

Vascular apoptosis-inducing protein 1 (VAP1) and VAP2 from C. atrox venom were crystallized in variety of different crystal forms. Diffraction data sets were obtained to 2.5 and 2.15 Å resolution for VAP1 and VAP2, respectively.
Keywords: vascular apoptosis-inducing proteins, apoptotic toxins, snake-venom metalloproteinases, ADAMs
Abstract
VAPs are haemorrhagic snake-venom toxins belonging to the reprolysin family of zinc metalloproteinases. In vitro, VAPs induce apoptosis specifically in cultured vascular endothelial cells. VAPs have a modular structure that bears structural homology to mammalian ADAMs (a disintegrin and metalloproteinases). VAP1 is a homodimer with a MW of 110 kDa in which the monomers are connected by a single disulfide bridge. VAP2 is homologous to VAP1 and exists as a monomer with a MW of 55 kDa. In the current study, several crystal forms of VAP1 and VAP2 were obtained using the vapour-diffusion method and diffraction data sets were collected using SPring-8 beamlines. The best crystals of VAP1 and VAP2 generated data sets to 2.5 and 2.15 Å resolution, respectively.
1. Introduction
Haemorrhagic snake venoms contain factors that induce apoptosis specifically in cultured vascular endothelial cells (Araki et al., 1993 ▶). The vascular apoptosis-inducing proteins VAP1 and VAP2 were originally isolated from the venom of the western diamondback rattlesnake Crotalus atrox (Masuda et al., 1997 ▶, 1998 ▶) and similar apoptotic toxins (VAPs) have been isolated from other snake venoms (Masuda et al., 2001 ▶; You et al., 2003 ▶; Trummal et al., 2005 ▶). VAP1 is a disulfide-bonded homodimeric protein with a molecular weight of 110 kDa and an isoelectric point of 8.5. VAP2 is an acidic single-chain protein with a molecular weight of 55 kDa and an isoelectric point of 4.5 (Masuda et al., 1997 ▶, 1998 ▶). VAP1 (Masuda et al., 2000 ▶) and VAP2 (S. Masuda, H. Hayashi & S. Araki, in preparation) are modular metalloproteinases with nucleotide-sequence homology to genes encoding the mammalian membrane-anchored metalloproteinases known as ADAMs. ADAMs are an emerging class of metalloproteinases whose function has been implicated in cell–cell and cell–matrix adhesion and signalling. They also appear to be associated with numerous diseases including arthritis, Alzheimer’s disease and cancer (White, 2003 ▶; Blobel, 2005 ▶; Seals & Courtneidge, 2003 ▶; Moss & Bartsch, 2004 ▶; Duffy et al., 2003 ▶).
Viperidae snake venoms contain a number of metalloproteinases, the snake-venom metalloproteinases (SVMPs), that induce local and systemic haemorrhage by disrupting the wall of the blood vessels in envenomed patients (Gutierrez et al., 2005 ▶). All known VAPs belong to the P-III class of SVMPs, which have been shown to be the most potent haemorrhagic toxins from snake venoms. The P-III SVMPs have a modular structure consisting of metalloproteinase (M), disintegrin (D) and cysteine-rich (C) domains (Fox & Serrano, 2005 ▶). SVMPs and ADAMs are members of the reprolysin group of zinc-dependent metalloproteinases, which together with astasins, serralysin and matrix metalloproteinases comprise the metzincin superfamily of metalloproteinases (Bode et al., 1993 ▶). All these enzymes share a signature consensus zinc-binding motif, HEXXHXXGXXH, in their catalytic region that defines proteins of the class, as well as a methionine-containing turn that serves as a structural base for the three active histidine residues (Bode et al., 1993 ▶).
The crystal structures of several SVMPs of the P-I class, which contain only an M domain, and of isolated domains of ADAMs have been determined. However, structures of SVMPs or ADAMs containing M, D and C domains have not been determined. To understand more about the structure of P-III SVMPs and ADAMs and how it relates to the molecular mechanism of VAP-induced apoptosis, we initiated the crystallographic analysis of VAP1 and VAP2. This is the first report of the crystallization and preliminary X-ray analysis of apoptotic SVMPs. Three-dimensional crystal structures of VAP1 derived from the two distinct crystal forms described in this report have recently been described (Takeda et al., 2006 ▶); the structural analysis of VAP2 is ongoing.
