

The crystal structure of S. aureus cytidine monophosphate kinase in complex with cytidine 5′-monophosphate has been determined.
Keywords: cytidine monophosphate kinase, Staphylococcus aureus, MRSA, cytidine 5′-monophosphate
Abstract
The crystal structure of Staphylococcus aureus cytidine monophosphate kinase (CMK) in complex with cytidine 5′-monophosphate (CMP) has been determined at 2.3 Å resolution. The active site reveals novel features when compared with two orthologues of known structure. Compared with the Streptococcus pneumoniae CMK solution structure of the enzyme alone, S. aureus CMK adopts a more closed conformation, with the NMP-binding domain rotating by ∼16° towards the central pocket of the molecule, thereby assembling the active site. Comparing Escherichia coli and S. aureus CMK–CMP complex structures reveals differences within the active site, including a previously unreported indirect interaction of CMP with Asp33, the replacement of a serine residue involved in the binding of CDP by Ala12 in S. aureus CMK and an additional sulfate ion in the E. coli CMK active site. The detailed understanding of the stereochemistry of CMP binding to CMK will assist in the design of novel inhibitors of the enzyme. Inhibitors are required to treat the widespread hospital infection methicillin-resistant S. aureus (MRSA), currently a major public health concern.
1. Introduction
Staphylococcus aureus has long been recognized as a major cause of hospital-acquired infection. It has become a considerable public health threat as a result of the increased incidence of drug resistance in this organism (Boyce et al., 2005 ▶). In 1947, S. aureus was the first bacterium in which penicillin resistance was found. Methicillin then became the antibiotic of choice, but over the past decade methicillin-resistant S. aureus has become endemic in hospitals worldwide (Boyce et al., 2005 ▶). This drug resistance has left antibiotics such as vancomycin as one of the few effective agents to treat serious MRSA infections. However, vancomycin-resistant S. aureus (VRSA) has now been identified: the first example was isolated in Japan in 1997 (Hiramatsu et al., 1997 ▶). It is clear that new drug therapies are required to treat S. aureus related infections.
Cytidine monophosphate kinase is a member of the nucleoside monophosphate kinase (NMK) family (Leipe et al., 2003 ▶). CMK has been suggested to be essential for the growth of two Gram-positive bacteria: Bacillus subtilis (Sorokin et al., 1995 ▶) and Streptococcus pneumoniae (Yu et al., 2003 ▶). The enzyme plays a crucial role in the biosynthesis of nucleotide precursors. Bacterial CMKs catalyze phosphoryl transfer from ATP specifically to CMP or dCMP. In eukaryotes, UMP/CMP kinase catalyzes the conversion of UMP and CMP to their diphosphate forms (Segura-Pena et al., 2004 ▶). The bacterial CMKs differ from other NMP kinases by possessing a 40-residue insert in the NMP-binding domains and short LID domains (Briozzo et al., 1998 ▶). These differences could be exploited in the development of safe novel antibacterials targeted specifically towards S. aureus CMK and/or closely related bacterial orthologues such as S. pneumoniae CMK (Yu et al., 2003 ▶) and B. subtilis CMK (Sorokin et al., 1995 ▶).
Here, we report the crystal structure of S. aureus CMK (SaCMK) in complex with CMP at a resolution of 2.3 Å. The SaCMK model reveals novel features of the active site when compared with two other orthologues of known structure. Comparison with the S. pneumoniae CMK (SpCMK) solution structure (Yu et al., 2003 ▶) shows that SaCMK adopts a more closed conformation with the NMP-binding domain rotating by ∼16° towards the central cavity of the molecule, thus assembling the active site. When comparing the SaCMK–CMP structure with Escherichia coli CMK (EcCMK) also in complex with CMP (Bertrand et al., 2002 ▶), we observe a previously unreported indirect interaction of Asp33 with CMP. There is an additional sulfate ion within the EcCMK substrate-binding site and Ala12 in SaCMK replaces a serine residue (14) in the corresponding position in EcCMK. This serine is known to play an important role in the binding of CDP (Briozzo et al., 1998 ▶).
