

Abstract
The congenital muscular dystrophies (CMD) constitute a clinically and genetically heterogeneous group of autosomal recessive myopathies. Patients show congenital hypotonia, muscle weakness and dystrophic changes on muscle biopsy. Mutations in four genes (FKT1, POMGnT1, POMT1, FKRP) encoding for putative glycosyltransferases have been identified in a subset of patients which is characterized by a deficient glycosylation of α-dystroglycan on muscle biopsy. FKRP mutations account for a broad spectrum of patients with muscular dystrophy, from a severe congenital form with or without mental retardation (MDC1C) to a much milder limb-girdle muscular dystrophy (LGMD2I). We identified two novel homozygous missense FKRP mutations, one, A455D, in six unrelated Tunisian patients and the other, V405L, in an Algerian boy. The patients, between the age of 3 and 12 years, presented with a severe form of MDC1C with calf hypertrophy, high serum creatine kinase levels. None had never walked. Two had cardiac dysfunction and one strabismus. They all had mental retardation, microcephaly, cerebellar cysts, and hypoplasia of the vermis. White matter abnormalities were found in five of them, mostly when cranial MRI was performed at a young age. These abnormalities were shown to regress in one case, as has been observed in FCMD patients. Identification of a new microsatellite close to FKRP allowed us to confirm the founder origin of the Tunisian mutation. These results suggest strongly that particular FKRP mutations in the homozygous state induce structural and clinical neurological lesions in addition to muscular dystrophy and relate MDC1C to other CMDs with abnormal protein glycosylation and disordered brain function.
Keywords: Abnormalities, Multiple; genetics; Cerebellum; abnormalities; Child; Child, Preschool; Family Health; Female; Founder Effect; Glycosylation; Haplotypes; Homozygote; Humans; Magnetic Resonance Imaging; Male; Mental Retardation; complications; genetics; Muscular Dystrophies; complications; congenital; genetics; Pedigree; Proteins; genetics; metabolism; Tunisia
Keywords: Congenital muscular dystrophy, FKRP gene, founder-haplotype, mental retardation, cerebellar cysts
INTRODUCTION
Congenital muscular dystrophies (CMD) constitute a heterogeneous family of autosomal recessively inherited diseases, presenting at birth or within the first few weeks of life with hypotonia, delayed motor development and dystrophic changes on skeletal-muscle biopsy (1). Clinical heterogeneity is evidenced by different patterns of motor involvement, variable serum creatine kinase (CK) levels and the presence or not of mental retardation and structural defects of central nervous system (CNS). At least one third of patients in western countries have a primary deficiency in the laminin α2 chain (merosin) resulting from mutations in the LAMA2 gene (2, 3, 4). This type of CMD, usually referred as merosin-deficient CMD, has now been classified as MDC1A and is always associated with diffuse white matter abnormalities, and occasionally cerebellar hypoplasia or abnormal lesions of the cerebral gyri (5, 6, 7). Epileptic seizures have been reported in a subset of these patients. A secondary deficiency of laminin α2 chain and of α-dystroglycan is found in some CMD forms including muscle-eye-brain disease (MEB), Walker-Warburg syndrome (WWS), Fukuyama CMD (FCMD), and MDC1C, mapped to chromosomes 1p32, 9q34, 9q31 and 19q13, respectively (8, 9, 10, 11). These CMDs are caused by defects in genes encoding for putative glycosyltransferases: POMGnT1 gene coding for an O-mannose glycosyltransferase (MEB) (8), POMT1 gene coding for O-mannosyltransferasel (WWS) (9), FKT1 gene encoding a protein of unknown function called fukutin (FCMD) (10), and FKRP, a homologue of the FKT1 gene (MDC1C) (11). Clinically, the first three types show severe symptoms and structural central nervous system involvement. MDC1C is characterized by marked elevation of serum creatine kinase (CK), early onset hypotonia, delayed or arrested motor development, muscle hypertrophy, and variable cardiomyopathy. However, no major CNS abnormalities have been described in initial reports even in the most severe forms (11, 12). Very recently, two homozygous FKRP mutations associated with mental retardation and cerebellar cysts have been identified in two Turkish patients (13). They were isolated cases and, therefore, it was not possible to conclude if particular FKRP gene mutations may constitute a subset of patients with a particular phenotype characterized by severe neurological involvement. To address this issue, we report two new FKRP mutations associated with CMD and mental retardation, one in a series of six unrelated Tunisian families and another in an Algerian case. All the cases had mental retardation and neuro-imaging showed a variable combination of CNS abnormalities including cerebellar cysts or atrophy with or without white matter lesions. These results suggest strongly that certain FKRP mutations induce structural and clinical neurological abnormalities in a subset of patients with MDC1C. It may help in better understanding of this entity and relates this type of CMD to others with abnormal protein glycosylation and structural CNS abnormalities. In addition, identification of a new microsatellite close to the FKRP gene was used to investigate the founder origin of the Tunisian mutation.
