

Diffraction data from E. coli RNase II crystals of wild type and of an inactive mutant and its SeMet-derivative form were obtained to 2.44 and 2.74 Å resolution, providing a set of preliminary phases. An improved purification protocol allowed higher reproducibility in the crystallization of the mutant form.
Keywords: RNase II, RNA degradation
Abstract
RNA degradation is important in the post-transcriptional control of gene expression. The processing, degradation and quality control of RNA is performed by many different classes of ribonucleases. Ribonuclease II (RNase II) is a 643-amino-acid enzyme that degrades single-stranded RNA from its 3′-end, releasing ribonucleoside 5′-monophosphates. RNase II was expressed both as the wild type and as a D209N mutant form. The latter was also produced as an SeMet derivative. The various protein forms were crystallized using the vapour-diffusion method. Wild-type RNase II was crystallized in two crystal forms, both of which belonged to space group P21. X-ray diffraction data were collected to 2.44 and 2.75 Å resolution, with unit-cell parameters a = 56.8, b = 125.7, c = 66.2 Å, β = 111.9° and a = 119.6, b = 57.2, c = 121.2 Å, β = 99.7°, respectively. The RNase II D209N mutant gave crystals that belonged to space group P65, with unit-cell parameters a = b = 86.3, c = 279.2 Å, and diffracted to 2.74 Å. Diffraction data from the mutant and its SeMet derivative enabled the determination of a partial Se-atom substructure by SIRAS.
1. Introduction
The levels of mRNA in a cell are a function of its synthesis and degradation. mRNA degradation is the result of the combined action of endonucleases and exonucleases (Regnier & Arraiano, 2000 ▶; Arraiano & Maquat, 2003 ▶). In Escherichia coli, RNase II and PNPase are the two main 3′ to 5′ exoribonucleases. RNase II is an Mg2+-dependent hydrolase that degrades single-stranded RNA, creating ribonucleoside 5′-monophosphate products. RNase II is the prototype of the RNB superfamily of exoribonucleases (Mian, 1997 ▶) acting as a non-specific ribonuclease that degrades single-stranded RNA processively. When the length of the single-stranded RNA substrate reaches ∼10 nucleotides it becomes more distributive and finally leaves an undigested core of 3–5 nucleotides. A single mutation in E. coli ribonuclease II has been shown to inactivate the enzyme without affecting RNA binding. This mutation, D209N, is at a highly conserved residue that has been proposed to be involved in metal-ion binding (Amblar & Arraiano, 2005 ▶). Although the mutant is totally inactive, it still retains the ability to bind the substrate (Amblar & Arraiano, 2005 ▶). Here, we describe the expression and purification of RNase II D209N and its SeMet derivative and the crystallization, heavy-atom derivatization attempts and preliminary diffraction data characterization of both wild-type (wt) and D209N mutant forms of RNase II.
2. Materials and methods
2.1. Protein expression and purification
RNase II was overexpressed in E. coli BL21 (DE3) and purified as previously described (Amblar & Arraiano, 2005 ▶): the protein was concentrated to 8.5 mg ml−1 using an Amicon Ultra centrifugal filter (molecular-weight cutoff 30 kDa; Millipore) in the presence of 3% glycerol, 1 mM MgCl2, 100 mM KCl and 100 mM Tris–HCl pH 8.0. The methods for expression and purification of the RNase II D209N mutant followed similar procedures, with the modifications outlined below.
For expression of RNase II D209N, the plasmid pMAA (Amblar & Arraiano, 2005 ▶) was transformed into E. coli strain BL21 (DE3) (Novagen). Cultures were grown in LB media supplemented with 150 µg ml−1 ampicillin at 310 K until an OD600 of 0.4 was reached. Expression was induced by the addition of 1 mM IPTG and the cultures were grown for a further 2 h at 310 K.