2. Experimental methods
2.1. Preparation and purification
The S. aureus CMK-encoding sequence was PCR-amplified and subcloned into the E. coli expression vector pET15b to yield the recombinant plasmid pMUT74. The plasmid was sequenced to ensure that no spurious mutations had been introduced during the PCR reaction. The plasmid pMUT53 was transformed into the E. coli expression strain Codon+ and transformants were selected on Luria agar supplemented with 100 µg ml−1 ampicillin and 35 µg ml−1 chloramphenicol. Cells transformed with pMUT74 were grown in an orbital incubator at 310 K in 500 ml volumes of Luria broth supplemented with 100 µg ml−1 ampicillin and 35 µg ml−1 chloramphenicol until an attenuance at 550 nm of 0.6. At this point, IPTG was added to a final concentration of 0.2 mg ml−1 and the cultures were incubated for a further 5 h at 310 K. Cells were then harvested by centrifugation and stored at 255 K until required.
The cells were sonicated in 50 mM potassium phosphate pH 7.2, 1 mM DTT, 1 mM benzamidine whilst stirring on an ice–water mixture. All subsequent steps were carried out at 277 K and 10 ml column fractions were collected. Following centrifugation, the cell-free supernatant was chromatographed on a 100 ml IMAC column charged to one-third capacity with Zn2+, equilibrated with 50 mM potassium phosphate pH 7.2, 1 mM DTT and then eluted with a 0.0–0.3 M imidazole gradient in 50 mM potassium phosphate pH 7.2, 1 mM DTT. Fractions containing the N-terminal His-tagged SaCMK were identified by SDS–PAGE, pooled appropriately and dialysed against 50 mM potassium phosphate pH 7.2, 1 mM DTT. After dialysis, the pool was filtered through a 0.45 µm filter and loaded onto a hydroxyapatite column equilibrated with 50 mM potassium phosphate pH 7.2, 1 mM DTT; the SaCMK eluted in the column flowthrough. Fractions containing SaCMK were again identified by SDS–PAGE and pooled appropriately. This procedure yielded approximately 480 mg of purified SaCMK from 25 g of cell paste.
2.2. Crystallization
Prior to crystallization, SaCMK was concentrated and buffer-exchanged for 20 mM Tris–HCl pH 7.4, 40 mM KCl and 0.1% sodium azide using Vivascience Vivaspin centrifugal concentrators. Pooled concentrates were then filtered through Amersham NAP 25 desalting columns and re-concentrated to 40 mg ml−1.
SaCMK was initially screened in crystallization trials with a total of ∼1440 droplets [with standard conditions consisting of Hampton, Wizard and Emerald kits for two different ligand complexes; one with adenosine 5′-(γ-thio)triphosphate (ATPγS) and CMP and the other with CMP alone]. The crystallization system developed at the Oxford Protein Production Facility was used for the initial screening phase (Walter et al., 2005 ▶). A Cartesian Technologies pipetting robot was used to set up nano-crystallizations consisting of 100 nl protein solution and 100 nl reservoir solution mixed as sitting drops in 96-well Greiner plates. Crystallization plates were placed in a TAP Homebase storage vault maintained at 295 K and imaged via a Veeco visualization system (Walter et al., 2005 ▶). Larger scale droplets were set up to optimize initial hits. The best diffracting crystals grew when a protein complex consisting of 15 mg ml−1 SaCMK, 2.5 mM CMP and 2.5 mM ATPγS was used with 1.6 M ammonium sulfate, 0.1 M HEPES pH 7.8, 2% PEG 200 as the precipitant. Crystals grew to a maximal length of 0.3–0.8 mm within 6 d (Fig. 1 ▶).
Figure 1.

A crystal of S. aureus CMK grown in a sitting drop by vapour diffusion. The crystal is approximately 0.2 mm in length. The crystals belong to the hexagonal space group P64.
2.3. Data collection and structure determination
Crystals of SaCMK were flash-cooled to 100 K (using 100% tacsimate as a cryoprotectant) and diffraction data were collected at beamline ID14EH1, ESRF (Grenoble, France). Indexing, integration and merging of data were carried out with the HKL (v.1.96.1) suite of programs (Otwinowski & Minor, 1997 ▶). Data-collection and refinement statistics are shown in Table 1 ▶.
Table 1. Statistics for crystallographic structure determination.
