

Locked nucleic acid (LNA) nucleotides are RNA analogues with a useful additional conformational constraint; the current investigation will provide the first crystallographic view of an all-LNA duplex.
Keywords: locked nucleic acids, LNA, tenascin-C, aptamer, TTA1
Abstract
The pharmacokinetic properties of an aptamer against the tumour-marker protein tenascin-C have recently been successfully improved by the introduction of locked nucleic acids (LNAs) into the terminal stem of the aptamer. Since it is believed that this post-SELEX optimization is likely to provide a more general route to enhance the in vitro and in vivo stability of aptamers, elucidation of the structural basis of this improvement was embarked upon. Here, the crystallographic and X-ray diffraction data of the isolated aptamer stem encompassed in a six-base-pair duplex both with and without the LNA modification are presented. The obtained all-LNA crystals belong to space group P41212 or P43212, with unit-cell parameters a = b = 52.80, c = 62.83 Å; the all-RNA crystals belong to space group R32, with unit-cell parameters a = b = 45.21, c = 186.97 Å, γ = 120.00°.
1. Introduction
Aptamers are nucleic acids that bind specific targets with high affinity and selectivity; they are similar to antibodies in function but are significantly smaller in molecular weight (typically 10–15 kDa; White et al., 2000 ▶). They are selected for specific target binding from a large library pool by the SELEX method (‘systematic evolution of ligands by exponential enrichment’; Tuerk & Gold, 1990 ▶; Ellington & Szostak, 1990 ▶). The structural binding properties as well as the therapeutic and analytical potential of aptamers have been reviewed exhaustively (Hermann & Patel, 2000 ▶; Nimjee et al., 2005 ▶; Tombelli et al., 2005 ▶; Rimmele, 2003 ▶). Their emerging therapeutic potential is best documented by the US Food and Drug Administration’s approval in December 2004 of the first aptamer drug, Macugen, which acts as a selective vascular endothelial growth-factor antagonist in the therapy of neovascular age-related macular degeneration (Gryziewicz, 2005 ▶). An example of the diagnostic potential of aptamers is provided by a tenascin-C-binding tumour marker, which has already been used in clinical trials (Hicke et al., 2001 ▶, 2006 ▶; Schmidt et al., 2004 ▶). A stability-enhancing component of this aptamer is the focus of the present communication. The target of this aptamer, tenascin-C, is a large hexameric human glycoprotein found in the extracellular matrix. Following expression during embryogenesis, this protein virtually disappears in the adult organism. However, its expression is sharply up-regulated in tissues undergoing remodelling processes including wound repair, neovascularization, inflammation and tumorigenesis (Hsia & Schwarzbauer, 2005 ▶). Based on the latter property, the original tenascin-C-binding aptamer (Hicke et al., 2001 ▶) was further developed into a marker for tumour imaging with radioisotopes by several modifications including a 5′-chelator for a technetium-99m label (Schmidt et al., 2004 ▶). The proposed secondary structure of the resulting 39-nucleotide aptamer, TTA1, is shown in Fig. 1 ▶(a). It is presumed to consist of three stem regions which are joined by two loops. Substitution experiments delineate stems II and III as the regions involved in binding, whereas stem I seems to be critical to the stability of the molecule (Hicke et al., 2001 ▶). As the molecular integrity is a crucial aspect of the application of aptamers, stem I was modified in order to enhance both the in vitro and the in vivo stability. The thermal in vitro stability, as assessed by the melting curves of duplexes formed from strand analogues of stem I with the sequence shown in Fig. 1 ▶(b), was found to follow the order 2′-F/2′-OMe < RNA/RNA ≤ 2′-OMe/2′-OMe < 2′-F/LNA < RNA/LNA = LNA/RNA < 2′-OMe/LNA < LNA/LNA (Schmidt et al., 2004 ▶). The highest in vitro stability was achieved by the introduction of locked nucleic acids (LNAs), which were first synthesized in the laboratories of T. Imanishi (Obika et al., 1997 ▶) and J. Wengel (Singh et al., 1998 ▶). The structure of this RNA analogue is depicted in Fig. 1 ▶(c). LNAs are known to drastically increase the thermal stability and nuclease resistance of oligonucleotides (Kurreck et al., 2002 ▶). These improvements are reflected in the altered in vivo properties of the correspondingly LNA-modified complete TTA1 aptamer, which include increased nucleolytic stability, slower blood clearance and excretion, improved tumour uptake, and a marginally improved binding affinity (EC50 = 2 nM; Schmidt et al., 2004 ▶). The beneficial effect of the incorporation of LNAs into non-binding stems of aptamers may be a more general way to enhance the in vitro as well as the in vivo stability of aptamers. In order to obtain structural details of this phenomenon, we have crystallized a six-base-pair duplex encompassing the TTA1 stem I both in the modified LNA/LNA form as well as in the standard RNA/RNA form and collected diffraction data to 1.95 and 2.60 Å resolution, respectively, applying synchrotron radiation.