Selenomethionine-derivatized D209N protein was produced by transforming the pMAA plasmid into the auxotrophic strain E. coli B834 (DE3). Cells were cultured in SelenoMet Minimal Media (Molecular Dimensions Ltd) according to the manufacturer’s instructions with the introduction of the following modifications. Briefly, a single colony was initially grown in LB medium (for about 8 h). 10 ml SelenoMet Minimal Medium containing l-methionine and Nutrient Mix (Molecular Dimensions Ltd) and supplemented with 150 µg ml−1 ampicillin was then inoculated with the culture in a 1:20 ratio. This new culture was grown overnight and centrifuged at 5000g and the pellet was washed with sterile minimal medium in order to eliminate any residual methionine. The pellet was then used to inoculate a 500 ml flask of SelenoMet minimal medium containing l-selenomethionine as directed by the manufacturer. The culture was grown to the point of induction, OD600 = 0.65, which was followed by addition of IPTG to 0.5 mM and growth for a further 1–2 h at 310 K.
Both native and selenomethionine-derivatized D209N proteins were purified as follows. The cells were harvested by centrifugation at 10 000g for 30 min and the bacterial pellets were resuspended in 500 mM NaCl and 50 mM Tris–HCl pH 8.0 (buffer A) plus 1 mM PMSF and 20 mM imidazole and the cells were lysed using a French press at 131 MPa. The cell lysate was incubated on ice for 1 h with 100 U benzonase (Novagen), the sample was clarified by centrifugation at 20 000g for 30 min and the supernatant was loaded onto a HisTrap-Chelating HP column (Amersham Biosciences). After washing with ten column volumes of buffer A plus 20 mM imidazole, the protein was gradient eluted with buffer A plus 500 mM imidazole. Fractions containing pure protein were selectively pooled and DTT was added to 1 mM; peaks I, II and III were pooled separately (Fig. 1 ▶). The purified protein was concentrated to 10 mg ml−1 in 20 mM Tris–HCl pH 8, 150 mM NaCl, 10% glycerol and 1 mM DTT.
Figure 1.

Expression and purification of the RNase II D209N mutant. (a) HisTrap chromatogram of RNase II D209N. (b) SDS–PAGE, 12% gel. The following samples were applied onto a 12% SDS–PAGE and stained with Coomassie blue: lane 1, molecular-weight markers; lane 2, whole cell homogenate from IPTG-induced cultures; lanes 3–4, flowthrough of unbound protein; lanes 5–9, fractionated protein after Ni–NTA purification. The IPTG-inducible band runs at the expected molecular weight of 74 kDa and can be purified to reasonable homogeneity by selective fractionation. The molecular weights of the markers are indicated in kDa on the left.
In anticipation that the 6×His tag at the N-terminus might interfere with crystal packing, it was removed in some of the batches. Cleavage took place using biotinylated thrombin in a cleavage capture kit (Novagen). Digestion was performed by overnight incubation at 293 K with 2.5 U thrombin per milligram of pure protein. Thrombin was subsequently removed using immobilized streptavidin beads, as described in the manufacturer’s instructions.
2.2. Crystallization
2.2.1. Wild-type RNase II
Crystallization trials were performed by the sitting-drop vapour-diffusion method using Hampton Research Crystal Screen 1 and Natrix 1 and Molecular Dimensions Crystal Strategy Screens 1 and 2. Crystals of purified RNase II were grown, with or without the His tag, in 24-well Linbro plates, mixing 1.5 µl protein solution and 1.5 µl reservoir solution and equilibrating the drops against 500 µl reservoir solution. Optimized crystals from initial batches of the wild-type (wt) enzyme, hereby called P21 form A crystals, were obtained as thin plates with dimensions of up to 0.4 × 0.15 × 0.08 mm in 22% PEG 4K, 0.6 M calcium acetate and 0.1 M Tris–HCl pH 8.5 at 293 K (Fig. 2 ▶ a). Subsequent batches were concentrated to 12 mg ml−1 in the presence of 2% glycerol, 100 mM NaCl, 20 mM Tris–HCl pH 7. These produced a different crystal form, referred to here as P21 form B, with dimensions of 0.1 × 0.05 × 0.02 mm and with optimized crystallization conditions of 25% PEG 8K and 0.2 M HEPES pH 7.5 at 277 K. Production of heavy-atom derivatives was attempted by soaking crystals for 1–2 d in crystallization solution complemented with metal salts and back-soaking the crystals for 3 min in crystallization solution containing 25%(v/v) glycerol. Alternatively, derivatization with heavy halides was also attempted by soaking the crystals for 1–2 min in crystallization solution complemented with halides and 25%(v/v) glycerol. Crystals soaked with salts of Pt, Au, I and Br were flash-cooled in liquid nitrogen and their diffraction examined and eventually collected if suitable diffraction resolution was achieved. Diffraction data sets were obtained from putative derivatives of Pt, Au and I, in addition to native data.