2. Methods
2.1. Purification
VAP1 and VAP2 were purified as described previously (Maruyama et al., 2005 ▶; Masuda et al., 1998 ▶) with some modifications. Briefly, crude C. atrox venom (Sigma–Aldrich, USA) was dissolved in buffer containing 10 mM Tris–HCl pH 7.0 and 10 mM NaCl and then applied onto a CM-Sepharose (Amersham Bioscience, USA) column equilibrated with the same buffer. VAP2 was eluted from the column with the above buffer, whereas VAP1 was eluted with buffer containing 10 mM Tris–HCl pH 7.0 and 50 mM NaCl.
The VAP1 was further purified on a hydroxylapatite column. The VAP1-containing CM-Sepharose fraction was first diluted with an equal amount of distilled water and then applied onto a hydroxylapatite column equilibrated with 25 mM sodium phosphate pH 7.0. VAP1 was eluted using buffer containing 50 mM sodium phosphate pH 7.0 and then concentrated using an Amicon Ultra membrane (Millipore) with a nominal molecular-weight limit (NMWL) of 50 000 Da. The final protein concentration was 6.5 mg ml−1. During the concentration step, the buffer was replaced with 10 mM Tris–HCl pH 7.0.
The VAP2-containing CM-Sepharose fraction was loaded onto a Resource Q (GM Healthcare) column equilibrated with 10 mM Tris–HCl pH 8.0 and 50 mM NaCl and then eluted with a gradient of NaCl. 55 kDa molecular-weight fractions, which were eluted at about 130 mM NaCl, were pooled and concentrated by Amicon Ultra with a 30 000 NMWL membrane. The final protein concentration was 3.8 mg ml−1 in buffer containing 10 mM Tris–HCl pH 8.0.
2.2. Initial crystallization screen
Initial screening for appropriate crystallization conditions for VAP1 and VAP2 was carried out using the sitting-drop vapour-diffusion method and Crystal Screen (Hampton Research, USA), with or without 63 µg ml−1 (almost twice the molar protein concentration) of the hydroxyamate inhibitor 3-(N-hydroxycarboxamide)-2-isobutyl-propanoyl-Trp-methylamide (GM6001, Calbiochem) in the protein solution. A volume of 0.3–0.5 µl protein solution was mixed with an equal amount of reservoir solution and droplets were allowed to equilibrate against 0.1 ml reservoir solution at 293 K.
2.3. Diffraction data collection
Crystals were cryoprotected, mounted in a nylon loop (Hampton Research, USA) or in a Lytho Loop (Protein Wave Corp., Japan) and immediately exposed to a stream of nitrogen gas at 100 K to flash-freeze the samples. The preliminary X-ray data were collected using an in-house X-ray diffractometer (Rigaku Micromax-007 X-ray generator with R-AXIS VII imaging-plate detector) and crystals that diffracted well were selected for data acquisition using the beamlines at SPring-8. All diffraction data sets were collected using undulator beamlines (BL41XU, BL45XU) at 100 K and diffraction images were processed using the HKL2000 software (Otwinowski & Minor, 1997 ▶).
3. Results
3.1. VAP1 crystals
3.1.1. Crystallization
VAP1 was reproducibly crystallized in two distinct crystal forms. Crystals were initially obtained using Crystal Screen solution No. 46, but these crystals diffracted poorly. Subsequently, droplets were prepared by mixing 1 µl protein solution and 1 µl reservoir solution containing 15% PEG 8000, 0.1 M sodium cacodylate pH 6.5 and then equilibrated against 1 ml reservoir solution. Within a couple of weeks, using the hanging-drop method, improved tetragonal crystals (form 1-1; Fig. 1 ▶ a) were obtained.
Figure 1.

VAP1 crystals. (a) Form 1-1. (b) Form 1-2. The scale bars indicate 0.1 mm.
Orthorhombic crystals (form 1-2; Fig. 1 ▶ b) were obtained using Additive Screen (Hampton Research, USA). The droplet was made by mixing 0.3 µl protein solution and 0.3 µl reservoir solution supplemented with one-fifth of the volume of 0.1 M cobalt(II) chloride (Additive Screen solution No. 4). The best crystals were obtained using the sitting-drop method after equilibration for 3 d against 0.1 ml of the same reservoir solution used to obtain form 1-1 crystals.
