

Abstract
Objective
Toll-like receptors (TLRs), a group of pathogen-associated microbial pattern recognition receptors, play an important role in innate immune signaling and are differentially regulated in chronic inflammatory diseases such as atherosclerosis. However, the involvement of TLRs in the progression of atherosclerosis is still unclear.
Methods and Results
TLR2 and apolipoprotein E double knockout (Tlr2−/−Apoe−/−) mice were generated and the progressive formation of atherosclerotic plaque in the aortas was examined in mice fed a normal chow diet. We demonstrate that inactivation of TLR2 resulted in reduced progression of atherosclerosis in both male and female Apoe−/− mice. Likewise, TLR2 deficiency resulted in a reduction in lipid accumulation and decreased macrophage recruitment to the aortic sinus as well as reduced monocyte chemoattractant protein-1 (MCP-1) levels. Furthermore, macrophages isolated from Tlr2−/−Apoe−/− mice demonstrated significantly reduced MCP-1 production upon stimulation with a TLR2 ligand. However, no differences in acetylated-low-density lipoprotein uptake and foam cell formation were observed in macrophages isolated from Tlr2−/−Apoe−/− mice as compared to Apoe−/− mice.
Conclusions
TLR2 plays a critical role in the progression of atherosclerosis in Apoe−/− mice, which is independent of dietary lipids and macrophage lipid uptake.
Keywords: atherosclerosis, Toll-like receptor, inflammation, macrophages
Atherosclerosis, formerly considered a lipid storage disease, actually involves ongoing inflammatory responses. Inflammation in the arterial vessel wall is considered to play an important role in the pathogenesis of atherosclerosis1, 2. Indeed, recent advances have established a fundamental role for inflammation in mediating all stages of this disease, from initiation through progression1. Activation of monocytes and macrophages is an important initial step in the cascades of events leading to many acute and chronic inflammatory diseases including atherosclerosis. It is believed that formation of foam cells, lipid-laden macrophages, is an early hallmark of atherosclerotic lesion formation1, 2. In humans, ongoing inflammatory reactions within coronary atherosclerotic plaques are increasingly thought to be crucial determinants of the clinical course of patients with coronary artery diseases2. Likewise, in a variety of animal models of atherosclerosis, signs of inflammation occur hand-in-hand with incipient lipid accumulation in the artery wall3, 4.
Toll-like receptors (TLRs) are a group of receptors that play a key role in innate immune signaling and initiating inflammatory responses. Ligation of these receptors initiates the activation of nuclear factor-κB (NF-κB) resulting in the expression of a wide array of inflammatory genes5, 6. The best studied of the TLRs are TLR2, and TLR4. Recent studies in humans have demonstrated that TLRs are expressed in macrophage-infiltrated atherosclerotic lesions7, 8. The Asp299Gly polymorphism in the human TLR4 gene has been associated with decreased risk for atherosclerosis in humans9. Elevated TLR expression has also been reported in the atheroma of murine models of atherosclerosis7, 8. Recent animal studies, employing hyperlipidemic mice fed a high fat diet, have shown that myeloid differentiation factor 88 (MyD88), TLR4 and TLR2 play an important role in atherosclerotic plaque accumulation10-12. These studies did not examine the temporal formation of atherosclerotic plaque and used mice placed on a high fat, cholesterol-rich diet.
Although it has been extensively reported that Apoe−/− mice fed a normal chow diet spontaneously develop atherosclerosis 13, 14, 15, high fat diet supplementation has been widely used because it can further accelerate atherosclerosis in Apoe−/− mice 13-15. However, it has been reported that high fat diet supplementation is associated with abnormal immunological responses underlying atherosclerosis. Severe hypercholesterolemia leads to the shifting of T helper cell responses from Th1 responses, which is commonly seen in atherosclerotic plaques16, to abnormal Th2 responses, in Apoe−/− mice17, 18. Since atherosclerotic plaque is dominated by Th1-type cytokines19, the excessively high circulating cholesterol levels in high fat diet fed Apoe−/− mice could potentially confound or mask immunological processes involved in atherosclerosis. This issue is particularly important in the immunological studies of atherosclerosis. In our study, to exclude the effects of dietary stresses on immunological responses underlying atherosclerosis, we used normal chow diet fed mice to investigate the role of TLR2 in the development of atherosclerosis.
Materials and Methods
Generation of Mice
Tlr2−/−Apoe−/− mice were generated from heterozygote intercrosses of the Apoe−/− mice (C57BL/6J background, The Jackson Laboratory) and the Tlr2−/− mice (C57BL/6J background, originally provided by S. Akira, Osaka University) and the double knock out mice were confirmed by genotyping. Age- and sex-matched Apoe−/− mice and C57BL/6J mice were obtained from the Jackson Laboratory. All mice were fed a normal chow diet and were cared for in accordance with Boston University Institutional Animal Care and Use Committee procedures. Total and free cholesterol concentrations in the sera of mice were determined by colorimetric assays according to the manufacturer's instructions (Wako Chemicals).