Figure 2.

RNase II crystals. (a) Thin plate-shaped crystals of wild-type RNase II from E. coli with dimensions 0.4 × 0.15 × 0.04 mm, crystallized in space group P21 (crystal form A). (b) Crystals of RNase II D209N with dimensions of 0.25 × 0.1 × 0.1 mm, crystallized in space group P65.
2.2.2. RNase II D209N
Initial crystallization conditions were screened with a nanodrop crystallization robot (Cartesian Technologies) and the standard conditions of the EMBL Partnership for Structural Biology High-Throughput Crystallization Robot (EMBL, Grenoble) using the sitting-drop vapour-diffusion method. Each drop was prepared by mixing 100 nl protein solution with an equal amount of crystallization solution and equilibrated by vapour diffusion against 100 µl crystallization solution in the reservoir at 293 K. Crystals were initially obtained in both 2.4 M ammonium sulfate pH 6–9 and 1.9–2.4 M sodium malonate pH 6–7 screens (Hampton Research). Such crystals grew to dimensions of 0.1 × 0.06 × 0.06 mm and diffracted to 3.5 Å resolution (data not shown).
After selective fractionation during the purification step (Fig. 1 ▶) and without removal of the N-terminal His tag, single crystals were obtained at 293 K both in 2.3–2.5 M ammonium sulfate, 0.1 M MES pH 6–9 or in 2.2–2.4 M sodium malonate pH 6–9 using sitting-drop vapour diffusion. However, crystallization only took place for pooled fractions I and II; even with further purification pooled fraction III proved recalcitrant to crystallization. Crystals for X-ray analysis were grown using the sitting-drop vapour-diffusion method by mixing 1.5 µl protein solution with 1.5 µl reservoir solution (2.4 M sodium malonate pH 6.0) and equilibrating against 500 µl reservoir solution at 293 K (Fig. 2 ▶ b). They were harvested in 2.5 M sodium malonate pH 6.0, which proved to be an adequate cryoprotectant. This procedure, in particular the chromatographic step, led to reproducible crystals, prompting a screen of heavy-atom derivatives by soaking crystals in metal-salt solutions (5 mM) for 2 d in the harvesting solution. Crystals were then back-soaked for 2 min in 2.5 M sodium malonate pH 6.0 and flash-cooled in liquid nitrogen. Diffraction data were collected from the native mutant, its SeMet derivative and other putative Hg, Au and Sm derivatives.
2.3. Crystallographic characterization and diffraction data collection, processing and analysis
X-ray data were collected at cryotemperatures using monochromatic synchrotron radiation (see Table 1 ▶ for further details). Data were indexed, processed and scaled with the HKL package (Otwinowski & Minor, 1997 ▶). The graphical user interface HKL2MAP (Pape & Schneider, 2004 ▶) was used with SHELXC (G. M. Sheldrick, personal communication) to scale and analyse isomorphous and/or anomalous data, using SHELXD (Schneider & Sheldrick, 2002 ▶) to locate heavy-atom substructures and SHELXE (Sheldrick, 2002 ▶) to validate the obtained solutions and produce experimental maps, which were examined with Xfit (McRee, 1999 ▶). Using the anomalous differences between RNase II D209N and its SeMet derivative, an Se substructure and the corresponding experimental maps were obtained by SIRAS (Fig. 3 ▶). Although the electron-density maps still do not allow a clear assignment of the polypeptide chain, one can recognize potential protein and solvent regions.
