

BphA3, a Rieske-type [2Fe–2S] ferredoxin, was crystallized by the hanging-drop vapour-diffusion method. A molecular-replacement calculation yielded a satisfactory solution.
Keywords: ferredoxins, electron transfer, Rieske-type [2Fe–2S] clusters
Abstract
BphA3, a Rieske-type [2Fe–2S] ferredoxin component of a biphenyl dioxygenase (BphA) from Pseudomonas sp. strain KKS102, was crystallized by the hanging-drop vapour-diffusion method. Two crystal forms were obtained from the same conditions. The form I crystal belongs to space group P21212, with unit-cell parameters a = 26.3, b = 144.3, c = 61.5 Å, and diffracted to 2.45 Å resolution. The form II crystal belongs to space group P212121, with unit-cell parameters a = 26.2, b = 121.3, c = 142.7 Å, and diffracted to 2.8 Å resolution. A molecular-replacement calculation using BphF as a search model yielded a satisfactory solution for both forms.
1. Introduction
The [2Fe–2S] clusters of ferredoxin components in aromatic ring-hydroxylating dioxygenases have a similar chemical structure to the Rieske proteins of the cytochrome bc 1 and cytochrome b 6 f complexes (Mason & Cammack, 1992 ▶). These [2Fe–2S] ferredoxins were therefore designated as Rieske-type ferredoxins. The [2Fe–2S] cluster of these proteins is coordinated by two His and two Cys residues.
BphA3 (MW 11.7 kDa, 109 amino acids) is a Rieske-type ferredoxin component of the biphenyl dioxygenase (BphA) derived from Pseudomonas sp. strain KKS102 (Kimbara et al., 1989 ▶). BphA is a multi-component enzyme composed of BphA1A2, BphA3 and BphA4. BphA1A2 is the terminal oxygenase component, catalyzing the addition of two hydroxyl groups to a biphenyl benzene ring. BphA1A2 requires two electrons for its catalytic activity. BphA4, an NADH-dependent ferredoxin reductase, and BphA3 constitute an electron-transfer system and transfer electrons from NADH to BphA1A2. Electron transfer of BphA begins with a hydride transfer from NADH to FAD in BphA4, resulting in two-electron-reduced BphA4. BphA3 then receives one electron from the reduced BphA4 and transfers the electron to BphA1A2. Because BphA3 shuttles between BphA4 and BphA1A2, redox-dependent affinity regulation of selection of a binding partner seems to play an important role in the electron transfer. It has been proposed that redox-linked conformational changes of ferredoxins are important in altering their affinity for their electron-acceptor proteins (Sevrioukova, 2005 ▶). In order to elucidate the molecular mechanism of affinity regulation in the electron transfer, we have studied the structure–function relationship of each component of BphA. We have thus far determined the crystal structures of BphA4 and BphA1A2 and have proposed their BphA3-interacting surfaces on the basis of their crystal structures (Senda et al., 2000 ▶; Furusawa et al., 2004 ▶). The crystal structure of BphA3 will therefore provide deeper insight into the molecular mechanism of affinity regulation in the electron-transfer system. Here, we report the crystallization of BphA3.
2. Methods and results
2.1. Protein purification and crystallization
BphA3 derived from Pseudomonas sp. strain KKS102 (hereafter referred to as BphA3) was overexpressed using Escherichia coli (BL21) and purified as described previously (Kimura et al., 2005 ▶). Briefly, BL21 containing the pCA3 expression plasmid was cultured at 310 K for 30 h after IPTG induction. The overexpressed BphA3 was purified by anion-exchange and gel-filtration chromatography. The purified BphA3 was the oxidized form, which was confirmed by the absorption spectrum, which showed the two characteristic peaks (325 and 460 nm) of the oxidized form of BphA3 (Kimura et al., 2005 ▶). The BphA3 was concentrated to 15.3 mg ml−1 in 10 mM potassium phosphate buffer pH 7.0, 1 mM dithiothreitol and 2%(w/v) glycerol and used for crystallization screening. The initial screening was performed by the hanging-drop vapour-diffusion method using Crystal Screens I and II (Hampton Research) at 293 K. A hanging drop was prepared by mixing 1.0 µl each of the protein and reservoir solutions and was equilibrated against 500 µl reservoir solution. The reservoir solution did not contain 2%(w/v) glycerol. Spherulites appeared from Crystal Screen II condition No. 28 (1.6 M sodium citrate pH 6.5) in 3 d. However, optimization of this condition did not yield a diffraction-quality crystal. After 1 y, we noticed that microcrystals had appeared from another condition, Crystal Screen I condition No. 4 (2.0 M ammonium sulfate, 0.1 M Tris–HCl pH 8.5). However, it was unclear when the crystals had appeared. This condition was optimized, resulting in the growth of diffraction-quality crystals. The best crystal was obtained by mixing 1.0 µl BphA3 (13.0 mg ml−1) and 1.0 µl optimized reservoir solution (2.55 M ammonium sulfate and 0.1 M Tris–HCl pH 8.5) at 277 K (Fig. 1 ▶). Needle-shaped crystals (approximately 0.2 × 0.02 × 0.02 mm) were obtained in three months. The most important factor in the optimization seemed to be the temperature. We obtained only very thin crystals at 293 K even when the same optimized reservoir solution was used.
Figure 1.

