

RuvA, a protein from M. tuberculosis H37Rv involved in recombination, has been cloned, expressed, purified and analysed by X-ray crystallography.
Keywords: RuvA, Mycobacterium tuberculosis, recombinational repair
Abstract
The process of recombinational repair is crucial for maintaining genomic integrity and generating biological diversity. In association with RuvB and RuvC, RuvA plays a central role in processing and resolving Holliday junctions, which are a critical intermediate in homologous recombination. Here, the cloning, purification and structure determination of the RuvA protein from Mycobacterium tuberculosis (MtRuvA) are reported. Analysis of the structure and comparison with other known RuvA proteins reveal an octameric state with conserved subunit–subunit interaction surfaces, indicating the requirement of octamer formation for biological activity. A detailed analysis of plasticity in the RuvA molecules has led to insights into the invariant and variable regions, thus providing a framework for understanding regional flexibility in various aspects of RuvA function.
1. Introduction
Recombinational repair is an important cellular process responsible for the generation of genetic diversity as well as for the maintainance of genomic integrity. A crucial step in recombination is the resolution of Holliday junctions produced from strand exchange between two homologous DNA helices (Kowalczykowski et al., 1994 ▶). Branch migration and resolution of a Holliday junction (HJ) into two recombinant DNA helices is promoted by RuvA, RuvB and RuvC proteins (Shinagawa & Iwasaki, 1996 ▶; West, 1996 ▶). Recombination mechanisms have been extensively studied in Escherichia coli, but it is unclear how far the mechanistic pathways elucidated in this species are applicable to other organisms, such as mycobacteria (McFadden, 1996 ▶; Muniyappa et al., 2000 ▶). Extrapolations of such a model would also have to account for both the reduced levels of homologous recombination and the higher levels of illegitimate recombination promoted by the tubercle bacillus.
Homologous recombination is also an important process that has been exploited to generate desired mutants in several model organisms (Muniyappa et al., 2000 ▶). In mycobacteria, however, difficulties in defining the phenotype of both wild-type and mutant strains have hampered genetic analysis of the organism (Shinagawa & Iwasaki, 1996 ▶). To facilitate genetic manipulation in mycobacteria, it is important to examine the roles of each of the components in the recombination pathway systematically. In this context, we have previously determined and analysed the crystal structure of the RecA protein from Mycobacterium tuberculosis (MtRecA) and M. smegmatis (MsRecA), a key component of the recombination machinery involved in strand exchange (Datta et al., 2000 ▶; Datta, Ganesh et al., 2003 ▶; Datta, Krishna et al., 2003 ▶; Krishna et al., 2006 ▶). Here, we report the crystal structure of the RuvA protein from M. tuberculosis (MtRuvA).
2. Materials and methods
2.1. Cloning, protein expression and purification
A 0.59 kbp DNA fragment containing the ruvA gene (Rv2593c) was amplified by PCR from the M. tuberculosis H37Rv Cosmid library with Pfu DNA polymerase (Stratagene), using the 5′ NdeI primer 5′-AGATATACATATGATCGCCTCGGTCCGCGGTGAGGTG-3′ and the 3′ BamHI primer 5′-AATTCGGATCCTCGGGCCTTCCCCAGCAACGACAA-3′. The bases in bold represent the NdeI and BamHI cleavage sites, respectively. The PCR amplicon was digested with NdeI and BamHI (NEB) and isolated using a Qiaquick PCR spin column (Qiagen). The product was ligated into a modified pET-28 vector containing a C-terminal six-His tag in frame with the BamHI restriction site using T4 DNA ligase (NEB) and transformed into BL21(DE3) (Novagen). The expressed protein contained the C-terminal tag GSHHHHHH, where GS is encoded by the BamHI restriction site (GGATCC). A 3 ml BL21(DE3) cell culture was tested for the expression of heterologous protein by binding to cobalt-chelated Talon superflow bead slurry (Clontech) and SDS–PAGE analysis.