Atherosclerotic Plaque Assessment
Apoe−/− female, Apoe−/− male, Tlr2−/−Apoe−/− male and Tlr2−/−Apoe−/− female mice (16 per group) fed a normal chow diet were used to determine atherosclerosis development. Half of the animals were sacrificed at five months of age and the remaining mice were sacrificed at seven month of age (n=8 mice/condition). Mice at the age of five and seven months were used to determine lesion progression as reported previously 20. The aorta of mice (male and female, Apoe−/− and Tlr2−/−Apoe−/−, n=8 each group) was harvested from the aortic valve to the iliac bifurcation, opened longitudinally, and stained with Sudan IV21. Digital micrographs were taken and the total area of atherosclerotic plaque in the aortic arch and the whole aorta was determined from on-screen images using IPLabs (Scanalytics, Inc.) by an observer blinded to the identity of the samples. In a separate experiment, cryosectioning of embedded aortic arch tissue (from five-month old Apoe−/− and Tlr2−/−Apoe−/− female mice, n=3 each group) was performed to evaluate histological and immunohistological features of atherosclerotic lesions. Cross-sections (7 μm thick) of the aortic sinus were stained with hematoxylin-eosin and oil red O (1% in 60% isopropanol). Detection of macrophages and MCP-1 in aortic cryosections was accomplished by using antibody to macrophage antigen F4/80 (1:50, Serotec) and mouse CCL2 (1:50, eBioscience) respectively, and ABC kit with biotinylated anti-rat (mouse absorbed) IgG as secondary antibody (Vecta Laboratory).
Reverse Transcription Polymerase Chain Reaction (RT-PCR)
The aorta harvested from five or seven-month old male Apoe−/− and Tlr2−/−Apoe−/− mice was cut into two parts at the distal root of left subclavian artery. The aortic arch (from aortic sinus to cut) and thoraco-abdominal aorta (from cut to iliac bifurcation) were collected. The tissue was homogenized and total RNA was extracted using an RNeasy kit (Qiagen). RT-PCRs were performed using specific primers for murine TLR2, TLR4, and β-actin using previously described conditions22.
Macrophage Stimulation
Since macrophage infiltration and foam cell formation occurs in the early stage (3 months of age) of atherosclerosis development in vivo13, 20, we used macrophages isolated from 12-week old for in vitro assays. Thioglycollate-elicited peritoneal macrophages cultured in RPMI 1640 medium were incubated with the TLR2 agonist, SLTA (0.02 or 2 μg/ml; InvivoGen), or the TLR4 agonist, Escherichia coli LPS (10 or 100 ng/ml; InvivoGen) for 24 hours. After challenge, cells and cell culture supernatants were collected for FACS and ELISA assay, respectively. Macrophages cultured in medium alone served as a negative control.
Flow Cytometry Analysis
Macrophages cultured in 12-well plates for 24 hours with S. aureus lipoteichoic acid (SLTA) (2 μg/ml) or LPS (100 ng/ml) were washed and incubated with Fc blocker (eBioscience), followed by FITC-labeled anti-TLR-2 (6C2), PE-labeled anti-TLR-4 (MTS510), or each isotype-matched antibody (eBioscience). The cells were washed and 10,000 events were analyzed by flow cytometry using FACScan flowcytometer (Becton Dickinson).
Cytokine and Chemokine Measurements
Concentrations of tumor necrosis factor α (TNF-α), interleukin 6 (IL-6,) and monocyte chemoattractant protein-1 (MCP-1) in the sera of each mouse, as well as the macrophage culture supernatant fluids, were determined using commercially available ELISA kits (BD Biosciences) as described per the manufacturer's instructions. Using these kits, the minimum detectable levels for TNF-α and IL-6 were 5 pg/ml and 3.8 pg/ml, respectively.
LDL Uptake and Foam Cell Formation
Thioglycollate-elicited peritoneal macrophages were isolated from 12 week-old male Apoe−/− and Tlr2−/−Apoe−/− mice and were cultured in RPMI 1640 medium containing 10% FBS. Macrophages cultured in 8-well chamber slides were incubated with 20 μg/ml DiI-labeled Acylated-LDL (Invitrogen) at 37°C for 24 hours. The cells were then washed 3 times with PBS and Ac-LDL uptake by macrophages was examined by fluorescence microscopy. Another set of DiI-labeled Ac-LDL-treated macrophages were stained with oil red O and examined by light microscopy to evaluate foam cell formation23.
Statistics
All statistical analyses were performed using Prism software (GraphPad Software Inc, San Diego). Data were first evaluated by Kolmogorov-Smirnov normality test to verify normal distribution. Two-tail Student t-test or one-way ANOVA with Tukey-Kramer test were performed and a value of P < 0.05 was considered significant. Two-way ANOVA was used for analysis of plaque area between two of the three factors (genotype, sex and age), and their interaction with each other. A value of P < 0.05 was considered significant.
