

Recombinant oligopeptidase B from T. brucei has been prepared and crystallized. Data were collected to 2.7 Å. Heavy-atom soaks and preparation of selenomethionine-substituted protein are in progress for structure determination by MAD or MIR.
Keywords: oligopeptidase B, Trypanosoma brucei, African trypanosomiasis, sleeping sickness
Abstract
African sleeping sickness, also called trypanosomiasis, is a significant cause of morbidity and mortality in sub-Saharan Africa. Peptidases from Trypanosoma brucei, the causative agent, include the serine peptidase oligopeptidase B, a documented virulence factor and therapeutic target. Determination of the three-dimensional structure of oligopeptidase B is desirable to facilitate the development of novel inhibitors. Oligopeptidase B was overexpressed in Escherichia coli as an N-terminally hexahistidine-tagged fusion protein, purified using metal-affinity chromatography and crystallized using the hanging-drop vapour-diffusion technique in 7%(w/v) polyethylene glycol 6000, 1 M LiCl, 0.1 M bis-tris propane pH 7.5. Diffraction data to 2.7 Å resolution were collected using synchrotron radiation. The crystals belong to space group P3121 or P3221, with unit-cell parameters a = b = 124.5, c = 249.9 Å. A complete data set to 2.7 Å was collected using synchrotron radiation.
1. Introduction
Oligopeptidase B (OPB) is a member of the prolyl oligopeptidase (POP) family of serine peptidases (Rawlings et al., 2006 ▶). OPB is present in Gram-negative bacteria, spirochetes and unicellular eukaryotes such as trypanosomes, but is not found in higher eukaryotes, with the exception of plants (Tsuji et al., 2004 ▶). Although the physiological role of OPB is unknown and its physiological substrates have not been identified, it has a documented role in the pathogenesis of South American and African trypanosomiasis. In Trypanosoma cruzi, the causative agent of Chagas’ disease (South American trypanosomiasis), OPB facilitates host-cell invasion (Burleigh et al., 1997 ▶; Caler et al., 1998 ▶) by generating a calcium signalling ligand (Burleigh & Andrews, 1995 ▶; Burleigh et al., 1997 ▶). OPB null-parasites are impaired with respect to their ability to generate calcium signals and infect mice or cultured mammalian cells (Caler et al., 1998 ▶). OPB appears to be directly involved in the virulence of the related T. brucei, the causative agent of African sleeping sickness. During infection, T. brucei OPB is released into the host bloodstream, where it is free to cleave regulatory peptides present in the host serum (Troeberg et al., 1996 ▶; Morty et al., 2001 ▶). Indeed, perturbed endocrine rhythms (Brandenberger et al., 1996 ▶) and the unusual cleavage of peptide hormones in the blood of T. brucei-infected rats (Tetaert et al., 1993 ▶) indicate a role for OPB in the disruption of host hormone metabolism during trypanosome infection. Diminished levels of regulatory peptides such as atrial natriuretic factor in hosts infected with T. brucei (Troeberg et al., 1996 ▶; Ndung’u et al., 1992 ▶) or Trypanosoma evansi (Morty, Pelle et al., 2005 ▶) provides further evidence for OPB involvement. The activity of T. brucei OPB is enhanced by reducing agents and diminished by thiol-blocking reagents (Morty et al., 1999 ▶; Morty, Shih et al., 2005 ▶), suggesting these agents may act as in vivo regulators of OPB activity.