3.1.2. X-ray analysis
For X-ray measurements, crystals of either crystal form were soaked in a solution containing 15% PEG 8000, 5% methanol, 20% xylitol and 0.1 M sodium cacodylate pH 6.5 for cryoprotection prior to flash-freezing. X-ray diffraction data were obtained by the oscillation method using beamline BL45XU and an oscillation angle of 0.75° per image. Data sets were collected using a CCD detector (Rigaku Jupiter) for crystal form 1-1 or an imaging-plate detector (Rigaku R-AXIS V) for crystal form 1-2. The unit-cell parameters and the data statistics for the two crystal forms are summarized in Table 1 ▶. The structures were determined at 2.5 Å resolution by the molecular-replacement method using the P-I SVMP acutolysin-C (PDB code 1qua) as a starting model (Takeda et al., 2006 ▶). The coordinates and the structure factors have been deposited in the PDB (2erq for form 1-1 and 2ero for form 1-2 crystals).
Table 1. Data-collection statistics for VAP1 crystals.
Values in parentheses are for the highest resolution shell. For each data set, a single crystal was used for measurement.
| Form 1-1 | Form 1-2 | |
|---|---|---|
| Space group | P41212 | P212121 |
| Unit-cell parameters | ||
| a (Å) | 93.9 | 86.7 |
| b (Å) | 93.9 | 93.3 |
| c (Å) | 244.8 | 137.7 |
| α = β = γ (°) | 90 | 90 |
| Beamline (detector) | BL45PX (Rigaku Jupiter) | BL45PX (Rigaku R-AXIS V) |
| Wavelength (Å) | 0.98 | 1.0 |
| Resolution (Å) | 50–2.50 (2.59–2.50) | 50–2.50 (2.59–2.50) |
| No. of unique reflections | 38868 (3773) | 38926 (3800) |
| Rmerge† | 0.084 (0.380) | 0.072 (0.369) |
| I/σ(I) | 18.7 (7.1) | 14.4 (2.9) |
| Completeness (%) | 99.7 (99.6) | 99.4 (98.8) |
| Redundancy | 12.7 | 3.91 |
| No. of molecules in ASU | 1 | 1 |
| Matthews value (Å3 Da−1) | 2.5 | 2.5 |
| Solvent content (%) | 51 | 51 |
R
merge = 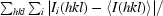
 , where I
i(hkl) is the ith intensity measurement of reflection hkl and 〈I(hkl)〉 is its average.
, where I
i(hkl) is the ith intensity measurement of reflection hkl and 〈I(hkl)〉 is its average.
3.2. VAP2 crystals
3.2.1. Crystallization
Five distinct crystal forms of VAP2 were analyzed by X-ray diffraction. The initial screening for VAP2 crystals was performed in the presence and absence of the inhibitor GM6001.
In the presence of GM6001, Crystal Screen solution No. 10 yielded crystals. With this as a starting condition, the pH of the mother liquor, the PEG concentration and molecular weight and the species and concentrations of salts and additives were optimized and four distinct crystal forms were obtained (forms 2-1, 2-2, 2-3 and 2-4). These four forms were only obtained in the presence of GM6001 and were never obtained in its absence. Monoclinic (form 2-1) and orthorhombic (form 2-2; Fig. 2 ▶ a) forms were obtained by the sitting-drop method under identical conditions as follows: droplets were made by mixing 0.5 µl protein solution with 0.5 µl reservoir solution containing 30% PEG 8000, 0.1 M ammonium acetate, 0.1 M sodium cacodylate pH 6.5 and were equilibrated against 0.1 ml reservoir solution. Tetragonal form crystals (form 2-3; Fig. 2 ▶ b) were obtained by adding a one-tenth volume of 1 M potassium chloride (Additive Screen solution No. 16) to the mother liquor and using a reservoir solution containing 30% PEG 8000, 0.1 M ammonium acetate, 0.1 M sodium acetate pH 4.6 with the same drop and reservoir volumes described above. Hexagonal crystals (form 2-4; Fig. 2 ▶ c) were obtained by the hanging-drop method using 1 ml of a reservoir solution containing 20% PEG 20 000, 0.2 M calcium acetate, 0.1 M sodium cacodylate pH 6.5. The droplet was made by mixing 1 µl protein solution and 1 µl reservoir solution supplemented with a one-fifth volume of 0.3 M glycyl-glycyl-glycine solution (Additive Screen solution No. 34).