PATIENTS AND METHODS
Patients
Seven unrelated patients belonging to 6 consanguineous Tunisian CMD families from Southern Tunisia (Patients 1–6) and one consanguineous Algerian CMD family (Patient 7) were studied. The diagnosis of CMD was made on the basis of hypotonia and weakness at birth or within the first months of life, and dystrophic changes on muscle biopsy. In addition, all patients had marked increase in serum creatine kinase (CK). Muscle biopsies were obtained from the deltoid muscle in five patients and from the tibialis anterior muscle in Patients 6 and 7. Brain magnetic resonance imaging (MRI) was available for Patients 1, 2, 3, 4, 6 and 7 and Patient 5 had a cerebral CT-Scan. Blood samples were collected from the members of these families after informed consent.
Immunohistochemistry
Unfixed frozen 8 μm sections were incubated with monoclonal antibodies binding specifically to the human laminin α2 chain 80 kDa fragment towards C-terminal region (MAB 1922 Chemicon International), and the human merosin 300 kDa fragment towards the amino-terminal region (NLC-merosin Novo-Castra) in all patients. In addition, muscle sections of Patients 6 and 7 were immunolabelled with α-dystroglycan antibodies (V1A4-1, 05298, Euromedex). All primary antibodies were applied for 1h and developed using an appropriate secondary antibody (FITC-conjugated Rabbit anti-Mouse Immunoglobulins DAKO).
Linkage and haplotype analysis
Genetic analysis was performed on genomic DNA extracted from blood leukocytes using a standard phenol-chloroform procedure (14). Microsatellites spanning the FKRP locus (D19S606, D19S412, D19S219 and fkrp 52) were studied. PCR amplification was carried out in 50 μl with 60 ng of genomic DNA, 1 μM of each primer, 125 μM dNTPs, 1.5 mM MgCl2, 5 mM KCl, 10 mM Tris-HCl, pH 8.8 and 1U of Taq DNA polymerase. PCR products were analyzed on 6 % denaturing polyacrylamide gels, transferred on to N+ Hybond membrane (Amersham Pharmacia Biotech) and hybridized with a poly AC probe labeled with α32P. The following primers were designed to amplify the new microsatellite, fkrp 52 (ID = ss12568442 in SNP NCBI database): 5′-TCTCCAAAAAACAACAACAAC-3′ and 5′-CTAGTGTTCTGGGACCTTT-3′. The sequence of the amplified fragment is available in the Third Party Annotation Section of the DDBJ/EMBL/Genbank databases under the accession number TPA: BK001438.
FKRP mutational analysis
A 1.8kb fragment containing the FKRP coding sequence of exon 4 and its bordering intronic sequences was amplified using 5 primer sets (Table 1). The overlapping PCR products were purified (Qiagen), and used for direct sequencing on both strands. Sequencing reactions were carried out using an ABI Prism Big Dye Terminator Cycle Sequencing Kit (Applied Biosystems). The 1364C>A mutation induces the loss of a NaeI restriction site. NaeI restriction pattern of the FKRP 4–5 fragment (421bp) was used to confirm the presence of the mutation in patients’s relatives and to screen 100 unrelated Tunisian, healthy individuals. Digestion of PCR products was performed according to the manufacturer’s instructions (New England Biolabs), followed by separation on 2% Nusieve gels. The 1213>T mutation identified in the FKRP 4–5 fragment was searched by direct sequencing in 50 Algerian and 100 French healthy control individuals.
Table 1.