The cells were grown as described by Studier (2005 ▶) with some modifications. The transformed cells were inoculated into 3 ml culture media (1 mM MgSO4, 0.5% glucose, the 17 amino acids Na–Glu, Asp, Lys–HCl, Arg–HCl, His–HCl, Ala, Pro, Gly, Thr, Ser, Gln, Asn, Val, Leu, Ile, Phe and Trp, each at 100 µg ml−1, a metal mix of 50 µM Fe, 20 µM Ca, 10 µM Mn, 10 µM Zn, 2 µM Co, Cu, Ni, Mo, Se and B and NPS with 5 mM phosphate, 5 mM Na, 2.5 mM K, 2.5 mM ammonium, 1.25 mM sulfate) and grown overnight at 310 K. From the seed culture, 0.5 ml was inoculated into 500 ml auto-induction media containing 1 mM MgSO4, a metal mix (the same as the seed culture), 0.5% glycerol, 0.5% glucose, 0.2% α-lactose, NPS (the same as the seed culture) and 35 µg ml−1 kanamycin. After the cells had been grown at 310 K until OD600 reached 0.5, cell growth was continued at 293 K for approximately 21 h until OD600 reached approximately 15 (as inferred from dilutions). The cells were harvested and stored at 193 K.
The cell pellet was lysed by sonication in 10 ml buffer A (20 mM Tris pH 8.0, 100 mM NaCl) per gram of cells for 10 min in 30 s pulses at 283 K. The cell debris was removed by centrifugation for 30 min at 38 000 rev min−1 using a Ti-60 rotor (Beckman). The supernatant was filtered through a 0.45 µm pore membrane and loaded onto a 5 ml Talon superflow cobalt-affinity column equilibrated with buffer A. After washing with 30 ml buffer A and 20 ml buffer B (20 mM Tris pH 8.0, 100 mM NaCl, 20 mM imidazole), the His-tagged ruvA was eluted from the column using buffer C (20 mM Tris–HCl pH 8.0, 100 mM NaCl and 300 mM imidazole). The eluted fraction was dialyzed against buffer D (20 mM Tris pH 8.0, 100 mM NaCl, 10 mM β-mercaptoethanol) and chromatographed on a Superdex-75 column (Amersham Biosciences) using buffer D for equilibration and elution. The peak fractions (monitored at OD280) were analyzed by SDS–PAGE and the pooled protein fractions were concentrated to 3 mg ml−1 using a Centricon Plus-20 (Millipore). The RuvA protein preparation, which had 97% purity as deduced by SDS–PAGE and MALDI–TOF mass spectroscopy (Applied Biosystem), was used for crystallization.
2.2. Crystallization and data collection
MtRuvA was crystallized at room temperature using the sitting-drop vapour-diffusion (IntelliPlate, Robbins Instruments) technique. Drops were made up of 0.5 µl of a 3 mg ml−1 protein stock and 0.5 µl reservoir solution. Protein stock was buffered in 20 mM Tris–HCl pH 8.0 with 100 mM sodium chloride and 10 mM β-ME. The reservoir contained 0.1 M sodium succinate pH 5.5 and 1.7 M ammonium sulfate. Crystals formed in a few days and were allowed to grow larger in size (150 × 150 × 50 µm) for up to a few months. Crystals belong to the tetragonal space group P42212, with unit-cell parameters a = b = 137.64, c = 88.97 Å and contained a single tetrameric molecule in the asymmetric unit. Diffraction screening and data collection were conducted on beamlines 5.0.1 and 5.0.2 at the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory. The diffraction data were processed with the program suite HKL-2000 (Otwinowski & Minor, 1997 ▶). Data-collection statistics are given in Table 1 ▶.
Table 1. X-ray crystal data, refinement and model statistics.
Values in parentheses are for the highest resolution shell (2.8–2.7 Å).
| Crystal data and data-collection statistics | |
| Space group | P42212 |
| Unit-cell parameters | |
| a (Å) | 137.64 |
| c (Å) | 88.97 |
| α = β = γ (°) | 90 |
| Resolution range (Å) | 45.0–2.7 |
| Matthews coefficient VM (Å3 Da−1) | 2.63 |
| Solvent content (%) | 53.2 |
| Subunits per ASU | 4 |
| Unique reflections | 23937 (2325) |
| Multiplicity | 7.8 (7.9) |
| Data completeness (%) | 99.9 (100) |
| Mean I/σ(I) | 14.1 (2.7) |
| Rmerge (%)† | 10.6 (51.2) |
| Refinement and model statistics | |
| R factor | 23.3 |
| Rfree‡ | 27.2 |
| Total No. of atoms per ASU | |
| Protein | 5269 |
| Water | 270 |
| Glycerol | 24 |
| R.m.s. deviations from ideal | |
| Bonds (Å) | 0.006 |
| Angles (°) | 1.3 |
| Dihedral angles (°) | 22.8 |
| Improper angles (°) | 2.67 |
| Ramachandran plot statistics (% of residues) | |
| Core regions | 86.6 |
| Allowed regions | 11.8 |
| Generously allowed regions | 1.6 |
| Disallowed regions | 0.0 |
R
merge = 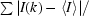
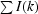 , where I(k) is the kth intensity measurement of a reflection, 〈I〉 is the average intensity value of that reflection and the summation is over all measurements.