Values in parentheses are for the outer resolution shell.
| Beamline | ID14EH1, ESRF |
| Wavelength (Å) | 0.9340 |
| Space group | P64 |
| Unit-cell parameters (Å) | a = b = 157.89, c = 76.23 |
| Resolution range (Å) | 30.0–2.3 (2.38–2.30) |
| Observations | 667447 (33099) |
| Unique reflections | 48287 (4810) |
| Average redundancy | 13.8 (6.9) |
| Completeness (%) | 100.0 (100.0) |
| I/σ(I) | 22.7 (3.4) |
| Rmerge† | 0.094 (0.597) |
| Refinement statistics | |
| Resolution range (Å) | 30.0–2.3 |
| Total No. of reflections | 48287 |
| No. of working reflections | 45687 |
| No. of test reflections | 2420 |
| R factor‡ | 0.211 |
| Rworking | 0.214 |
| Rfree | 0.261 |
| No. of atoms | |
| Protein | 5091 |
| Water | 334 |
| Others | 90 |
| R.m.s. bond-length deviation (Å) | 0.0083 |
| R.m.s. bond-angle deviation (°) | 1.27 |
| Mean B factor (Å2) | |
| Main chain | 40 |
| Side chain | 48 |
| Waters | 43 |
| Ligands§ | 38 |
| R.m.s. backbone B-factor deviation¶ | 4.6 |
R
merge = 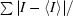
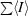 .
.
R factor = 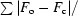
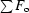 .
.
CMP, SO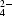 and PEG.
and PEG.
R.m.s. deviation between B factors for bonded main-chain atoms.
A molecular-replacement solution was found with the CNS suite of programs (Brünger et al., 1998 ▶) using the modified PDB entry 1kdo (Bertrand et al., 2002 ▶), i.e. E. coli CMK in complex with CMP, as a search model. The NMP-binding, LID and CORE domains (Briozzo et al., 1998 ▶) were treated as separate ‘rigid bodies’ during molecular replacement. The initial model underwent two rounds of rigid-body refinement followed by simulated annealing. Iterative cycles of conjugate-gradient minimization with subsequent individual B-factor refinement in CNS and manual model rebuilding with O (Jones et al., 1991 ▶) were then run.
Structural superpositions of the CMK orthologues were performed with SHP (Stuart et al., 1979 ▶).
3. Results and discussion
3.1. Overall and domain structure
There are three SaCMK molecules within the asymmetric unit, containing residues 3–218 of the protein. When compared with molecule A, both molecules B and C have a root-mean-square deviation (r.m.s.d.) of just 0.1 Å for the superposition of 216 pairs of Cα atoms. Therefore, the three protein molecules are essentially identical in structure. The N-terminal methionine and lysine residues are not observed and the C-terminal lysine appears to be disordered. Electron-density maps show unambiguous F
o − F
c density for CMP in all three molecules of the asymmetric unit; these were added to the model during the later stages of refinement (Fig. 2 ▶
a). In addition, the asymmetric unit contains three SO ions, one PEG molecule and 334 water molecules. The SaCMK model was evaluated using PROCHECK (Laskowski et al., 1993 ▶), which indicated that 90% of the final model’s residues lie within the most favoured regions of a Ramachandran plot and 10% within the additionally allowed regions (Ramachandran et al., 1963 ▶).
Figure 2.

(a) Stereo diagram showing the F o − F c annealed electron-density map with the CMP molecule omitted, contoured at 5σ. The figure was prepared using CNS (Brünger et al., 1998 ▶), BOBSCRIPT (Esnouf, 1999 ▶) and RASTER3D (Merritt & Bacon, 1997 ▶). (b) Atom nomenclature for CMP. The figure, showing a stick representation of CMP, was prepared using PyMOL (DeLano, 2002 ▶).