Results
Generation of Tlr2−/−Apoe−/− Mice
To examine the role of TLR2 on the formation of atherosclerosis, Tlr2−/−Apoe−/− mice were generated from heterozygote intercrosses of the Apoe−/− and Tlr2−/− mice. Generation of Tlr2−/−Apoe−/− mice was confirmed by genotyping of mice tail DNA by PCR (supplemental Fig. 1). The Tlr2−/−Apoe−/− mice showed similar body weight as the age-matched Apoe−/− mice (supplemental Fig. 2), and gross differences between Tlr2−/−Apoe−/− mice and their counterparts were not observed. Sex- and age-matched Tlr2−/−Apoe−/− and Apoe−/− mice were fed a normal chow diet and were euthanized at the specified time for subsequent analysis.
TLR2 Deficiency Reduces Atherosclerotic Plaque Formation
Analysis of the aortic arch (Fig.1A and C) and whole aorta (supplemental Fig. 3A and B) revealed an overall reduction of atherosclerotic plaque area in Tlr2−/−Apoe−/− mice as compared to that in Apoe−/− mice at five-months of age. In addition, female Apoe−/− mice displayed more plaque accumulation than their genotype-matched male. The trend that Tlr2−/−Apoe−/− mice accumulated less atherosclerotic plaque than the age- and sex-matched Apoe−/− mice was more pronounced in seven-month old mice (Fig. 1B and D). Similar to our observation at the age of five-months, seven-month old mice presented with plaque accumulation predominantly in the aortic arch region and female Apoe−/− mice displayed more plaque accumulation than their male counterparts (supplemental Fig. 3).
Figure 1. TLR2 deficiency reduces atherosclerosis in the Apoe−/− mice.
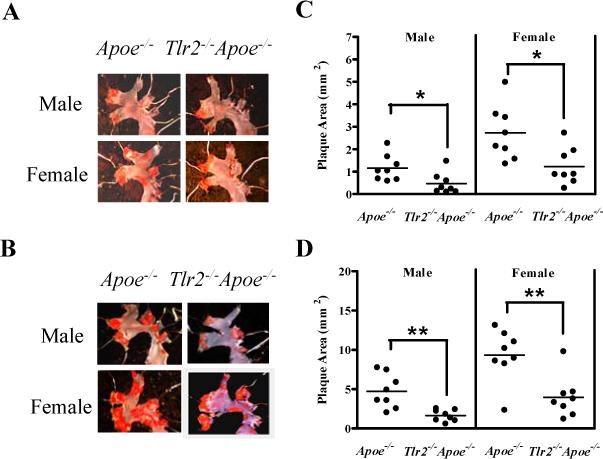
After dissection, aortas were stained with Sudan IV to assess plaque area in the entire aorta. Representative micrographs of plaque accumulation of five-month old mice (A) and seven-month old mice (B) are shown. Quantified plaque area in aortic arch of five-month old mice (C) and seven-month old mice (D) are shown. Each point represents a single mouse and the horizontal line represents mean plaque area for each group (n = 8). ** P < 0.005; * P < 0.02 by two tail Student t-test.
TLR2 Plays An Important Role in Progression in Atherosclerotic Plaque Accumulation
The progression of plaque accumulation in the whole aorta (supplementary Fig. 3) was further analyzed by two-way ANOVA analysis (Table) with emphasis on the interaction between genotype and age in both male and female mice. In five-month old mice, Tlr2−/−Apoe−/− mice displayed 58.4 % or 51.34 % reduction of the plaque area in male and female mice, respectively. In seven-month old mice, the reduction was more pronounced. Tlr2−/−Apoe−/− mice displayed 69.5 % or 65.1 % reduction of the plaque area in male and female mice, respectively. Two-way ANOVA revealed that plaque accumulation was dependent on both genotype and age and they were significantly interacted. These results indicate that the effects of genotype are influenced by age. TLR2-dependent progression of plaque accumulation was more evident in seven-month old mice as compared to five-month old mice in both the male and female groups. In the context of a normal chow diet, no significant differences were observed in serum cholesterol levels between Tlr2−/−Apoe−/− mice and Apoe−/− mice (supplemental Fig. 4), indicating that the reduction in atherosclerosis plaque formation in TLR2-deficient mice was independent of systemic lipid levels.
TLR2 Deficiency Results in Diminished Expression of Atherosclerosis Markers in Plaques
To further examine the effect of TLR2 on the composition of atherosclerotic plaques, histochemical and immunohistochemical analysis of aortic sinus lesions were performed. Compared with Apoe−/− mice, Tlr2−/−Apoe−/− mice exhibited a marked reduction in intimal thickening in the aortic sinus lesions (Fig. 2A and B). Aortic sinus plaques of Tlr2−/−Apoe−/− mice exhibited less neutral lipid accumulation (Fig. 2C) than that of Apoe−/− mice. Compared with Apoe−/− mice, Tlr2−/−Apoe−/− mice also demonstrated less macrophage infiltration (Fig. 2D) and MCP-1 expression (Fig. 2E) in the endothelium and sub-endothelial lesions of aorta sinus plaque. No background staining was observed for the isotype control IgG (data not shown). Taken together, these data indicate that TLR2 deficiency resulted in reduced plaque formation, less neutral lipid accumulation, macrophage recruitment and MCP-1 expression in the aortic sinus.
Figure 2. TLR2 deficiency reduces expression of atherosclerosis markers in the aortic sinus.