, where I(k) is the kth intensity measurement of a reflection, 〈I〉 is the average intensity value of that reflection and the summation is over all measurements.
5% of reflections were used for the R free calculations.
2.3. Structure solution and refinement
The structure of MtRuvA was determined by molecular replacement using AMoRe (Navaza, 1994 ▶) with one of the tetramers of the M. leprae RuvA molecule (PDB code 1bvs) as the initial search model and was refined at 2.7 Å resolution using CNS (Brünger et al., 1998 ▶). The starting model was subjected to rigid-body refinement, treating the three domains of each subunit of the protein as rigid groups. This was followed by cycles of positional refinement and simulated annealing. Both 2F o − F c and F o − F c electron-density maps were generated from calculated phases based on the roughly refined model at this stage. The atomic model was built using the program O (Jones et al., 1991 ▶) in an iterative manner until convergence of R and R free. The grouped B factors in main chains and side chains for the protein residues were refined and the bulk-solvent correction was applied. Clear electron densities exist for four glycerol molecules. 374 O atoms were added to the model based on densities above 2.5σ and 0.8σ in F o − F c and 2F o − F c maps, respectively. Of these, those which were within 4 Å of any protein atom, numbering 270, were retained in the coordinate list as probable water O atoms. The electron density corresponding to residues 133–145 and 193–195 was hardly discernible even at the final stage of refinement, presumably owing to structural disorder. The final R and R free were 23.3 and 27.3, respectively. The stereochemical quality of the structures was validated using PROCHECK (Laskowski et al., 1993 ▶). The salient results of validation along with refinement statistics are given in Table 1 ▶. Superposition of structures was carried out using ALIGN (Cohen, 1997 ▶). The plasticity of the molecule was calculated domain-wise using the program ESCET (Schneider, 2002 ▶). Figures were prepared by using the programs MOLSCRIPT (Kraulis, 1991 ▶) and RASTER3D (Merritt & Bacon, 1997 ▶).
3. Results and discussion
Purified MtRuvA could be crystallized as single crystals, from which diffraction data were collected and processed to 2.7 Å. Structure determination, carried out through molecular replacement with MlRuvA as the search model, provided an acceptable solution, revealing one tetramer in the asymmetric unit.
3.1. Monomer and tetramer
MtRuvA exhibits sequence identities of 76% with MlRuvA, 31% with EcRuvA and 37% with TthRuvA (Thermus thermophilus RuvA). Despite the lack of high sequence conservation across the phyla, the overall structures of the protein from all these species closely resemble one another. Each MtRuvA asymmetric unit contains one RuvA tetramer, in which each subunit exhibits a tripartite domain architecture, consisting of three domains with distinct functional features (Rafferty et al., 1996 ▶, 1998 ▶; Yamada et al., 2002 ▶; Roe et al., 1998 ▶; Nishino et al., 1998 ▶) as illustrated in Fig. 1 ▶: a 63-residue (1–63) N-terminal domain I, which comprises a six-stranded β-barrel with a Greek-key motif, mainly involved in the tetramerization with a central fourfold axis, a 70-residue domain II (64–133) made up of a helical bundle, consisting of five α-helices including two helix–hairpin–helix (HhH) motifs, which contributes to DNA recognition, and a smaller 50-residue (146–196) domain III, made up of a three-helix bundle, involved in RuvB binding. A 13-residue flexible linker connects domains II and III, the precise functional roles are not as yet fully characterized. Each tetramer has a non-crystallographic fourfold symmetry comparable to that observed in all other RuvA structures. The symmetry, however, is near-perfect when only domains I and II are considered. A considerable departure from fourfold symmetry is exhibited by domain III, indicative of its relative flexibility.
Figure 1.