The overall structure of the SaCMK–CMP complex is illustrated in Fig. 3 ▶. The secondary-structure elements of SaCMK include nine α-helices and eight β-strands. Until recently, all members of the NMP kinase family for which three-dimensional structures were known consisted of three domains (Yan & Tsai, 1999 ▶). This is no longer true as the recently published structures of UMP kinases from E. coli (Briozzo et al., 2005 ▶) and Pyrococcus furiosus (Marco-Marin et al., 2005 ▶) are not comprised of three domains. However, the three classical domains of NMP kinases are found in SaCMK. The parallel β-sheet consisting of strands β1–β2 and β6–β8 flanked by α-helices α1, α6 and α9 make up the CORE domain that contains a highly conserved phosphate-binding loop (P-loop; residues 10–16). The small LID domain, containing residues 155–168 (i.e. including the C-terminal and N-terminal halves of α7 and α8, respectively), closes over the phosphate donor and bears functionally essential residues, whilst the NMP-binding domain (residues 33–100) comprises α-helices α2–α5, the antiparallel β-sheet β3–β5 and the intervening loop regions. This latter domain is responsible for the recognition and binding of the CMP or dCMP substrate (Bertrand et al., 2002 ▶). The three domains pack together to form a central pocket for substrate-binding and enzyme catalysis. In order to prevent unwanted ATP hydrolysis in the absence of substrate, the kinases disassemble their active site. On the binding of substrate, an induced-fit movement occurs, repositioning the catalytically important residues (Schulz et al., 1990 ▶; Krell et al., 2001 ▶; Bertrand et al., 2002 ▶).
Figure 3.

A cartoon representation of the overall structure of the SaCMK–CMP complex. The protein structure is coloured according to secondary-structure element: α-helices are coloured red, β-strands are yellow and loop regions are green. The figure was prepared using PyMOL.
Compared with other NMP kinases, CMKs have a large insert in the NMP-binding domain (Briozzo et al., 1998 ▶). In SaCMK, the NMP-binding insert is from Leu57 to Tyr97. The insert contains α-helices α3–α5 and the antiparallel β-sheet β3–β5. The NMP-binding insert is thought to play a role in dCMP phosphorylation (Bertrand et al., 2002 ▶).
3.2. The SaCMK NMP-binding site
The cytosine base of CMP binds near the ends of β2 and β6, with the α-phosphate group pointing towards the P-loop located between β1 and α1. The P-loop has a typical mononucleotide-binding motif, Gly(10)-X-X-Ala-X-Gly-Lys(16). This loop forms a large anion hole in which a sulfate ion is bound. This sulfate ion is observed in a similar position in several other NMP kinase structures, such as E. coli CMK (Briozzo et al., 1998 ▶; Bertrand et al., 2002 ▶), yeast GMK (Stehle & Schulz, 1992 ▶) and bovine AMK (Diederichs & Schulz, 1991 ▶). The position of the SO ion corresponds to the β-phosphate group of ATP (Diederichs & Schulz, 1991 ▶).
The NMP-binding pocket is characterized by a generally hydrophobic surface together with a number of charged residues projecting into the pocket. The lipophilic part of the the cavity is formed by the highly conserved CMK residues Gly35 and Gly125 [identified using BLAST (Altschul et al., 1990 ▶), results not shown] and the semi-conserved residues Val95 and Ala99, which in some orthologues are replaced by an alanine and a serine, respectively. Ser96, part of the NMP-binding insert, forms a hydrogen bond with Asp180 and is thought to play a critical role in the phosphorylation of dCMP (Bertrand et al., 2002 ▶).
The CMP molecule makes a number of key interactions with several highly conserved residues projecting into the NMP-binding pocket, namely Asp33, Tyr38, Arg39, Arg105, Arg126, Asp127, Arg176, Asp180 and Arg183 and the semi-conserved Thr34 (Fig. 4 ▶). These interactions include (i) Arg39 and Arg126 hydrogen bonding with the α-phosphate of the CMP, (ii) Arg105 interacting with both the cytosine moiety and α-phosphate, (iii) the aromatic ring of Tyr38 stacking against the cytosine ring, (iv) Thr34, Asp127 and Arg183 hydrogen bonding to the cytosine, (v) Arg176 and Asp180 hydrogen bonding with the hydroxyl groups of the ribose ring and (vi) the previously unreported indirect interaction of Asp33 with the α-phosphate of CMP by forming 2.8 and 3.4 Å hydrogen bonds with the ordered water molecules W4 and W2 through its OD1 and OD2 hydroxyl groups, respectively. A detailed list of contacts made between the CMP molecule and SaCMK can be found in Table 2 ▶.
Figure 4.