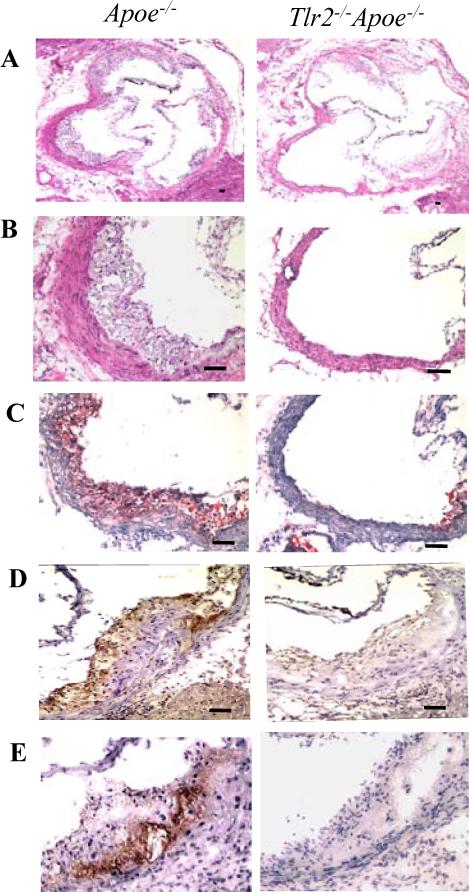
Representative staining of aortic sinus plaques from Apoe−/− and Tlr2−/−Apoe−/− 5 month old female mice. Sections were stained with H&E (A, ×40, B, ×200 magnification), oil red O for neutral lipid (C, ×200), macrophage marker F4/80 (D, ×200) and mouse MCP-1 (E, ×200). The scale bar represents 100 μm.
Evaluation of Aortic TLR Expression and Inflammatory Mediator Levels in Apoe−/− and Tlr2−/−Apoe−/− Mice
To confirm there were no effects on TLR4 expression in the Tlr2−/−Apoe−/− mice, RT-PCR was performed on aortic tissues. Constitutive TLR2 gene expression was detected in the aorta of Apoe−/− mice, but not in Tlr2−/−Apoe−/− mice. The aortic arch, which is the predominant site of atherosclerotic lesions in Apoe−/− mice, exhibited higher levels of TLR2 gene transcripts than that of the thoraco-abdominal part of the aorta (Fig. 3A), in agreement with increased plaque observed in these regions in Apoe−/− mice. Similar levels of TLR4 gene expression was observed in the aorta of Apoe−/− and Tlr2−/−Apoe−/− mice, indicating deletion of TLR2 does not affect TLR4 expression in the aorta of Apoe−/− mice.
Figure 3. Evaluation of TLR expression and serum MCP-1 levels in Apoe−/− and Tlr2−/−Apoe−/− mice.
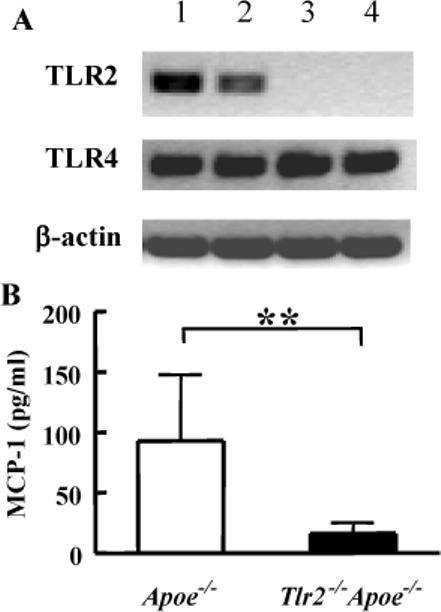
(A) Representative RT-PCR from the aortic arch and thoraco-abdominal aorta of Apoe-/ and Tlr2−/−Apoe−/− 5 month old male mice (similar results obtained from all genotype-matched mice, n=3/group). Lane 1, aortic arch of Apoe−/− mice; lane 2, thoraco-abdominal aorta of Apoe−/− mice; lane 3, aortic arch of Tlr2−/−Apoe−/− mice; lane 4, thoraco-abdominal aorta of Tlr2−/−Apoe−/− mice. (B) MCP-1 concentration in Apoe−/− and Tlr2−/−Apoe−/− sera of 5 month old male mice. Values expressed as mean ± SD (n = 8), ** P < 0.01 by two-tail Student's t-test.
To examine whether deficiency of TLR2 has any effect on systemic inflammatory cytokine and chemokine production, ELISAs were performed to measure serum levels of TNF-α, IL-6 and MCP-1. TNF-α and IL-6 were not detected in the serum of Apoe−/− and Tlr2−/−Apoe−/− mice (data not shown). However, MCP-1 was detected at significantly lower levels in Tlr2−/−Apoe−/− mice than Apoe−/− mice (Fig. 3B).
