 27356 (4028) |
| Completeness (%) | 93.3 (95.4) |
| Redundancy | 3.0 |
| Rmerge† (%) | 14.0 (57.6) |
| Average I/σ(I) | 9.1 (1.9) |
| R factor‡ (%) | 37.8 |
| Rfree§ (%) | 42.1 |
R
merge =
× 100, where I is the intensity of a reflection and 〈I〉 is the average intensity.
R factor =
| × 100.
R free is calculated from a randomly selected 5% of data for cross-validation.
3.3. Structure solution for MMTase and YMTase
The crystal structure of MMTase was solved by the molecular-replacement method using the program MOLREP (Vagin & Teplyakov, 1997 ▶). The search model was based on the DVMTase structure (PDB code 1r6a) in which all non-conserved residues had been manually mutated to Ala. The four independent MMTase molecules located as the molecular-replacement solution in the crystallographic asymmetric unit were subjected to rigid-body refinement to optimize both their orientation and position and partially refined (R = 34.9%, R free = 41.2% at 2.8 Å resolution) using REFMAC5 (Winn et al., 2001 ▶). Inspection of difference Fourier maps at this stage showed a strong residual density feature compatible with one SAM molecule that was not present in the search model.
Similarly, for YMTase, the structure was solved using the molecular-replacement program MOLREP (Vagin & Teplyakov, 1997 ▶) using the polypeptide chain of MMTase as a search model. The molecular-replacement solution (three independent YMTase molecules) was subjected to rigid-body refinement and partially refined (R = 37.8%, R free = 42.1% at 2.7 Å resolution) using REFMAC5 (Winn et al., 2001 ▶). In this case also, a prominent electron-density difference peak suggested the location of an active-site-bound SAM molecule.
Inspection of the preliminary MMTase and YMTase models, as well as consideration of their aligned sequences within the MTase family, indicates that residues Lys62, Asp147, Lys184 and Glu220 (MMTase numbering) specifically characterize the active-site region in both enzymes. Moreover, SAM appears to be held in MMTase and YMTase via a network of hydrogen bonds and van der Waals contacts reminiscent of the described consensus interactions found in DVMTase. Such structural properties suggest that MMTase and YMTase can both be ascribed to the 2′OMTase subgroup.
Acknowledgments
This work was supported by the EU IP Project Vizier (CT 2004-511960). Part of the activity was supported by the Italian Ministry for University and Scientific Research FIRB grant ‘Biologia Strutturale’. MB is grateful to CIMAINA (University of Milano) and to Fondazione CARIPLO (Milano, Italy) for continuous support.
References
- Ahola, T. & Kaariainen, L. (1995). Proc. Natl Acad. Sci. USA, 92, 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers, T. J., Hans, C. S., Galler, R. & Rice, C. M. (1990). Annu. Rev. Microbiol.44, 649–688. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Egloff, M. P., Benarroch, D., Selisko, B., Romette, J. L. & Canard, B. (2002). EMBO J.21, 2757–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre-D’Amare, A. R. & Burley, S. K. (1994). Structure, 25, 357–359. [DOI] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Parker, R. & Song, H. (2004). Nature Struct. Biol.11, 121–127. [DOI] [PubMed]
- Shuman, S. (2001). Prog. Nucleic Acid Res. Mol. Biol.66, 1–40. [DOI] [PubMed] [Google Scholar]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025. [Google Scholar]
- Wengler, G. (1993). Virology, 197, 265–273. [DOI] [PubMed] [Google Scholar]
- Winn, M. D., Isupov, M. N. & Murshudov, G. N. (2001). Acta Cryst. D57, 122–133. [DOI] [PubMed] [Google Scholar]
- Zulauf, M. & D’Arcy, A. (1992). J. Cryst. Growth, 122, 102–106.