Stereo diagram showing the superposition of some of the CMP-interacting residues and ordered water molecules of SaCMK–CMP (coloured by element) and E. coli CMK–CMP (blue) complexes. Residue numbers in parentheses are the corresponding residues in the E. coli orthologue. The hydrogen-bonding network is represented as grey dashed lines. A full list of the interactions made between SaCMK and CMP can be found in Table 2 ▶. The figure was prepared using BOBSCRIPT and RASTER3D.
Table 2. Interactions between SaCMK and CMP (molecule A).
The distances were determined using the CONTACT program (Collaborative Computational Project, Number 4, 1994 ▶) with 0–3.6 Å distance limits.
| CMP atom | SaCMK/water atom | Distance (Å) |
|---|---|---|
| C2 | Arg105 NH1 | 3.5 |
| Arg105 NH2 | 3.4 | |
| C4 | Thr34 OG1 | 3.4 |
| Tyr38 CE1 | 3.6 | |
| Arg126 N | 3.2 | |
| C5 | Thr34 OG1 | 3.2 |
| Wat1 O | 3.5 | |
| C6 | Tyr38 CD1 | 3.5 |
| C2* | Asp180 OD1 | 3.4 |
| Asp180 OD2 | 3.3 | |
| C3* | Asp180 OD2 | 3.4 |
| C5* | Arg176 NH2 | 3.5 |
| N1 | Tyr38 CD1 | 3.5 |
| N3 | Tyr38 CE1 | 3.4 |
| Arg105 NH2 | 3.0 | |
| Arg126 CB | 3.5 | |
| N4 | Thr34 OG1 | 2.9 |
| Gly125 C | 3.5 | |
| Gly125 CA | 3.1 | |
| Arg126 N | 2.9 | |
| Asp127 OD1 | 3.2 | |
| O2 | Arg105 CZ | 3.3 |
| Arg105 NH1 | 2.7 | |
| Arg105 NH2 | 3.1 | |
| Arg183 CZ | 3.2 | |
| Arg183 NH1 | 2.8 | |
| Arg183 NH2 | 2.8 | |
| O2* | Ser96 CA | 3.6 |
| Asp180 CG | 3.3 | |
| Asp180 OD1 | 2.7 | |
| Asp180 OD2 | 3.3 | |
| O3* | Arg176 NE | 3.4 |
| Arg176 NH2 | 3.0 | |
| Asp180 OD2 | 2.7 | |
| Wat3 O | 3.6 | |
| O1P | Arg126 NH1 | 3.3 |
| Arg126 NH2 | 3.3 | |
| Wat1 O | 3.3 | |
| Wat2 O | 2.8 | |
| O2P | Arg39 CZ | 3.5 |
| Arg39 NE | 2.8 | |
| Arg39 NH2 | 3.4 | |
| Wat4 O | 2.7 | |
| O3P | Arg39 NH2 | 3.1 |
| Arg126 NH2 | 3.6 | |
| Arg176 NH2 | 2.5 | |
| Wat5 O | 2.8 |
All of the interactions described above serve to position and orientate the O3P hydroxyl of the CMP molecule ready for nucleophilic attack by the γ-phosphate of ATP.
3.3. Structural comparison of S. aureus CMK with two orthologues
The three-dimensional structures of two other bacterial CMKs have been determined. These are (i) the solution structure of S. pneumoniae CMK (Yu et al., 2003 ▶) and (ii) the crystal structure of unliganded E. coli CMK and in a complex with CDP (Briozzo et al., 1998 ▶), as well as in complex with CMP, dCMP, 2′,3′-dideoxy-CMP (ddCMP) and cytosine β-d-arabinofuranoside 5′-monophosphate (AraCMP) (Bertrand et al., 2002 ▶).
The S. aureus and S. pneumoniae CMKs share a sequence identity of 51% (Fig. 5 ▶ a). The structures superimpose with an r.m.s.d. of 1.9 Å for 141 pairs of Cα atoms. The SaCMK and SpCMK structures closely resemble one another. However, whereas the LID domains adopt similar conformations, the NMP-binding domain of the apo SpCMK adopts a more ‘open’ conformation compared with the corresponding domain in the SaCMK–CMP structure. Thus, the SaCMK NMP-binding domain, which comprises residues responsible for the recognition and binding of CMP, is rotated by ∼16° towards the central pocket of the molecule, thereby assembling the active site (Fig. 5 ▶ b). This movement is a consequence of the binding of CMP.