Macrophage Responses Following Stimulation with Defined TLR Agonists in Tlr2−/−Apoe−/− Mice
Growing evidence has indicated that defined TLR ligands not only are able to activate TLR mediated pro-inflammatory signaling, but also can regulate TLR expression24, 25. Based on these observations, we performed in vitro experiments to evaluate macrophage responses to the TLR2 agonist, S. aureus lipoteichoic acid (SLTA) and the TLR4 agonist, E. coli lipoplysaccharide (LPS). By FACS analysis we observed that macrophages from Apoe−/− mice cultured with SLTA significantly up-regulated TLR2 expression. As expected, Tlr2−/−Apoe−/− mice failed to regulate TLR2 due to deletion of this gene (Fig. 4A). Similar levels of TLR4 expression were observed in Apoe−/− and Tlr2−/−Apoe−/− mouse macrophages, with or without the TLR2 agonist stimulation (Fig. 4B). This suggests that TLR2 is up-regulated in response to a TLR2 pathogen-associated microbial product and that in Apoe−/− mice or Tlr2−/−Apoe−/− mice, TLR4 expression is not regulated via TLR2 signaling pathway. Consistent with this observation, macrophages isolated from Tlr2−/−Apoe−/− mice exhibited a significant deficiency of MCP-1 (Fig. 4C), and TNF-α and IL-6 (data not shown) production in response to SLTA stimulation.
Figure 4. Loss of TLR2 up-regulation and macrophage response following stimulation with TLR agonists in Apoe−/− Tlr2−/− mice.
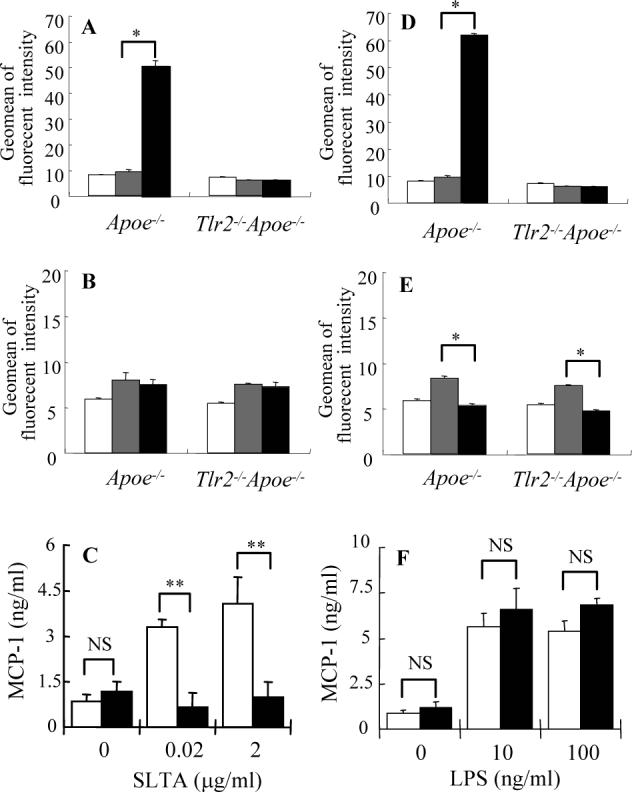
Macrophages harvested from 12-week old Apoe−/− and Tlr2−/−Apoe−/− mice were cultured with 2 μg/ml of SLTA (black bars) (A, B, C), 100 ng of E. coli LPS (black bars) (D, E, F) or medium alone (gray bars) (A-D) for 24 hours. TLR2 (A, B) and TLR4 (C, D) surface expression was determined by FACS analysis using specific antibodies and isotype controls (white bars) (A-D). Values expressed as mean ± SD (n = 3), * P < 0.01 compared SLTA or E. coli LPS stimulation with medium alone by one-way ANOVA with Tukey-Kramer test. Production of MCP-1 by Apoe−/− (white bars) or Tlr2−/−Apoe−/− (black bars) mouse macrophages in response to SLTA (C) or LPS (F) stimulation was examined by ELISA. Values expressed as mean ± SD (n = 3), ** P < 0.001; NS = not significant by two-tail Student's t-test.
Stimulation of macrophages with E. coli LPS significantly up-regulated TLR2 expression in Apoe−/− mice (Fig. 4D), which is consistent with previous findings26. This trend was not observed in Tlr2−/−Apoe−/− mice (Fig. 4D). However, no significant difference on TLR4 down-regulation in macrophages was observed in Apoe−/− and Tlr2−/−Apoe−/− mice (Fig. 4E). Macrophages isolated from Apoe−/− and Tlr2−/−Apoe−/− mice also exhibited similar levels of MCP-1 production in response to LPS stimulation (Fig. 4F).
These results, together with the in vivo data, suggest that TLR2 deficiency is associated with reduced MCP-1 production in response to a TLR2 ligand. This may contribute to the decreased inflammatory cell infiltration and plaque formation observed in Tlr2−/−Apoe−/− mice.