Enzymes of the prolyl oligopeptidase family display distinct substrate specificities (Polgár, 2002 ▶; Rea & Fülöp, 2006 ▶). As expected from the family name, some recognize and cleave adjacent to proline residues. In contrast, OPB catalyzes the hydrolysis of oligopeptides exclusively at the carboxyl side of arginine or lysine residues. There is a strong preference for cleavage after pairs of basic residues, which represent a marker for processing enzymes in eukaryotes (Polgár, 1997 ▶). OPB also exhibits a specific carboxypeptidase activity (Hemerly et al., 2003 ▶). Prolyl oligopeptidase was the first enzyme from this family to be structurally characterized (Fülöp et al., 1998 ▶). The active site was found to lie within the cavity of the enzyme, buried between the catalytic α/β-hydrolase domain and the regulatory β-propeller domain. This provided an explanation for the observed oligopeptidase activity, whereby small peptides of up to 30 residues can access the active site whilst proteins and larger peptides are excluded. The crystal structures of dipeptidyl peptidase IV (Rasmussen et al., 2003 ▶) and acylaminoacyl peptidase (Bartlam et al., 2004 ▶) revealed a similar two-domain α/β-hydrolase fold and β-propeller topology, although the number of propeller blades can vary. A similar overall structure is anticipated for OPB. A three-dimensional model of Escherichia coli OPB has been built based on the crystal structure of porcine POP (Gérczei et al., 2000 ▶). The model predicted an S2 binding site comprised of Asp460 and Asp462, which interact with the positively charged side chain of the preferred P2 Arg or Lys. However, the S1 binding site was placed in an unlikely non-ionic environment, which is inconsistent with the preferred basic P1 residue. In a mutagenesis study carried out on Salmonella enterica OPB, nine acidic residues that are conserved in OPB but absent in POP were converted to their corresponding POP residues (Morty et al., 2002 ▶). In agreement with the E. coli OPB model, Asp460 and Asp462 were implicated in binding the P2 residue. A pair of Glu residues (Glu576 and Glu578) apparently interact with the P1 residue. Glu576 occupies the same position as POP residue Trp595 in a sequence alignment, which is known to interact with and provide specificity for a proline in the P1 position (Fülöp et al., 1998 ▶). OPB activity is particularly sensitive to ionic strength, which is consistent with electrostatic interactions between enzyme carboxylate groups and positively charged substrate residues (Polgár, 1997 ▶). Eliminating the positive charge of the P1 arginine using a structurally similar citrulline derivative is accompanied by a large decrease in activity, illustrating the importance of the positive charge to catalysis (Polgár, 1999 ▶). A kinetic deuterium isotope effect is not observed and catalysis is accompanied by a positive activation entropy (Polgár, 1999 ▶). This suggests that like POP, OPB catalysis involves rate-limiting conformational changes and the release of ordered water molecules upon substrate binding (Polgár, 1999 ▶).
OPB inhibitors exhibit trypanocidal activity, making them important potential chemotherapeutic targets (Morty et al., 1998 ▶). Whilst homology modelling of OPB based on the POP crystal structure is useful, the crystal structure of T. brucei OPB (81 kDa, 715 amino acids) would be more likely to explain the structure–function relationships that would benefit structure-based inhibitor design. The active form of this enzyme has previously been shown to be monomeric (Morty, Shih et al., 2005 ▶). Moraxella lacunata OPB has been crystallized, but the structure has not yet been published (Yoshimoto et al., 1995 ▶).
2. Materials and methods
2.1. Overexpression and purification
A pET19b expression construct containing the T. brucei OPB gene (GeneID 3665223; Morty et al., 1999 ▶) was used to transform E. coli BL21 (λDE3) that also contained the pRARE plasmid for supplementation of rare tRNAs. This construct expresses the enzyme preceded by the N-terminal histidine tag sequence MGHHHHHHHHHHSSGHIDDDDKHMLEDP. Bacteria were grown at 310 K in 2 l LB medium containing 100 mg ml−1 ampicillin and 35 mg ml−1 chloramphenicol. Protein expression was induced by the addition of IPTG to 1 mM and growth continued at 293 K for 18 h. The cells were harvested by centrifugation at 6000g for 15 min and resuspended in 50 mM HEPES pH 8.0, 0.5 M NaCl, 25 mM imidazole (buffer A) prior to sonication and clarification of the extract by centrifugation at 20 000g for 30 min. The cell extract was applied to a nickel-Sepharose column equilibrated in buffer A. The column was washed with buffer A, followed by buffer B (50 mM HEPES pH 8.0, 0.5 M NaCl, 100 mM imidazole), after which the enzyme was eluted with buffer C (50 mM HEPES pH 8.0, 0.5 M NaCl, 500 mM imidazole). Fractions containing enzyme were combined, concentrated in a 20 ml Centricon 10 000 molecular-weight cutoff centrifugal concentrator, applied onto a 5 ml Hi-trap desalting column equilibrated with 20 mM Tris–HCl pH 8.5 and eluted in this buffer. OPB eluting from this column appeared to be greater than 99% pure by SDS–PAGE and was concentrated to 12 mg ml−1 prior to crystallization.