Table 1. Statistics of diffraction data collection.
Values in parentheses are for the highest resolution shell.
| RNase II, form A | RNase II, form B | RNase II D209N | RNase II D209N, Se derivative | |
|---|---|---|---|---|
| X-ray source | ESRF, ID13 | ESRF, ID14-2 | ESRF, ID14-2 | ESRF, ID14-2 |
| Temperature (K) | 100.0 | 100.0 | 100.0 | 100.0 |
| Space group | P21 | P21 | P65 | P65 |
| Unit-cell parameters (Å, °) | a = 56.8, b = 125.7, c = 66.2, β = 111.9 | a = 119.6, b = 57.2, c = 121.2, β = 99.7 | a = b = 86.3, c = 279.2 | a = b = 86.3, c = 278.5 |
| Wavelength (Å) | 0.975 | 0.933 | 0.933 | 0.933 |
| Crystal mosaicity (°) | 0.29–0.81 | 0.46–0.70 | 0.23–0.31 | 0.20–0.44 |
| Resolution (Å) | 43.94–2.44 (2.53–2.44) | 59.10–2.75 (3.85–2.75) | 51.00–2.74 (2.85–2.74) | 58.20–3.50 (3.56–3.50) |
| Data completeness (%) | 98.9 (98.2) | 97.1 (94.4) | 99.9 (100.0) | 99.9 (100.0) |
| No. of unique reflections | 31569 | 40858 | 30737 | 14887 |
| Redundancy | 7.3 | 3.0 | 9.2 | 17.3 |
| No. of rejected outliers | 2607 [1.1%] | 817 [2.0%] | 336 [0.1%] | 166 [0.06%] |
| I/σ(I) | 23.4 (5.2) | 13.6 (1.8) | 28.6 (3.4) | 23.5 (8.4) |
| Rp.i.m.† (%) | 3.2 (17.4) | 4.6 (32.6) | 2.5 (24.8) | 3.5 (11.0) |
| Rr.i.m.‡ (%) | 8.7 (46.3) | 8.2 (57.8) | 8.0 (70.0) | 14.7 (44.5) |
| Rsym§ (%) | 8.0 (42.3) | 6.8 (52.6) | 7.6 (64.2) | 14.1 (43.7) |
R
p.i.m. = 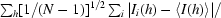
 , where I is the observed intensity and 〈I〉 is the average intensity of multiple observations of symmetry-related reflections. Calculated with the program RMERGE (Weiss, 2001 ▶).
, where I is the observed intensity and 〈I〉 is the average intensity of multiple observations of symmetry-related reflections. Calculated with the program RMERGE (Weiss, 2001 ▶).
R
r.i.m. =  , where I is the observed intensity and 〈I〉 is the average intensity of multiple observations of symmetry-related reflections. Calculated with the program RMERGE (Weiss, 2001 ▶).
, where I is the observed intensity and 〈I〉 is the average intensity of multiple observations of symmetry-related reflections. Calculated with the program RMERGE (Weiss, 2001 ▶).
R
sym = 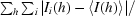 , where I is the observed intensity and 〈I〉 is the average intensity of multiple observations of symmetry-related reflections. Calculated with SCALEPACK (Otwinowski & Minor, 1997 ▶).
, where I is the observed intensity and 〈I〉 is the average intensity of multiple observations of symmetry-related reflections. Calculated with SCALEPACK (Otwinowski & Minor, 1997 ▶).
Figure 3.