TLR2 Does Not Alter Macrophage Ac-LDL Uptake and Foam Cell Formation
Macrophage foam cell formation is believed to play an important role in the development of atherosclerosis3. To assess if Tlr2−/−Apoe−/− macrophages have a defect in low density lipoprotein (LDL) uptake, peritoneal macrophages from Tlr2−/−Apoe−/− and Apoe−/− mice were cultured with DiI-Ac-LDL. Fluorescence microscopy and oil red O staining revealed that macrophages from Apoe−/− and Tlr2−/−Apoe−/− mice displayed similar levels of Ac-LDL uptake (Fig. 5Aa) and foam cell formation (Fig. 5Ab). Apoe−/− and Tlr2−/−Apoe−/− mice macrophages exhibited similar levels of oil red O positive staining (Fig. 5B), indicating that TLR2 is not directly involved in macrophage Ac-LDL uptake and foam cell formation.
Figure 5. TLR2 does not affect Ac-LDL uptake and foam cell formation.
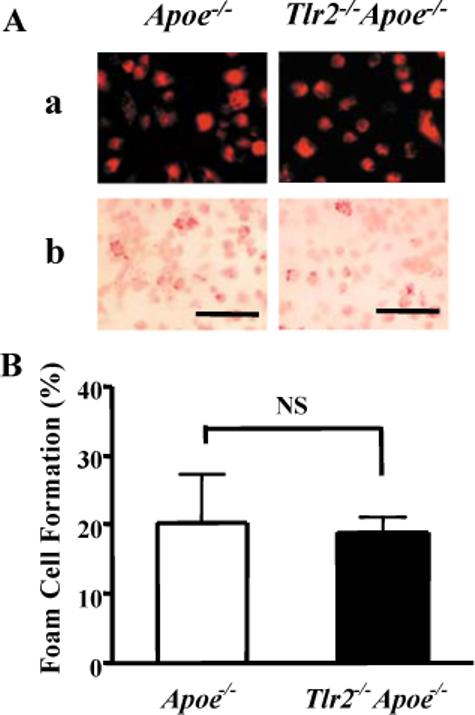
Peritoneal macrophages harvested from 12-week old Apoe−/− and Tlr2−/−Apoe−/− male mice were incubated with 20 μg/ml DiI-labeled Ac-LDL at 37°C for 24 hours. (A) Ac-LDL uptake and foam cell formation was evaluated by fluorescence microscopy (a) and oil red O stain by light microscopy (b), respectively. The scale bar represents 100 μm. (B) The percentage of foam cells was determined by counting oil red O positive stained cells collected from three mice in ten random 200× magnification microscopic fields. Values expressed as mean ± SD, NS = not significant by two-tail Student's t-test.
Discussion
In this study, we investigated the association between TLR2 and atherosclerosis using an apoE knockout mouse model. Mice fed a normal chow diet were used in this study to exclude the effect of dietary stresses on immunological reactions in atherosclerosis. We demonstrate that inactivation of TLR2 resulted in reduced progression of atherosclerotic plaque formation in both male and female Apoe−/− mice independent of dietary lipids. Likewise, TLR2 deficiency resulted in a reduction in lipid accumulation and decreased macrophage recruitment to the aortic sinus as well as reduced MCP-1 levels as compared with Apoe−/− mice. Furthermore, macrophages isolated from Tlr2−/−Apoe−/− mice demonstrated significantly reduced MCP-1 production upon stimulation with a TLR2 ligand. Taken together these results indicate that TLR2 signaling plays an important role in the development of atherosclerosis, probably through an increase in macrophage recruitment.
The role of macrophages and chemokines, especially MCP-1 on development of atherosclerosis has recently become a point of great interest27, 28. Clinically, significantly elevated serum MCP-1 levels have been observed in patients with coronary artery atherosclerosis27, 28. MCP-1 expression has been detected in human and experimental atherosclerosis, suggesting an active role for this molecule in monocyte recruitment29, 30. Inactivation of MCP-1, or its receptor, CCR2, resulted in markedly reduced macrophage accumulation and attenuated the progression of dietary-induced atherosclerosis in mice31, 32. Additionally, bone marrow transplantation studies revealed that over-expression of MCP-1 in macrophages led to increased foam cell formation and atherosclerosis in the irradiated hypercholesterolemic mice33, 34. These results together with the data presented in this study support a critical role for MCP-1 in the progression of atherosclerosis and suggest that low level of MCP-1 caused by TLR2 deficiency contributes to the reduced progression of atherosclerosis.
The role of TLRs in lipid uptake has been investigated in recent studies. Hoebe et al.34 reported that TLR2 interacts with CD36 and regulates specific TLR2 ligand interactions, most specifically diacylglyceride recognition. It is well established that modified LDL including Ac- and oxidized (ox)-LDL are taken up via scavenger receptors including type A scavenger receptor (SR-A) and CD3635, 36. Interestingly, Tamura et al.37 reported that in addition to SR-A and CD36, scavenger receptor expressed by endothelial cells-1 (SREC-1) plays a role in macrophage Ac-LDL accumulation, particularly when cells are activated by LPS. As LPS is a defined TLR4 ligand and it has previously been reported that hyperlipidemic mice deficient in TLR4 present with reduced atherosclerosis11, it is tempting to speculate that multiple TLRs may interact with multiple scavenger receptors and this level of recognition could represent a mechanism by which TLRs affect atherosclerotic plaque accumulation. However our studies indicate that TLR2 does not influence Ac-LDL uptake by murine macrophages.