Figure 1.

(a) Predicted secondary structure of the tenascin-C-binding aptamer with the presumed three helical regions I, II and III (indicated), of which only the latter two are thought to be directly involved in binding. For in vivo imaging, the aptamer has been functionalized with 2′-OMe and 2′-F nucleotides (light and dark grey, respectively), a thymidine cap at the 3′-end and a MAG2 chelate, which is a Cys-Gly-Gly motif conjugated via a hexyl-amino linker, at the 5′-end (Hicke et al., 2001 ▶; Schmidt et al., 2004 ▶). (b) LNA (and RNA) sequence of the crystallized stem I following the design described in Schmidt et al. (2004 ▶). (c) Diagram highlighting the additional covalent 2′-O,4′-C-methylene bridge in the β-d-ribofuranosyl moiety of locked nucleic acids (LNAs).
2. Materials and methods
2.1. Oligonucleotide synthesis and purification
Two LNA oligonucleotides with sequences 5′-ACTCCC-3′ and 5′-GGGAGT-3′ were synthesized on an Applied Biosystems 394 DNA/RNA synthesizer by solid-phase phosphoramidite chemistry with building blocks obtained from Proligo (Hamburg, Germany; in the LNA the building block cytosine is 5-methylated). Oligonucleotides were purified on a Resource 3 ml column (Amersham Biosciences) in the DMT-on mode with a Beckmann HPLC System equipped with a System Gold 168 detector and System Gold 126 solvent modules. Following detritylation, the oligonucleotides were purified again on a Waters XTerra RP18 column (5 µm, 3.9 × 150 mm) using a triethylammonium acetate (TEAA)/acetonitrile (ACN) buffer system (solvent A, 50 mM TEAA; solvent B, 80% ACN in solvent A). Two RNA oligonucleotides with sequences 5′-ACUCCC-3′ and 5′-GGGAGU-3′ were synthesized by CureVac GmbH, Tübingen, Germany.
2.2. Hybridization and crystallization
To obtain the LNA/LNA duplex 5′-ACTCCC-3′/5′-GGGAGT-3′, the two strands, each at a concentration of 0.5 mM, were hybridized in water. Analogously, the two strands of the RNA/RNA duplex 5′-ACUCCC-3′/5′-GGGAGU-3′ were hybridized in water at a concentration of 0.5 mM each. After heating the respective mixtures to 363 K, they were allowed to cool to room temperature for the formation of duplexes. The resulting solutions were used in subsequent crystallization experiments.
The initial LNA screening was performed with the 48 conditions of the Natrix formulation from Hampton Research (CA, USA), applying the sitting-drop vapour-diffusion technique at 294 K in 96-well CrystalQuick Lp plates (Greiner Bio-One, Germany). A 1 µl sample of 0.5 mM LNA/LNA duplex 5′-ACTCCC-3′/5′-GGGAGT-3′ in water was mixed with 1 µl reservoir solution and equilibrated against 80 µl reservoir solution. The crystallization condition with 50 mM 2-N-morpholinoethanesulfonic acid (MES) pH 5.6, 10 mM magnesium chloride and 2.0 M lithium sulfate as precipitant was selected as the starting point for an optimization in which the lithium sulfate concentration was varied between 2.0 and 2.25 M in increments of 0.05 M, applying the hanging-drop vapour-diffusion technique in 24-well Linbro Plates (ICN Biomedicals Inc., Ohio, USA). A 1 µl sample of 0.5 mM LNA/LNA duplex in water was mixed with 1 µl reservoir solution and equilibrated against 1 ml reservoir solution at 294 K. 50 mM MES pH 5.6, 10 mM magnesium chloride and 2.1 M lithium sulfate yielded the particularly large crystal used for data collection and shown in Fig. 2 ▶(a).
Figure 2.