PCR primers used to amplify the coding sequence of the FKRP gene located within exon 4
| Name | Position on mRNA (GI: 15866719) | Forward primer | Reverse primer | Product |
|---|---|---|---|---|
| FKRP 4-1 | -170ATG/517 | 5′CAAAGCTGAAACCAAATAGGGA3′ | 5′GCTGGGCTGGGTCTTGCTG3′ | 401 bp |
| FKRP 4-2 | 422/830 | 5′GCCCCCGTGTCACCGTCCT3′ | 5′TAGCGGGCGGTCCACTCTC3′ | 408 bp |
| FKRP 4-3 | 660/1170 | 5′TGTGGCCCTAGTACCTGATG3′ | 5′GCGCGTGGTCTCCTTGTTG3′ | 510 bp |
| FKRP 4-4 | 1112/1558 | 5′TAGTGAGCTGGGAAGGCGG3′ | 5′CATTGCGGGGGTAGAAGGG3′ | 446 bp |
| FKRP 4-5 | 1500/1921 | 5′ GCAGTACAGCGAAAGC AACC3′ | 5′GCCTTCTCTCATGCTCTCCT3′ | 421 bp |
RESULTS
Clinical and complementary findings
The main clinical features of the Tunisian (Patients 1 to 6) and Algerian patients are summarized in Table 2.
Table 2.
Summary of the clinical features of the seven patients (CK creatine kinase, ND no data)
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Sex | F | M | F | F | F | F | M |
| Age of onset | Birth | Birth | Birth | 4 months | 2 months | Birth | Birth |
| Current age (years) | 7 | 6 | 6 | 5 | 6 | 12 | 3 |
| Serum CK | 4,400 IU/I | 5,500 IU/I Sat | 7,330 IU/I | 2,978 IU/I | 2,600 IU/I | 4,997 IU/I | 1,370 IU/I |
| Maximal motor milestone | Sat unsupported | unsupported | Sat unsupported | Sat unsupported | Sat unsupported | Sat unsupported | Sat unsupported |
| Contractures | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Hypertrophy | Calf | Calf | No | Calf | No | Tongue | Calf |
| Mental Retardation | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Head circumference | − 2 SD | − 2 SD | − 2 SD | − 2 SD | − 2 SD | − 2 SD | − 2 SD |
| White Matter Changes | Yes (3 years) | Yes (3 years) | No (6 years) | No (5 years) | Yes (3 years) | Yes (1 year)/+/−(4 years)/No (12 years) | Yes (2.5 years) |
| Cerebellar Cysts | Yes | Yes | Yes | Yes | ND | Yes | Yes |
| Hypoplasia of vermis | Yes | Yes | Yes | No | ND | Yes | Yes |
| Microcephaly | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Eye involvement | No | Strabismus | No | No | No | Ophthalmoplegia | ND |
| left ventricular | Left ventricular | ||||||
| Cardiac function | Normal | Normal | ND | hypertrophy | ND | dilatation | Normal |
| Respiratory function | Normal | Normal | Normal | Normal | Normal | Mechanical ventilation | Normal |
| Scoliosis | Yes | Yes | No | Yes | No | Vertebral arthrodesis | No |
Patients 1, 2 and 3
These patients have previously been described (15); they presented a severe form of CMD with mental retardation and cerebellar cysts. The state of these patients worsened progressively. For Patient 1, brain MRI performed at ages of 3 and 6 showed a significant reduction of the white matter abnormalities with age (Figure 1). In contrast, the hypoplasia of the pons and cerebellar cysts remained unchanged.
Figure 1.
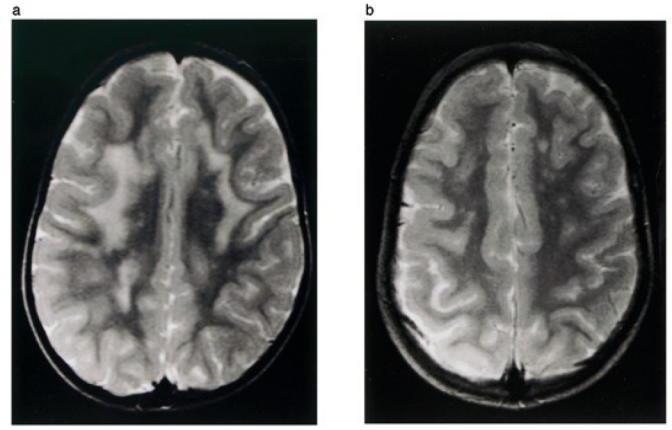
T2-Weigted axial MRI of the brain of Patient 1 shows abnormal high white matter signal intensities at age of 3 (a) and regression of these abnormalities on follow-up examination at the age of 6 (b).