Two recent studies have reported that genetic deficiency of MyD88, a TLR signaling adaptor molecule, reduces aortic atherosclerosis (∼ 60%) in Apoe−/− mice fed high fat diet10, 11. Deletion of TLR4 was also shown to result in reduced atherosclerosis in Apoe−/− mice (∼ 24% reduction). Our results are in agreement with these studies and further demonstrated an involvement for TLR2 in older mice fed a normal chow diet. In a recent study published while we were performing these studies, a role for TLR2 in atherosclerosis in a low-density lipoprotein receptor (LDLR)-null mice fed a high fat diet was reported12. These authors also demonstrated a role for bone marrow-derived cells (such as macrophages) in TLR2 mediated atherosclerosis formation. The studies here are distinct from these previous studies in that we 1) Used different mouse genetic backgrounds and thus different model systems, 2) Characterized the effect of TLR2 deficiency on the progression of atherosclerosis in Apoe−/− mice, and its association with local and systemic macrophages responses, in particular, MCP-1, 3) Assessed the sex-bias of the TLR2 mutation on atherosclerosis, and 4) Used a normal chow diet mice model. Our study demonstrated that TLR2 signaling plays an important role in progression of atherosclerosis and that the contribution of TLR2 was more apparent in older mice in both male and female groups. Most importantly our results indicate that an efficient innate immune defense system is associated with inflammatory progression of atherosclerosis and that this TLR2 dependent progression is independent of dietary lipids and macrophage LDL uptake.
TABLE.
Involvement of TLR2 in the progression of atherosclerotic plaque formation
| aorta plaque area (mm2) |
||||
|---|---|---|---|---|
| 5 month | 7 month | interaction | ||
| Male | Apoe−/− | 1.32 ± 0.55 | 6.20 ± 3.05* | P = 0.0046 |
| Tlr2−/−Apoe−/− | 0.55 ± 0.51 | 1.89 ± 0.84*† | ||
| (58.4%) | (69.5%) | |||
| Female | Apoe−/− | 3.10 ± 1.51 | 13.05 ± 4.62* | P = 0.0017 |
| Tlr2−/−Apoe−/− | 1.59 ± 0.85 | 4.56 ± 2.84*† | ||
| (51.34%) | (65.1%) | |||
Interaction; interaction between genotype and age.
Statistical analysis was performed by Two-way ANOVA.
P<0.001 significant between 5 and 7-month old mice with same genotype.
P<0.001 significant between Apoe−/− and Tlr2−/−Apoe−/− mice with same age.
(); reduction of % ratio in Tlr2−/−/Apoe−/− against Apoe−/− mice.
Acknowledgement
This study was supported by NIH Public Health Service grants HL080387 (CAG) and DE014774 (FCG). We would like to thank Dr. Lisa Sullivan (Boston University School of Public Health) for her expert assistance with statistical analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Reference
- 1.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105(9):1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 2.Hansson G. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352(16):1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 3.Skalen K, Gustafsson M, Rydberg EK, et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002;417(6890):750–754. doi: 10.1038/nature00804. [DOI] [PubMed] [Google Scholar]
- 4.Leitinger N. Oxidized phospholipids as modulators of inflammation in atherosclerosis. Curr Opin Lipidol. 2003;14(5):421–430. doi: 10.1097/00041433-200310000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17(1):1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 6.O'Neill LA. DisSARMing Toll-like receptor signaling. Nat Immunol. 2006;7(10):1023–1025. doi: 10.1038/ni1006-1023. [DOI] [PubMed] [Google Scholar]
- 7.Xu XH, Shah PK, Faure E, et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001;104(25):3103–3108. doi: 10.1161/hc5001.100631. [DOI] [PubMed] [Google Scholar]
- 8.Edfeldt K, Swedenborg J, Hansson GK, et al. Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation. 2002;105(10):1158–1161. [PubMed] [Google Scholar]
- 9.Kiechl S, Lorenz E, Reindl M, et al. Toll-like receptor 4 polymorphisms and atherogenesis. N Engl J Med. 2002;347(3):185–192. doi: 10.1056/NEJMoa012673. [DOI] [PubMed] [Google Scholar]
- 10.Bjorkbacka H, Kunjathoor VV, Moore KJ, et al. Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat Med. 2004;10(4):416–421. doi: 10.1038/nm1008. [DOI] [PubMed] [Google Scholar]
- 11.Michelsen KS, Wong MH, Shah PK, et al. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A. 2004;101(29):10679–10684. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest. 2005;115(11):3149–3156. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang SH, Reddick RL, Piedrahita JA, et al. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258(5081):468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 14.Plump AS, Smith JD, Hayek T, et al. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71(2):343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 15.Zhang SH, Reddick RL, Burkey B, et al. Diet-induced atherosclerosis in mice heterozygous and homozygous for apolipoprotein E gene disruption. J Clin Invest. 1994;94(3):937–945. doi: 10.1172/JCI117460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frostegard J, Ulfgren AK, Nyberg P, et al. Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis. 1999 Jul;145(1):33–43. doi: 10.1016/s0021-9150(99)00011-8. [DOI] [PubMed] [Google Scholar]