(a) Crystal of the all-LNA form of the TTA1 stem I 6-mer (dimensions of ∼1.4 × 0.2 × 0.2 mm); (b) fragile all-RNA crystals grown under the same conditions that were unsuitable for X-ray diffraction; (c) RNA crystals suitable for X-ray diffraction obtained under optimized conditions (dimensions typically over 0.5 mm in length and less than 0.05 mm in diameter).
The initial RNA screening was also performed with the Natrix formulation applying the sitting-drop vapour-diffusion technique at 294 K in 96-well CrystalQuick Lp plates. A 1 µl sample of 0.5 mM RNA/RNA duplex 5′-ACUCCC-3′/5′-GGGAGU-3′ in water was mixed with 1 µl reservoir solution and equilibrated against 80 µl reservoir solution. However, the crystals obtained were very fragile and were not suitable for X-ray diffraction. This included crystals that grew in a comparable time window under the conditions detailed above for the LNA. Much better RNA crystals could be obtained with conditions found by screening with the Nucleic Acid Mini Screen (Hampton Research), applying the hanging-drop vapour-diffusion technique at 294 K in 24-well Linbro Plates; a 1 µl sample of 0.5 mM RNA/RNA duplex in water was mixed with 1 µl reservoir solution and equilibrated against 1 ml reservoir solution. A condition consisting of 40 mM sodium cacodylate pH 6.0, 12 mM spermine–HCl, 80 mM sodium chloride and 10%(v/v) 2-methyl-2,4-pentanediol (MPD) yielded crystals in approximately one week at 294 K which were used for data collection.
2.3. Crystallographic data collection and evaluation
Prior to data collection, LNA crystals were cryoprotected in 50 mM MES pH 5.6, 10 mM magnesium chloride, 2.1 M lithium sulfate and 20%(v/v) glycerol and flash-frozen. RNA crystals were flash-frozen in 40 mM sodium cacodylate pH 6.0, 12 mM spermine–HCl, 80 mM sodium chloride and 10%(v/v) MPD. X-ray diffraction data were recorded at the X13 consortium beamline (DESY/HASYLAB, Hamburg). For the LNA/LNA duplex, a high-resolution data set from 50 to 1.95 Å was collected at a wavelength of 0.8075 Å. For the RNA/RNA duplex, a data set from 20 to 2.60 Å was collected at a wavelength of 0.8063 Å. Data processing and calculation of the space group and unit-cell parameters were performed with the HKL2000 package (Otwinowski & Minor, 1997 ▶).
3. Results and discussion
3.1. Crystallization
Fig. 1 ▶ shows the predicted secondary structure of TTA1 and the sequence of the crystallized LNA variant of stem I (following the design described in the study of Schmidt et al., 2004 ▶). Since LNA is an RNA analogue with an additional covalent 2′-O,4′-C-methylene cross-link in the ribofuranose moiety designed to ‘lock’ it in the C3′-endo conformation (Fig. 1 ▶ c; Obika et al., 1997 ▶; Petersen & Wengel, 2003 ▶; Singh et al., 1998 ▶), we also crystallized the native RNA counterpart of the TTA1 stem I for a meaningful comparison. We are not aware that a comparison of the crystallization behaviour of LNA compared with RNA duplexes has been reported before and found these to be notably different. In both cases, about 8% of the inital Natrix screen conditions yielded crystals, but only 4% yielded both LNA and RNA crystals. The conditions optimized to obtain LNA crystals (described above; Fig. 2 ▶ a) did not yield RNA crystals that were suitable for X-ray diffraction. RNA crystals thus obtained were very fragile and disintegrated upon handling (Fig. 2 ▶ b). However, with conditions that had been specifically optimized, more robust RNA crystals could be obtained that grew more slowly (Fig. 2 ▶ c). In summary, the optimized LNA crystallization conditions were not equally well suited to crystallization of the corresponding RNA variant. LNA crystals typically appeared overnight and several crystals grew to the size of the crystallization drop. This phenomenon mirrors the gain in stability and rigidity as exemplified by the increase of the melting temperature by increments of approximately 50 K between the RNA and LNA variants (Schmidt et al., 2004 ▶).