Domain architecture of the RuvA tetramer. A schematic representation of the sequence is shown in the top panel. The four subunits are labelled A, B, C and D. The three domains in each subunit, DI, DII and DIII, are shown in subunit A. The key functional motifs are highlighted (Rafferty et al., 1996 ▶, 1998 ▶; Yamada et al., 2002 ▶; Roe et al., 1998 ▶; Nishino et al., 1998 ▶). The residue numbers are based on the MtRuvA sequence. Variant (red) and invariant (blue) regions of the tetrameric RuvA deduced from an analysis of MtRuvA, MlRuvA, EcRuvA and TthRuvA are illustrated.
3.2. Octamerization
The MtRuvA tetramer in the asymmetric unit and its twofold symmetry equivalent constitute an octamer resembling those of MlRuvA (Roe et al., 1998 ▶) and TthRuvA (Yamada et al., 2002 ▶). Helix 117–126 from each of the four subunits of one tetramer is found to interact with its counterpart in the second tetramer. These interactions involving eight helices are primarily through salt bridges between conserved arginine and glutamic acid residues, which have also been noted previously in the discussion on the MlRuvA octamer (Roe et al., 1998 ▶). A conserved hydrophobic residue, most often a leucine, making van der Waals interactions with the same residue from the other subunit is also found at the octamer interface. The conserved arginine, the glutamic acid and the leucine all belong to the HhH motif II of domain II. In addition to these, Ser78 from motif I of domain II also appears to be within interaction distance of its counterpart at the interface across the tetramers. This position too is predominantly occupied by polar uncharged amino acids in other RuvAs. A similar interaction could also be inferred from the MlRuvA structure. Although the electrostatic interactions in the interface are, in general, similar in the octamers of MtRuvA, MlRuvA and TthRuvA, the tetramers are slightly farther away from each other in TthRuvA. The distance between the centroids of the two tetramers is 37.6, 38.2 and 40.1 Å, respectively, in MtRuvA, MlRuvA and TthRuvA.
3.3. Plasticity of the molecule
An appreciation of the structural flexibility within the RuvA molecule is of great importance in order to understand its function thoroughly. The relatively variant and invariant regions of the molecules have been delineated employing different available crystal structures using an error-inclusive structure comparison and evaluation tool (ESCET). Two monomers from M. tuberculosis and M. leprae, three independent monomers from E. coli, including one from the molecule bound to DNA, and one monomer from T. thermophilus were included in the calculations. Calculations were performed independently for each domain. The cutoff parameter was carefully chosen as 2.7σ such that neither too few nor too many residues were delineated as flexible regions, in the same manner as has been performed by us previously in relation to the structures of the single-stranded binding protein, ribosome recycling factor from M. tuberculosis (Saikrishnan, Manjunath et al., 2005 ▶; Saikrishnan, Kalapala et al., 2005 ▶) and Dps from M. smegmatis (Roy et al., 2004 ▶).
The above analysis enabled a delineation of the invariant and variable regions in the molecule (Fig. 1 ▶). Domain I has distinct segments of invariant and variable residues approximately divided by the barrel axis making up the OB (oligonucleotide/oligosaccharide-binding) fold of the domain. The N-terminal loop of domain I is flexible, whereas the first three β-strands are rigid. The helix that caps the barrel is flexible. The last two strands in domain I are partly flexible. The flexible regions of these strands are also involved in the subunit–subunit interface of the tetramer. Consequently, it appears that the intersubunit interface consists of an invariant and a variable region, where the invariant segment of one subunit interacts with the variable segment of the other. In the second domain, the first HhH motif is variable, whereas the second motif is mainly invariant, with only the rear end identified as variable. These two motifs host the residues that are involved in the HJ binding. A significant alteration in the subunit orientations upon DNA binding has been reported previously for this domain (Ariyoshi et al., 2000 ▶). Domain III is surprisingly predominantly invariant, suggesting that the movements of the domain that accompany RuvB binding are largely rigid-body movements.
Supplementary Material
PDB reference: RuvA, 2h5x, r2h5xsf
Acknowledgments
We thank the staff at ALS beamlines 5.0.1 and 5.0.2 for assistance during data collection. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, Materials Science Division of the US Department of Energy under Contract No. DE-AC03-76SF00098 at Lawrence Berkeley National Laboratory. Computations were carried out at the Supercomputer Education and Research Centre at the Indian Institute of Science, and the Bioinformatics Centre and Graphics Facility, supported by the Department of Biotechnology (DBT). Financial assistance from the DBT is acknowledged. MV is supported by a Distinguished Biotechnology Award of the DBT. RP is a CSIR research fellow.