Crystal of BphA3 from Pseudomonas sp. strain KKS102 (form I).
2.2. X-ray data collection and processing
For X-ray diffraction measurements at 100 K, the crystals were cryoprotected by adding 4 µl of reservoir solution containing 30%(w/v) trehalose to the droplet. After 2–5 min, the crystals were flash-cooled in liquid nitrogen. Diffraction data were collected using an ADSC Quantum 210 CCD detector at beamline NW12 of the Photon Factory (Tsukuba, Japan). The diffraction data were processed and scaled using the programs DENZO and SCALEPACK as implemented in the HKL2000 package (Otwinowski & Minor, 1997 ▶). Data processing revealed that two different crystal forms, forms I and II, were obtained from the same crystallization conditions (Table 1 ▶). The Matthews coefficients (V M; Matthews, 1968 ▶) for crystal forms I and II were calculated to be 2.5 Å3 Da−1, corresponding to a solvent content of 50.3%, and 2.4 Å3 Da−1, corresponding to a solvent content of 48.8%, respectively. The data-collection statistics are given in Table 1 ▶.
Table 1. Data-collection statistics.
Values in parentheses are for the outermost resolution shell.
| Crystal | Form I | Form II |
|---|---|---|
| Wavelength (Å) | 0.97799 | 0.97800 |
| Space group | P21212 | P212121 |
| Unit-cell parameters (Å) | a = 26.3, b = 144.3, c = 61.5 | a = 26.2, b = 121.3, c = 142.7 |
| Resolution limits (Å) | 50.0–2.45 (2.54–2.45) | 50.0–2.70 (2.80–2.70) |
| Observations | 57202 | 69252 |
| Unique reflections | 9119 | 13423 |
| Completeness (%) | 98.2 (81.6) | 99.4 (99.5) |
| Average I/σ(I) | 8.8 (3.9) | 6.2 (3.3) |
| Average redundancy | 6.3 (4.6) | 5.1 (5.0) |
| Rsym† | 0.119 (0.424) | 0.131 (0.505) |
R
sym = 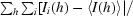
 , where I
i is the ith measurement and 〈I(h)〉 is the weighted mean of all measurements of I(h).
, where I
i is the ith measurement and 〈I(h)〉 is the weighted mean of all measurements of I(h).
2.3. Structure determination
The crystal structure of BphA3 was determined by the molecular-replacement method using the program MOLREP (Vagin & Teplyakov, 1997 ▶) from the CCP4 program suite (Collaborative Computational Project, Number 4, 1994 ▶). A molecular-replacement calculation using BphF (Colbert et al., 2000 ▶) as a search model yielded a satisfactory solution for both crystal forms. BphF has 71% amino-acid sequence identity to BphA3. As predicted from the V M values, form I and II crystals have two and four BphA3 molecules in the asymmetric unit, respectively. Packing analysis showed that there were no close contacts between the subunits in both cases. We collected 2.0 Å resolution diffraction data from a form I crystal at BL5A of the Photon Factory (Fig. 2 ▶). Crystallographic refinement at 2.0 Å resolution is in progress.
Figure 2.

X-ray diffraction pattern of the BphA3 crystal (crystal form I). The diffraction data were collected at beamline BL5A of the Photon Factory using an ADSC Quantum 315 CCD detector.
Acknowledgments
We thank Professor M. Fukuda for his continuous support of this work. This study was supported in part by the New Energy and Trial Technology Development Organization (NEDO) of Japan.
References
- Colbert, C. L., Couture, M. M., Eltis, L. D. & Bolin, J. T. (2000). Structure, 8, 1267–1278. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Furusawa, Y., Nagarajan, V., Tanokura, M., Masai, E., Fukuda, M. & Senda, T. (2004). J. Mol. Biol.342, 1041–1052. [DOI] [PubMed] [Google Scholar]
- Kimbara, K., Hashimoto, T., Fukuda, M., Koana, T., Takagi, M., Oishi, M. & Yano, K. (1989). J. Bacteriol.171, 2740–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, S., Kikuchi, A., Senda, T., Shiro, Y. & Fukuda, M. (2005). Biochem. J.388, 869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason, J. R. & Cammack, R. (1992). Annu. Rev. Microbiol.46, 277–305. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Senda, T., Yamada, T., Sakurai, N., Kubota, M., Nishizaki, T., Masai, E., Fukuda, M. & Mitsui, Y. (2000). J. Mol. Biol.304, 397–410. [DOI] [PubMed] [Google Scholar]
- Sevrioukova, I. F. (2005). J. Mol. Biol.347, 607–621. [DOI] [PubMed] [Google Scholar]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025. [Google Scholar]