3.2. Crystallographic data
Table 1 ▶ presents a comparison of the crystallographic characteristics of the all-LNA and all-RNA forms of the TTA1 stem I duplex obtained under the individually optimized conditions. While the LNA crystallizes in space group P41212 or P43212 (corresponding to two molecules per asymmetric unit in the enantiomorph space groups, with a Matthews coefficient V M of 2.75 Å3 Da−1), the RNA crystallizes in space group R32 (with a Matthews coefficient calculated to be 2.45 Å3 Da−1 considering two molecules per asymmetric unit). A data set was collected to 1.95 Å for the LNA and to 2.60 Å for the RNA. The respective data sets were processed in the resolution ranges 50–1.95 Å with an overall R merge of 4.5% and 20–2.60 Å with an overall R merge of 6.1%. Molecular-replacement calculations are presently in progress using the CCP4-associated programs AMoRe, MOLREP and Phaser. A variety of structures are being tested, including a heptameric helix derived from the tRNAAla acceptor stem (PDB code 434d; Mueller et al., 1999 ▶), a dodecameric helix containing the Escherichia coli Shine–Dalgarno sequence (PDB code 1sdr; Schindelin et al., 1995 ▶) and also an artificial palindromic homodecamer, the sequence of which contains a single LNA monomer in each strand of DNA (PDB code 1i5w). The latter is to the best of our knowledge the only crystallographic report of LNA in any oligonucleotide setting (Egli et al., 2001 ▶). Also important for the analysis, although dissimilar in sequence, are the NMR structures of a nonameric RNA/LNA heteroduplex varying in the balance of LNA to DNA content in the LNA strand (PDB codes 1hhw, 1hhx and 1h0q; Petersen et al., 2002 ▶; Nielsen et al., 2004 ▶). Partially homologous to the sequence of this study and of particular interest in this context is the recent crystal structure of a decameric all-RNA/all-HNA hybrid, as hexitol nucleic acids (HNAs) also enhance duplex stability by means of conformationally restrained sugar moieties (PDB code 2bj6; Maier et al., 2005 ▶).
Table 1. Data-collection and processing statistics of the all-LNA and all-RNA forms of the TTA1 stem I crystals.
Values in parentheses are for the highest resolution shell.
| LNA | RNA | |
|---|---|---|
| Beamline | X13 consortium beamline (DESY/HASYLAB) | X13 consortium beamline (DESY/HASYLAB) |
| Wavelength (Å) | 0.80750 | 0.80630 |
| Space group | P41212 or P43212 | R32 |
| Unit-cell parameters (Å, °) | a = b = 52.80, c = 62.83 | a = b = 45.21, c = 186.97, γ = 120.00 |
| Matthews coefficient VM (Å3 Da−1) | 2.75 | 2.45 |
| Measured reflections | 119435 | 16840 |
| Unique reflections | 6887 | 2467 |
| Resolution range (Å) | 50–1.95 | 20–2.60 |
| Completeness (%) | 99.3 (100) | 99.4 (100) |
| Multiplicity (%) | 17.3 (17.3) | 6.8 (7.0) |
| Rmerge† (%) | 4.5 (40.0) | 6.1 (40.1) |
| Average I/σ(I) | 51.0 (6.8) | 22.6 (5.3) |
R
merge = 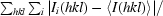
 , where I
i(hkl) and 〈I(hkl)〉 are the observed individual and mean intensities of a reflection with indices hkl, respectively,
, where I
i(hkl) and 〈I(hkl)〉 are the observed individual and mean intensities of a reflection with indices hkl, respectively, 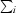 is the sum over the individual measurements of a reflection with indices hkl and
is the sum over the individual measurements of a reflection with indices hkl and  is the sum over all reflections.
is the sum over all reflections.
The structure of the LNA variant of the TTA1 stem I will provide the first crystallographic view of an LNA/LNA duplex. This is a timely investigation because LNAs are now widely used for an ever-increasing number of applications (Jepsen et al., 2004 ▶; Jepsen & Wengel, 2004 ▶; Kauppinnen et al., 2006 ▶; Petersen & Wengel, 2003 ▶). Structural investigations in particular have a role in paving the way to a deliberate stability tuning by balancing the properties of RNA and DNA with LNA.
Acknowledgments
This work was funded in the RiNA Network for RNA Technologies by the Federal Ministry of Education and Research, the City of Berlin and the European Regional Development Fund. We are grateful to Schering AG for making the synthesis facility available and to the Fonds der Chemischen Industrie (Verband der Chemischen Industrie eV) and the National Foundation for Cancer Research, USA for additional support.
References
- Egli, M., Minasov, G., Teplova, M., Kumar, R. & Wengel, J. (2001). J. Chem. Soc., Chem. Commun., pp. 651–652.