References
- Ariyoshi, M., Nishino, T., Iwasaki, H., Shinagawa, H. & Morikawa, K. (2000). Proc. Natl Acad. Sci. USA, 97, 8257–8262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Cohen, G. E. (1997). J. Appl. Cryst.30, 1160–1161. [Google Scholar]
- Datta, S., Ganesh, N., Chandra, N. R., Muniyappa, K. & Vijayan, M. (2003). Proteins, 50, 474–485. [DOI] [PubMed] [Google Scholar]
- Datta, S., Krishna, R., Ganesh, N., Chandra, N. R., Muniyappa, K. & Vijayan, M. (2003). J. Bacteriol.185, 4280–4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta, S., Prabu, M. M., Vaze, M. B., Ganesh, N., Chandra, N. R., Muniyappa, K. & Vijayan, M. (2000). Nucleic Acids Res.28, 4964–4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kowalczykowski, S. C., Dixon, D. A., Eggleston, A. K., Lauder, S. D. & Rehrauer, W. M. (1994). Microbiol. Rev.58, 401–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis, P. J. (1991). J. Appl. Cryst.24, 946–950. [Google Scholar]
- Krishna, R., Manjunath, G. P., Kumar, P., Surolia, A., Chandra, N. R., Muniyappa, K. & Vijayan, M. (2006). Nucleic Acids Res.34, 2186–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291. [Google Scholar]
- McFadden, J. (1996). Mol. Microbiol.21, 205–211. [DOI] [PubMed] [Google Scholar]
- Merritt, E. A. & Bacon, D. J. (1997). Methods Enzymol.277, 505–524. [DOI] [PubMed]
- Muniyappa, K., Vaze, M. B., Ganesh, N., Sreedhar Reddy, M., Guhan, N. & Venkatesh, R. (2000). Microbiology, 146, 2093–2095. [DOI] [PubMed] [Google Scholar]
- Navaza, J. (1994). Acta Cryst. A50, 157–163. [Google Scholar]
- Nishino, T., Ariyoshi, M., Iwasaki, H., Shinagawa, H. & Morikawa, K. (1998). Structure, 6, 11–21. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Rafferty, J. B., Ingleston, S. M., Hargreaves, D., Artymiuk, P. J., Sharples, G. J., Lloyd, R. G. & Rice, D. W. (1998). J. Mol. Biol.278, 105–116. [DOI] [PubMed] [Google Scholar]
- Rafferty, J. B., Sedelnikova, S. E., Hargreaves, D., Artymiuk, P. J., Baker, P. J., Sharples, G. J., Mahdi, A. A., Lloyd, R. G. & Rice, D. W. (1996). Science, 274, 415–421. [DOI] [PubMed] [Google Scholar]
- Roe, S. M., Barlow, T., Brown, T., Oram, M., Keeley, A., Tsaneva, I. R. & Pearl, L. H. (1998). Mol. Cell, 2, 361–372. [DOI] [PubMed] [Google Scholar]
- Roy, S., Gupta, S., Das, S., Sekar, K., Chatterji, D. & Vijayan, M. (2004). J. Mol. Biol.339, 1103–1113. [DOI] [PubMed] [Google Scholar]
- Saikrishnan, K., Kalapala, S. K., Varshney, U. & Vijayan, M. (2005). J. Mol. Biol.345, 29–38. [DOI] [PubMed] [Google Scholar]
- Saikrishnan, K., Manjunath, G. P., Singh, P., Jeyakanthan, J., Dauter, Z., Sekar, K., Muniyappa, K. & Vijayan, M. (2005). Acta Cryst. D61, 1140–1148. [DOI] [PubMed] [Google Scholar]
- Schneider, T. R. (2002). Acta Cryst. D58, 195–208. [DOI] [PubMed] [Google Scholar]
- Shinagawa, H. & Iwasaki, H. (1996). Trends Biochem. Sci.21, 107–111. [PubMed] [Google Scholar]
- Studier, F. W. (2005). Protein Expr. Purif.41, 207–234. [DOI] [PubMed] [Google Scholar]
- Yamada, K., Miyata, T., Tsuchiya, D., Oyama, T., Fujiwara, Y., Ohnishi, T., Iwasaki, H., Shinagawa, H., Ariyoshi, M., Mayanagi, K. & Morikawa, K. (2002). Mol. Cell, 10, 671–681. [DOI] [PubMed] [Google Scholar]
- West, S. C. (1996). J. Bacteriol.178, 1237–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: RuvA, 2h5x, r2h5xsf