Figure 5.

(a) An alignment of the amino-acid sequence of SaCMK with those of CMK from E. coli (EcCMK) and S. pneumoniae (SpCMK), drawn with ESPRIPT (Gouet et al., 1999 ▶). Residues are boxed according to an alignment made with ClustalW (Thompson et al., 1994 ▶; SwissProt IDs P63807, Q97PK6 and P0A6I0 for SaCMK, SpCMK and EcCMK sequences, respectively). Residues that are strictly conserved have a red background, residues that are well conserved are indicated by red lettering and the remainder are in black. The secondary-structure elements of SaCMK are presented at the top: α-helices by squiggles and β-strands by arrows. The positions of the P-loop, the NMP-binding domain and the LID domain of SaCMK are indicated below the alignment. (b) A ribbon representation showing the superposition of the SaCMK–CMP (coloured red), EcCMK–CMP (blue) and apo SpCMK (green) complexes. The figure was prepared using PyMOL.
The CMKs from S. aureus and E. coli share a sequence identity of 38% (Fig. 5 ▶ a). Compared with both EcCMK and SpCMK, SaCMK has a two-residue deletion in β-strand β1 and a four-residue deletion in the loop region between α-helices α2 and α3. Also, SaCMK has a single aspartate residue insertion at position 70 compared with EcCMK. As expected, the SaCMK–CMP structure most closely resembles the EcCMK–CMP complex structure, with an r.m.s.d. of 1.4 Å for the superposition of 198 pairs of Cα atoms. The CORE, LID and NMP-binding domains of the E. coli and S. aureus CMK orthologues all adopt very similar conformations. Indeed, the architecture of the NMP-binding site is well conserved (Fig. 4 ▶). The significant differences between the SaCMK and EcCMK structures within the active-site cavity are as follows. (i) Thr34 is conservatively substituted for a serine residue in EcCMK. However, the same hydrogen-bonding interaction with the N4 amine group of the cytosine moiety of CMP is maintained by the threonine OG1 and serine OG hydroxyl groups of S. aureus and E. coli CMKs, respectively. (ii) There is an additional sulfate ion in the EcCMK substrate-binding site. (iii) Ala12 in SaCMK is replaced by a serine (Ser14) in EcCMK. Most orthologues have an alanine or glycine residue at this position, while some have a serine (identified using BLAST; results not shown). In the EcCMK–CDP crystal structure it was shown that the hydroxyl group of Ser14 forms a strong 2.6 Å hydrogen bond with the β-phosphate of CDP (Briozzo et al., 1998 ▶). This suggests that CDP may bind more tightly to EcCMK than SaCMK.
In conclusion, knowledge of the detailed stereochemistry of substrate binding to CMK is clearly important in the use of structure-based approaches to design novel inhibitors targeting the CMP-binding site. Design of such inhibitors is needed as part of efforts to develop new antibacterial drugs for treating MRSA infections, which currently cause many deaths in hospitalized patients and are thus a major public health concern.
Supplementary Material
PDB reference: CMK–CMP complex, 2h92, r2h92sf
Acknowledgments
We thank Dr K. Harlos for assistance with the use of the in-house X-ray facilities and Dr R. Esnouf and Ms J. Dong for computer support. We also thank the staff of the ESRF ID14 EH1 Beamline. Work at Oxford and Newcastle was funded by Arrow Therapeutics.