2.2. Crystallization
Initial crystallization trials were carried out at the Oxford Protein Production Facility using the sitting-drop vapour-diffusion method with 96-well plates (Greiner Bio-One Ltd, UK) containing 0.05 ml reservoir solutions from the available block screens (Walter et al., 2005 ▶). Specifically, these were Hampton Research Crystal Screens I and II, Wizard Screens I and II, Hampton Research PEG/Ion screen, Hampton PEG 6000 and Ammonium Sulfate Grid Screens, Hampton Natrix and Cryo Screens, Hampton PEG/Li, NaCl, MPD and ‘Quik Screen’ Phosphate Grid screen and the PACT HT96 screen from Molecular Dimensions. Drops consisted of 0.1 µl protein and 0.1 µl reservoir solution. Crystals appeared after 3 d in condition B6 of the Hampton PEG/LiCl grid screen [10%(w/v) PEG 6000, 1 M LiCl, 0.1 M Bicine pH 9.0; Fig. 1 ▶]. This condition was optimized using the hanging-drop vapour-diffusion method using 24-well Linbro plates containing 0.2 ml reservoir solution. Drops consisted of 1 µl protein and 1 µl reservoir solution. Crystals deemed suitable for diffraction studies were grown in a solution containing 7%(w/v) PEG 6000, 1 M LiCl, 0.1 M bis-tris propane pH 7.5.
Figure 1.

Photograph of T. brucei OPB crystals obtained in 0.1 µl sitting drops containing 10%(w/v) polyethylene glycol 6000, 1.0 M LiCl, 0.1 M Bicine pH 9.0. The crystals are 0.2 mm in the longest dimension.
3. X-ray diffraction analysis
Crystals were picked up from the crystallization drop using a nylon loop and transferred into a solution containing 7%(w/v) PEG 6000, 1 M LiCl, 0.1 M bis-tris propane pH 7.5 and 30% glycerol for a few seconds for cryoprotection. Crystals were then placed in a cryostream, cooled to 100 K and stored in liquid nitrogen until needed for data collection. A complete data set was collected to a resolution of 2.7 Å on the microfocus beamline ID13 at the European Synchrotron Radiation Facility using a MAR CCD detector with an oscillation angle of 1° and a crystal-to-detector distance of 141 mm. Owing to radiation damage, the beam was focused on a fresh part of the crystal after every 15° of data. A typical diffraction image is shown in Fig. 2 ▶. All data were indexed, integrated and scaled using the HKL suite of programs (Otwinowski & Minor, 1997 ▶). The crystals belong to the trigonal space group P3121 or P3221, with unit-cell parameters a = b = 124.5, c = 249.9 Å. The Matthews probability calculation suggests the presence of three molecules in the asymmetric unit, with a V M value of 2.33 Å3 Da−1 and a solvent content of 47%. Data-collection and processing statistics for the highly redundant (10.4-fold) data set are shown in Table 1 ▶. A self-rotation function did not reveal the presence of a noncrystallographic rotational axis. T. brucei OPB shares only 25% sequence identity with porcine prolyl oligopeptidase, which resulted in the failure of molecular replacement using the prolyl oligopeptidase coordinates (Fülöp et al., 1998 ▶) as a search model. Heavy-atom soaks and preparation of selenomethionine-substituted protein are in progress for structure determination using MAD or MIR.
Figure 2.

A typical diffraction image of a T. brucei OPB crystal collected on beamline ID13 at the ESRF using a MAR CCD detector. The resolution at the edge is 2.3 Å.
Table 1. Data-collection and processing statistics.
Values in parentheses are for the highest resolution shell.
| Synchrotron-radiation source | ESRF ID13 |
| Detector | MAR CCD |
| Wavelength (Å) | 0.976 |
| Space group | P3121 or P3221 |
| Unit-cell parameters | |
| a = b (Å) | 124.1 |
| c (Å) | 249.3 |
| Molecules per ASU | 3 |
| Matthews coefficient (Å3 Da−1) | 2.88 |
| Solvent convent (%) | 57 |
| Resolution range (Å) | 50–2.7 (2.8–2.7) |
| Total observations | 647583 |
| Unique reflections | 62450 |
| Average I/σ(I) | 9.6 (1.6) |
| Rsym† (%) | 0.188 (0.959) |
| Completeness (%) | 99.8 (100.0) |
R
sym = 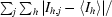
 , where I
h,j is the jth observation of reflection h and 〈I
h〉 is the mean intensity of that reflection.
, where I
h,j is the jth observation of reflection h and 〈I
h〉 is the mean intensity of that reflection.
Acknowledgments
We are grateful for access and user support at the synchrotron facilities of ESRF, Grenoble. DR and CH thank the BBSRC for the award of a studentship.