- 17.Zhou X, Paulsson G, Stemme S, et al. Hypercholesterolemia is associated with a T helper (Th) 1/Th2 switch of the autoimmune response in atherosclerotic apo E-knockout mice. J Clin Invest. 1998;101(8):1717–1725. doi: 10.1172/JCI1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robertson AK, Zhou X, Strandvik B, et al. Severe hypercholesterolaemia leads to strong Th2 responses to an exogenous antigen. Scand J Immunol. 2004;59(3):285–293. doi: 10.1111/j.0300-9475.2004.01403.x. [DOI] [PubMed] [Google Scholar]
- 19.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6(7):508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 20.Reddick RL, Zhang SH, Maeda N. Atherosclerosis in mice lacking apo E. Evaluation of lesional development and progression. Arterioscler Thromb. 1994;14(1):141–147. doi: 10.1161/01.atv.14.1.141. [DOI] [PubMed] [Google Scholar]
- 21.Tangirala RK, Rubin EM, Palinkski W. Quantitation of atherosclerosis in murine models: correlation between lesions in the aortic origin and in the entire aorta, and differences in the extent of lesions between sexes in LDL receptor-deficient and apolipoprotein E-deficient mice. J Lipid Res. 1995;36:2320–2328. [PubMed] [Google Scholar]
- 22.Miyamoto T, Yumoto H, Takahashi Y, Davey M, Gibson FC, Genco CA. Pathogen-accelerated atherosclerosis occurs early after exposure and can be prevented via immunization. Infect. Immun. 2006;74:1376–1380. doi: 10.1128/IAI.74.2.1376-1380.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qi M, Miyakawa H, Kuramitsu HK. Porphyromonas gingivalis induces murine macrophage foam cell formation. Microb Pathog. 2003;35(6):259–267. doi: 10.1016/j.micpath.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 24.Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. 2002;168(2):554–561. doi: 10.4049/jimmunol.168.2.554. [DOI] [PubMed] [Google Scholar]
- 25.Yumoto H, Chou HH, Yakahashi Y, Davey M, Gibson FC, Genco CA. Sensitization of human aortic endothelial cells to lipopolysaccharide via regulation of Toll-like receptor 4 by bacterial fimbria-dependent invasion. Infect. Immun. 2005;73(12):8050–8059. doi: 10.1128/IAI.73.12.8050-8059.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang T, Lafuse WP, Zwilling BS. Regulation of toll-like receptor 2 expression by macrophages following Mycobacterium avium infection. J Immunol. 2000;165(11):6308–6313. doi: 10.4049/jimmunol.165.11.6308. [DOI] [PubMed] [Google Scholar]
- 27.Charo IF, Taubman MB. Chemokines in the pathogenesis of vascular disease. Circ Res. 2004;95(9):858–866. doi: 10.1161/01.RES.0000146672.10582.17. [DOI] [PubMed] [Google Scholar]
- 28.Martinovic I, Abegunewardene N, Seul M, et al. Elevated monocyte chemoattractant protein-1 serum levels in patients at risk for coronary artery disease. Circ J. 2005;69(12):1484–1489. doi: 10.1253/circj.69.1484. [DOI] [PubMed] [Google Scholar]
- 29.Yla-Herttuala S, Lipton BA, Rosenfeld ME, et al. Expression of monocyte chemoattractant protein 1 in macrophage-rich areas of human and rabbit atherosclerotic lesions. Proc Natl Acad Sci U S A. 1991;88(12):5252–5256. doi: 10.1073/pnas.88.12.5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelken NA, Coughlin SR, Gordon D, et al. Monocyte chemoattractant protein-1 in human atheromatous plaques. J Clin Invest. 1991;88(4):1121–1127. doi: 10.1172/JCI115411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gosling J, Slaymaker S, Gu L, et al. MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein B. J Clin Invest. 1999;103(6):773–778. doi: 10.1172/JCI5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boring L, Gosling J, Cleary M, et al. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394(6696):894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 33.Aiello RJ, Bourassa PA, Lindsey S, et al. Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19(6):1518–1525. doi: 10.1161/01.atv.19.6.1518. [DOI] [PubMed] [Google Scholar]
- 34.Hoebe K, Georgel P, Rutschmann S, et al. CD36 is a sensor of diacylglycerides. Nature. 2005;433(7025):523–527. doi: 10.1038/nature03253. [DOI] [PubMed] [Google Scholar]
- 35.Kunjathoor VV, Febbraio M, Podrez EA, et al. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem. 2002;277(51):49982–49988. doi: 10.1074/jbc.M209649200. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki H, Kurihara Y, Takeya M, et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature. 1997;386(6622):292–296. doi: 10.1038/386292a0. [DOI] [PubMed] [Google Scholar]
- 37.Tamura Y, Osuga J, Adachi H, et al. Scavenger receptor expressed by endothelial cells I (SREC-I) mediates the uptake of acetylated low density lipoproteins by macrophages stimulated with lipopolysaccharide. J Biol Chem. 2004;279(30):30938–30944. doi: 10.1074/jbc.M313088200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.