References
- Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990). J. Mol. Biol.215, 403–410. [DOI] [PubMed] [Google Scholar]
- Bertrand, T., Briozzo, P., Assairi, L., Ofiteru, A., Bucurenci, N., Munier-Lehmann, H., Golinelli-Pimpaneau, B., Barzu, O. & Gilles, A. M. (2002). J. Mol. Biol.315, 1099–1110. [DOI] [PubMed] [Google Scholar]
- Boyce, J. M., Cookson, B., Christiansen, K., Hori, S., Vuopio-Varkila, J., Kocagoz, S., Oztop, A. Y., Vandenbroucke-Grauls, C. M., Harbarth, S. & Pittet, D. (2005). Lancet Infect. Dis.5, 653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briozzo, P., Evrin, C., Meyer, P., Assairi, L., Joly, N., Barzu, O. & Gilles, A. M. (2005). J. Biol. Chem.280, 25533–25540. [DOI] [PubMed] [Google Scholar]
- Briozzo, P., Golinelli-Pimpaneau, B., Gilles, A. M., Gaucher, J. F., Burlacu-Miron, S., Sakamoto, H., Janin, J. & Barzu, O. (1998). Structure, 6, 1517–1527. [DOI] [PubMed] [Google Scholar]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- DeLano, W. L. (2002). The PyMOL Molecular Visualization System. http://www.pymol.org.
- Diederichs, K. & Schulz, G. E. (1991). J. Mol. Biol.217, 541–549. [DOI] [PubMed] [Google Scholar]
- Esnouf, R. M. (1999). Acta Cryst. D55, 938–940. [DOI] [PubMed] [Google Scholar]
- Gouet, P., Courcelle, E., Stuart, D. I. & Metoz, F. (1999). Bioinformatics, 15, 305–308. [DOI] [PubMed] [Google Scholar]
- Hiramatsu, K., Hanaki, H., Ino, T., Yabuta, K., Oguri, T. & Tenover, F. C. (1997). J. Antimicrob. Chemother.40, 135–136. [DOI] [PubMed] [Google Scholar]
- Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Krell, T., Maclean, J., Boam, D. J., Cooper, A., Resmini, M., Brocklehurst, K., Kelly, S. M., Price, N. C., Lapthorn, A. J. & Coggins, J. R. (2001). Protein Sci.10, 1137–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291. [Google Scholar]
- Leipe, D. D., Koonin, E. V. & Aravind, L. (2003). J. Mol. Biol.333, 781–815. [DOI] [PubMed] [Google Scholar]
- Marco-Marin, C., Gil-Ortiz, F. & Rubio, V. (2005). J. Mol. Biol.352, 438–454. [DOI] [PubMed] [Google Scholar]
- Merritt, E. A. & Bacon, D. J. (1997). Methods Enzymol.277, 505–524. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 207–326. [DOI] [PubMed]
- Ramachandran, G. N., Ramakrishnan, C. & Sasisekharan, V. (1963). J. Mol. Biol.7, 95–99. [DOI] [PubMed] [Google Scholar]
- Schulz, G. E., Muller, C. W. & Diederichs, K. (1990). J. Mol. Biol.213, 627–630. [DOI] [PubMed] [Google Scholar]
- Segura-Pena, D., Sekulic, N., Ort, S., Konrad, M. & Lavie, A. (2004). J. Biol. Chem.279, 33882–33889. [DOI] [PubMed] [Google Scholar]
- Sorokin, A., Serror, P., Pujic, P., Azevedo, V. & Ehrlich, S. D. (1995). Microbiology, 141, 311–319. [DOI] [PubMed] [Google Scholar]
- Stehle, T. & Schulz, G. E. (1992). J. Mol. Biol.224, 1127–1141. [DOI] [PubMed] [Google Scholar]
- Stuart, D. I., Levine, M., Muirhead, H. & Stammers, D. K. (1979). J. Mol. Biol.134, 109–142. [DOI] [PubMed] [Google Scholar]
- Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994). Nucleic Acids Res.22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, T. S., Diprose, J. M., Mayo, C. J., Siebold, C., Pickford, M. G., Carter, L., Sutton, G. C., Berrow, N. S., Brown, J., Berry, I. M., Stewart-Jones, G. B., Grimes, J. M., Stammers, D. K., Esnouf, R. M., Jones, E. Y., Owens, R. J., Stuart, D. I. & Harlos, K. (2005). Acta Cryst. D61, 651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, H. & Tsai, M. D. (1999). Adv. Enzymol. Relat. Areas Mol. Biol.73, 103–134. [DOI] [PubMed] [Google Scholar]
- Yu, L., Mack, J., Hajduk, P. J., Kakavas, S. J., Saiki, A. Y., Lerner, C. G. & Olejniczak, E. T. (2003). Protein Sci.12, 2613–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: CMK–CMP complex, 2h92, r2h92sf