References
- Bartlam, M., Wang, G., Yang, H., Gao, R., Zhao, X., Xie, G., Cao, S., Feng, Y. & Rao, Z. (2004). Structure, 12, 1481–1488. [DOI] [PubMed] [Google Scholar]
- Brandenberger, G., Buguet, A., Spiegel, K., Stanghellini, A., Muanga, G., Bogui, P. & Dumas, M. (1996). J. Biol. Rhythms, 11, 258–267. [DOI] [PubMed] [Google Scholar]
- Burleigh, B. A. & Andrews, N. W. (1995). J. Biol. Chem.270, 5172–5180. [DOI] [PubMed] [Google Scholar]
- Burleigh, B. A., Caler, E. V., Webster, P. & Andrews, N. W. (1997). J. Cell Biol.136, 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caler, E. V., Vaena, S., Haynes, P. A., Andrews, N. W. & Burleigh, B. A. (1998). EMBO J.17, 4975–4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fülöp, V., Böcskei, Z. & Polgár, L. (1998). Cell, 94, 161–70. [DOI] [PubMed] [Google Scholar]
- Gérczei, T., Keserü, G. M. & Náray-Szabó, G. (2000). J. Mol. Graph. Model.18, 7–17. [DOI] [PubMed] [Google Scholar]
- Hemerly, J. P., Oliveira, V., Del Nery, E., Morty, R. E., Andrews, N. W., Juliano, M. A. & Juliano, L. (2003). Biochem. J.373, 933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morty, R. E., Fülöp, V. & Andrews, N. W. (2002). J. Bacteriol.184, 3329–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morty, R. E., Lonsdale-Eccles, J. D., Mentele, R., Auerswald, E. A. & Coetzer, T. H. (2001). Infect. Immun.69, 2757–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morty, R. E., Lonsdale-Eccles, J. D., Morehead, J., Caler, E. V., Mentele, R., Auerswald, E. A., Coetzer, T. H., Andrews, N. W. & Burleigh, B. A. (1999). J. Biol. Chem.274, 26149–26156. [DOI] [PubMed] [Google Scholar]
- Morty, R. E., Pelle, R., Vadasz, I., Uzcanga, G. L., Seeger, W. & Bubis, J. (2005). J. Biol. Chem.280, 10925–10937. [DOI] [PubMed] [Google Scholar]
- Morty, R. E., Shih, A. Y., Fülöp, V. & Andrews, N. W. (2005). FEBS Lett.579, 2191–2196. [DOI] [PubMed] [Google Scholar]
- Morty, R. E., Troeberg, L., Pike, R. N., Jones, R., Nickel, P., Lonsdale-Eccles, J. D. & Coetzer, T. H. T. (1998). FEBS Lett.433, 251–256. [DOI] [PubMed] [Google Scholar]
- Ndung’u, J. M., Wright, N. G., Jennings, F. W. & Murray, M. (1992). Parasitol. Res.78, 553–556. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods. Enzymol.276, 307–326. [DOI] [PubMed]
- Polgár, L. (1997). Proteins, 28, 375–379. [PubMed] [Google Scholar]
- Polgár, L. (1999). Biochemistry, 38, 15548–15555. [DOI] [PubMed] [Google Scholar]
- Polgár, L. (2002). Cell. Mol. Life Sci.59, 349–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen, H. B., Branner, S., Wiberg, F. C. & Wagtmann, N. (2003). Nature Struct. Biol.10, 19–25. [DOI] [PubMed] [Google Scholar]
- Rawlings, N. D., Morton, F. R. & Barrett, A. J. (2006). Nucleic Acids Res.34, 270–272. [DOI] [PMC free article] [PubMed]
- Rea, D. & Fülöp, V. (2006). Cell Biochem. Biophys.44, 349–365. [DOI] [PubMed] [Google Scholar]
- Tetaert, D., Soudan, B., Huet-Duvillier, G., Degand, P. & Boersma, A. (1993). Int. J. Pept. Protein Res.41, 147–152. [DOI] [PubMed] [Google Scholar]
- Troeberg, L., Pike, R. N., Morty, R. E., Berry, R. K., Coetzer, T. H. T. & Lonsdale-Eccles, J. D. (1996). Eur. J. Biochem.238, 728–736. [DOI] [PubMed] [Google Scholar]
- Tsuji, A., Yuasa, K. & Matsuda, Y. (2004). J. Biochem. (Tokyo), 136, 673–681. [DOI] [PubMed] [Google Scholar]
- Walter, T. S., Diprose, J. M., Mayo, C. J., Siebold, C., Pickford, M. G., Carter, L., Sutton, G. C., Berrow, N. S., Brown, J., Berry, I. M., Stewart-Jones, G. B., Grimes, J. M., Stammers, D. K., Esnouf, R. M., Jones, E. Y., Owens, R. J., Stuart, D. I. & Harlos, K. (2005). Acta Cryst. D61, 651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto, T., Tabira, J., Kabashima, T., Inoue, S. & Ito, K. (1995). J. Biochem. (Tokyo), 117, 654–660. [DOI] [PubMed] [Google Scholar]