- Ellington, A. D. & Szostak, J. W. (1990). Nature (London), 346, 818–822. [DOI] [PubMed] [Google Scholar]
- Gryziewicz, L. (2005). Adv. Drug Deliv. Rev.57, 2092–2098. [DOI] [PubMed] [Google Scholar]
- Hermann, T. & Patel, D. J. (2000). Science, 287, 820–825. [DOI] [PubMed] [Google Scholar]
- Hicke, B. J., Marion, C., Chang, Y. F., Gould, T., Lynott, C. K., Parma, D., Schmidt, P. G. & Warren, S. (2001). J. Biol. Chem.276, 48644–48654. [DOI] [PubMed] [Google Scholar]
- Hicke, B. J., Stephens, A. W., Gould, T., Chang, Y. F., Lynott, C. K., Heil, J., Borkowski, S., Hilger, C. S., Cook, G., Warren, S. & Schmidt, P. G. (2006). J. Nucl. Med.47, 668–678. [PubMed] [Google Scholar]
- Hsia, H. C. & Schwarzbauer, J. E. (2005). J. Biol. Chem.280, 26641–26644. [DOI] [PubMed] [Google Scholar]
- Jepsen, J. S., Sorensen, M. D. & Wengel, J. (2004). Oligonucleotides, 14, 130–146. [DOI] [PubMed] [Google Scholar]
- Jepsen, J. S. & Wengel, J. (2004). Curr. Opin. Drug Discov. Devel.7, 188–194. [PubMed] [Google Scholar]
- Kauppinnen, S., Vester, B. & Wengel, J. (2006). RNA Towards Medicine, edited by V. A. Erdmann, J. Brosius & J. Barciszewski, pp. 405–422. Berlin: Springer.
- Kurreck, J., Wyszko, E., Gillen, C. & Erdmann, V. A. (2002). Nucleic Acids Res.30, 1911–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier, T., Przylas, I., Strater, N., Herdewijn, P. & Saenger, W. (2005). J. Am. Chem. Soc.127, 2937–2943. [DOI] [PubMed] [Google Scholar]
- Mueller, U., Schübel, H., Sprinzl, M. & Heinemann, U. (1999). RNA, 5, 670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, K. E., Rasmussen, J., Kumar, R., Wengel, J., Jacobsen, J. P. & Petersen, M. (2004). Bioconjug. Chem.15, 449–457. [DOI] [PubMed] [Google Scholar]
- Nimjee, S. M., Rusconi, C. P. & Sullenger, B. A. (2005). Annu. Rev. Med.56, 555–583. [DOI] [PubMed] [Google Scholar]
- Obika, S., Nanbu, D., Hari, Y., Morio, K., Ishida, T. & Imanishi, T. (1997). Tetrahedron Lett.38, 8735–8738.
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Petersen, M., Bondensgaard, K., Wengel, J. & Jacobsen, J. P. (2002). J. Am. Chem. Soc.124, 5974–5982. [DOI] [PubMed] [Google Scholar]
- Petersen, M. & Wengel, J. (2003). Trends Biotechnol.21, 74–81. [DOI] [PubMed] [Google Scholar]
- Rimmele, M. (2003). Chembiochem, 4, 963–971. [DOI] [PubMed] [Google Scholar]
- Schindelin, H., Zhang, M., Bald, R., Fürste, J. P., Erdmann, V. A. & Heinemann, U. (1995). J. Mol. Biol.249, 595–603. [DOI] [PubMed] [Google Scholar]
- Schmidt, K. S., Borkowski, S., Kurreck, J., Stephens, A. W., Bald, R., Hecht, M., Friebe, M., Dinkelborg, L. & Erdmann, V. A. (2004). Nucleic Acids Res.32, 5757–5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, S. K., Nielsen, P., Koshkin, A. K., Olsen, C. E. & Wengel, J. (1998). J. Chem. Soc., Chem. Commun., pp. 455–456.
- Tombelli, S., Minunni, M. & Mascini, M. (2005). Biosens. Bioelectron.20, 2424–2434. [DOI] [PubMed] [Google Scholar]
- Tuerk, C. & Gold, L. (1990). Science, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- White, R. R., Sullenger, B. A. & Rusconi, C. P. (2000). J. Clin. Invest.106, 929–934. [DOI] [PMC free article] [PubMed] [Google Scholar]