Figure 2.

VAP2 crystals. (a) Form 2-2, (b) form 2-3, (c) form 2-4 and (d) form 2-5 crystals. The scale bars indicate 0.1 mm.
In the absence of GM6001, crystals were obtained with Crystal Screen solution No. 46, but these crystals yielded poor diffraction data. To improve the quality of the crystals, several additives were screened. Monoclinic crystals (form 2-5; Fig. 2 ▶ d) were obtained by adding a one-tenth volume of 40% n-propanol solution (Additive Screen solution No. 90) to the reservoir solution (final composition 4% n-propanol, 16.2% PEG 8000, 0.18 M calcium acetate, 0.09 M sodium cacodylate pH 6.5). A mixture of 0.5 µl protein solution and 0.5 µl reservoir solution was equilibrated against 0.1 ml reservoir solution. These form 2-5 crystals were only obtained in the absence of GM6001 and were never obtained in its presence.
3.2.2. X-ray analysis
The mother liquors of the form 2-2 and 2-3 crystals were suitable for freezing; all others were first cryoprotected. For form 2-1 and 2-4 crystals, 20% glycerol was added to the reservoir solution for cryoprotection. For form 2-1, the cryogenic solution was added gradually to the crystal droplet in order to avoid cracking induced by osmotic shock. Crystal form 2-5 was rinsed in a solution containing 15% PEG 8000, 5% methanol, 20% xylitol and 0.1 M sodium cacodylate pH 6.5 and then immediately flash-frozen at 100 K. Because these crystals were extremely thin and fragile, they were mounted in a LithoLoop, an etched Mylar film, to prevent bending of the crystal.
All diffraction data sets for the VAP2 crystals were acquired using the oscillation method and beamline BL41XU (the oscillation angle was 1.0° for all data sets) at a wavelength of 1.0 Å and data were collected using an ADSC Quantum 310R detector. The unit-cell parameters and statistics for the data sets are summarized in Table 2 ▶. The estimated number of molecules in the asymmetric unit for each crystal form was obtained by a preliminary molecular-replacement method using MOLREP from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶) and the metalloproteinase (M) and cysteine-rich (C) domains of VAP1 (Takeda et al., 2006 ▶) as the starting models. Structural analyses of these crystals along with the molecular-replacement phases are ongoing.
Table 2. Data-collection statistics for VAP2 crystals.
Values in parentheses are for the highest resolution shell. For each data set, a single crystal was used for measurement.
| Form 2-1 | Form 2-2 | Form 2-3 | Form 2-4 | Form 2-5 | |
|---|---|---|---|---|---|
| GM6001 | + | + | + | + | − |
| Space group | P21 | P212121 | P41 | P6322 | C2 |
| Unit-cell parameters | |||||
| a (Å) | 56.9 | 57.7 | 60.7 | 156.8 | 220.7 |
| b (Å) | 138.0 | 118.2 | 60.7 | 156.8 | 79.5 |
| c (Å) | 59.2 | 138.5 | 257.9 | 95.6 | 58.7 |
| α (°) | 90 | 90 | 90 | 90 | 90 |
| β (°) | 91.5 | 90 | 90 | 90 | 91.7 |
| γ (°) | 90 | 90 | 90 | 120 | 90 |
| Beamline (detector) | BL41XU (ADSC Quantum 310R CCD detector) | ||||
| Wavelength (Å) | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| Resolution (Å) | 50–2.15 (2.23–2.15) | 50–2.50 (2.59–2.50) | 50–3.20 (3.31–3.2) | 50–3.80 (3.94–3.80) | 50–2.70 (2.80–2.70) |
| No. of unique reflections | 48664 (4428) | 33288 (2925) | 15097 (1437) | 7169 (682) | 26911 (2313) |
| Rmerge† | 0.081 (0.196) | 0.089 (0.321) | 0.091 (0.360) | 0.117 (0.397) | 0.085 (0.231) |
| I/σ(I) | 9.8 (4.6) | 10.3 (3.7) | 10.9 (4.0) | 8.4 (6.5) | 10.1 (5.5) |
| Completeness (%) | 98.1 (89.5) | 98.6 (88.4) | 99.5 (95.7) | 99.8 (99.9) | 95.9 (82.5) |
| Redundancy | 3.3 | 6.5 | 7.0 | 19.2 | 3.4 |
| No. of molecules in ASU | 2 | 2 | 2 | 1 | 2 |
| Matthews value (Å3 Da−1) | 2.4 | 2.4 | 2.5 | 3.1 | 2.7 |
| Solvent content (%) | 49 | 49 | 50 | 60 | 54 |
R
merge =
, where I
i(hkl) is the ith intensity measurement of reflection hkl and 〈I(hkl)〉 is its average.