RNase II D209N SIRAS statistics. (a) As a zero signal, 〈d′′/sig〉 is about 0.8 (SHELXC output documentation), the SeMet diffraction data (blue) is shown to contain anomalous signal until its resolution cutoff, in contrast to the mutant native data (red). (b) The Se site-occupancy profile indicates that the asymmetric unit contains only one RNase II molecule, as RNase II contains 16 Met residues in its sequence and residual occupancies of 0.2 indicate noise peaks (Schneider & Sheldrick, 2002 ▶). (c) The space-group ambiguity was solved by SHELXE as the density-modification ‘map contrast’ statistics (Sheldrick, 2002 ▶) assuming space group P65 (red) clearly converged to a higher value when compared with that of its hand-inverted counterpart P61 (blue).
3. Discussion and conclusions
For the inactive mutant RNase II D209N, purification by selective fractionation of the protein in the gradient elution provided a source of homogeneous protein that enabled improved reproducibility of crystals relative to the wild-type forms, which had been purified using a previous protocol (Amblar & Arraiano, 2005 ▶). The heterogeneity shown in the elution peak suggests the possibility of RNA binding during the purification step of the D209N mutant. In terms of crystal reproducibility and the final quality of the different crystal forms of RNase II, we could not establish any correlation with protein batches that included or excluded the N-terminal 6×His tag.
Two monoclinic crystal forms of wt RNase II and a hexagonal form of its D209N mutant were obtained. Attempts to solve the phase problem using any of the monoclinic native data sets in conjunction with putative heavy-atom derivatives have remained unsuccessful to date. The RNase II D209N mutant produced crystals belonging to space group P61 or P65. The diffraction data of the mutant and its SeMet derivative enabled the determination of an Se substructure and a set of experimental phases by SIRAS (Fig. 3 ▶). As the SeMet-derivative data were collected for phasing purposes, the resolution was cut off at 3.5 Å, near its anomalous signal reliability limit (Fig. 3 ▶ a). The number of sites in the heavy-atom substructure shows that the crystal contains one protein molecule in the asymmetric unit (Fig. 3 ▶ b), corresponding to a calculated solvent content of 65%. The hexagonal space-group ambiguity was solved by SHELXE, as both the ‘map contrast’ (Fig. 3 ▶ c) and ‘map connectivity’ statistics calculated for space group P65 clearly converge to higher values than those calculated for the alternative P61 space-group possibility. Additionally, the ‘map connectivity’ converged to 0.915, indicating that the phase problem may have been solved as it is above the 0.9 threshold (Sheldrick, 2002 ▶). Improvements of the solution of the phase problem in order to obtain the RNase II structure are in progress.
Acknowledgments
We would like to thank the staff of the High-Throughput Crystallization Laboratory of the EMBL Grenoble Outstation for crystallization screens and the ESRF maintenance and operation of the beamlines the staff at beamlines ID-13 and ID-14 for technical support. This work was supported by grants from Fundação para a Ciência e Tecnologia.
References
- Amblar, M. & Arraiano, C. M. (2005). FEBS J.272, 363–374. [DOI] [PubMed] [Google Scholar]
- Arraiano, C. M. & Maquat, L. E. (2003). Mol. Microbiol.49, 267–276. [DOI] [PubMed] [Google Scholar]
- McRee, D. E. (1999). J. Struct. Biol.125, 156–165. [DOI] [PubMed] [Google Scholar]
- Mian, I. S. (1997). Nucleic Acids Res.25, 3187–3195. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pape, T. & Schneider, T. R. (2004). J. Appl. Cryst.37, 843–844. [Google Scholar]
- Regnier, P. & Arraiano, C. M. (2000). Bioessays, 22, 235–244. [DOI] [PubMed] [Google Scholar]
- Schneider, T. R. & Sheldrick, G. M. (2002). Acta Cryst. D58, 1772–1779. [DOI] [PubMed] [Google Scholar]
- Sheldrick, G. M. (2002). Z. Kristallogr.217, 644–650.
- Weiss, M. S. (2001). J. Appl. Cryst.34, 130–135. [Google Scholar]