Acknowledgments
We thank Mariko Tomisako for her help in crystallization experiments and the staff of SPring-8 for assistance with data acquisition. This work was partly supported by Grant Nano-001 for Research on Advanced Medical Technology from the Ministry of Health, Labour and Welfare of Japan and by grants from the Takeda Science Foundation, from the Kao Foundation for Arts and Science and from the Senri Life Science Foundation.
References
- Araki, S., Ishida, T., Yamamoto, T., Kaji, K. & Hayashi, H. (1993). Biochem. Biophys. Res. Commun.190, 148–153. [DOI] [PubMed] [Google Scholar]
- Blobel, C. P. (2005). Nature Rev. Mol. Cell Biol.6, 32–43. [DOI] [PubMed]
- Bode, W., Gomis-Ruth, F. X. & Stockler, W. (1993). FEBS Lett.331, 134–140. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Duffy, M. J., Lynn, D. J., Lloyd, A. T. & O’Shea, C. M. (2003). Thromb. Haemost.89, 622–631. [PubMed] [Google Scholar]
- Fox, J. W. & Serrano, S. M. (2005). Toxicon, 45, 969–985. [DOI] [PubMed] [Google Scholar]
- Gutierrez, J. M., Rucavado, A., Escalante, T. & Diaz, C. (2005). Toxicon, 45, 997–1011. [DOI] [PubMed] [Google Scholar]
- Maruyama, J., Hayashi, H., Miao, J., Sawada, H. & Araki, S. (2005). Toxicon, 46, 1–6. [DOI] [PubMed] [Google Scholar]
- Masuda, S., Araki, S., Yamamoto, T., Kaji, K. & Hayashi, H. (1997). Biochem. Biophys. Res. Commun.235, 59–63. [DOI] [PubMed] [Google Scholar]
- Masuda, S., Hayashi, H. & Araki, S. (1998). Eur. J. Biochem.253, 36–41. [DOI] [PubMed] [Google Scholar]
- Masuda, S., Hayashi, H., Atoda, H., Morita, T. & Araki, S. (2001). Eur. J. Biochem.268, 3339–3345. [DOI] [PubMed] [Google Scholar]
- Masuda, S., Ohta, T., Kaji, K., Fox, J. W., Hayashi, H. & Araki, S. (2000). Biochem. Biophys. Res. Commun.278, 197–204. [DOI] [PubMed] [Google Scholar]
- Moss, M. L. & Bartsch, J. W. (2004). Biochemistry, 43, 7227–7235. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Seals, D. F. & Courtneidge, S. A. (2003). Genes Dev.17, 7–30. [DOI] [PubMed] [Google Scholar]
- Takeda, S., Igarashi, T., Mori, H. & Araki, S. (2006). EMBO J.25, 2388–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trummal, K., Tonismagi, K., Siigur, E., Aaspollu, A., Lopp, A., Sillat, T., Saat, R., Kasak, L., Tammiste, I., Kogerman, P., Kalkkinen, N. & Siigur, J. (2005). Toxicon, 46, 46–61. [DOI] [PubMed] [Google Scholar]
- White, J. M. (2003). Curr. Opin. Cell Biol.15, 598–606. [DOI] [PubMed] [Google Scholar]
- You, W. K., Seo, H. J., Chung, K. H. & Kim, D. S. (2003). J. Biochem. (Tokyo), 134, 739–749. [DOI] [PubMed] [Google Scholar]