Patient 4
This patient was a 5 year-old girl. She had a severe form of CMD with mental retardation. EMG was myopathic and muscle biopsy showed severe dystrophic features. Cranial MRI at age 5 revealed only cerebellar cysts without any white matter or other cerebellar abnormality (Figure 2). Echocardiogram showed left ventricular hypertrophy.
Figure 2.
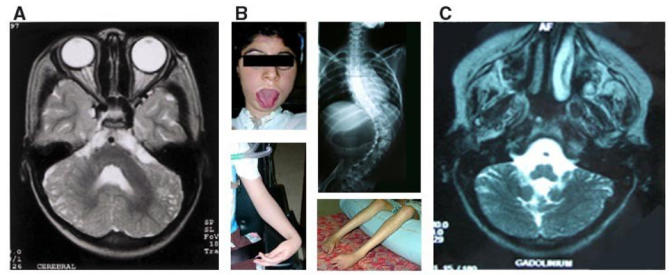
A- Transverse T2-Weighted MRI image from Patient 4 showing cerebellar cysts.
B- Clinical features of Patient 6: severe facial weakness and tongue enlargement, wrist and flexor finger contractures without elbow contractures, severe distal amyotrophy, and spinal deformity.
C- Transverse T2-Weighted MRI image from Patient 6 showing hypoplasia of cerebellar vermis and cerebellar cysts.
Patient 5
This patient was a 6 year-old girl. She presented since birth a severe form of CMD with axial and facial weakness and had mental retardation. The ophthalmological examination was normal. Brain CT scan had been performed at the age of three years and showed white matter abnormalities (not shown).
Patient 6
Patient 6 was a twelve year-old mentally retarded girl who had an older brother with congenital muscle dystrophy associated with encephalopathy who had died at the age of three due to a pneumopathie. She was the oldest child of this series and was the most severely affected patient. She never walked. She showed a severe facial weakness, progressive weakness, macroglossia, limited ocular excursion and scoliosis which was treated surgically. She was mechanically ventilated by age 10 because of severe restrictive respiratory insufficiency. At 12 years she was only able to perform minimal movements of the fingers which were contracted in flexion (Figure 2) and a mild cardiac involvement was detected, with a reduced shortening fraction and a normal ejection fraction. She did not speak a word before 5 years of age. At 12 years, she was able to say only single simple words of two or three syllables (Verbal IQ: 50) and her brain MRI showed cerebral atrophy, very white matter abnormalities and hypoplasia of cerebellar vermis and cerebellar cysts (Figure 2). White matter abnormal signal was very diffuse in early life and regressed progressively.
Patient 7
This patient was a 3 year-old mentally retarded boy. He presented since birth with severe form of CMD. EMG showed myopathic features and the muscle biopsy of the tibialis muscle showed severe dystrophic features. ECG, EEG and ophthalmological examination were normal. Brain MRI revealed cerebellar cysts, megacisterna with hypoplasia of the vermis and an abnormally high periventricular intensity on T2 weighed images. At the age of three years, he was microcephalic and could say only a few words.
Immunocytochemistry
All the patients showed histological changes characteristic of muscular dystrophy, and reduced immunohistochemical staining of laminin α2 chain using antibodies against the 80 and the 300 kDa fragments; α-dystroglycan expression was found to be markedly reduced in the only two samples available (data not shown).
Mutation screening of the FKRP gene
After exclusion of the CMD loci (FCMD, MEB, MDC1A and MDC1B) in Families 1–7 (data not shown), all patients were found to be homozygous for the markers surrounding the FKRP locus (D19S219, D19S412, D19S606) on chromosome 19q13.3, suggesting homozygous mutations in FKRP (Figure 5, data not shown for Family 7). Sequencing of the FKRP coding region revealed the same homozygous transversion, 1364C>A, in the six Tunisian patients inducing the change of alanine 455 to a negatively charged aspartic acid (A455D) (Figure 3). We took advantage of the loss of a NaeI restriction site induced by this mutation to rule it out as a non-pathogenic polymorphism. This variant was not found in 200 control chromosomes. Using NaeI digestion and direct sequencing, the mutation was found to segregate with the disease in an autosomal recessive fashion in the six consanguineous families (Figures 4 and 5). A second homozygous transversion 1213G>T was identified in the Algerian patient changing valine 405 to a leucine (V405L). This change was not found by direct sequencing in 50 Algerian and 100 French healthy control individuals.
Figure 5.
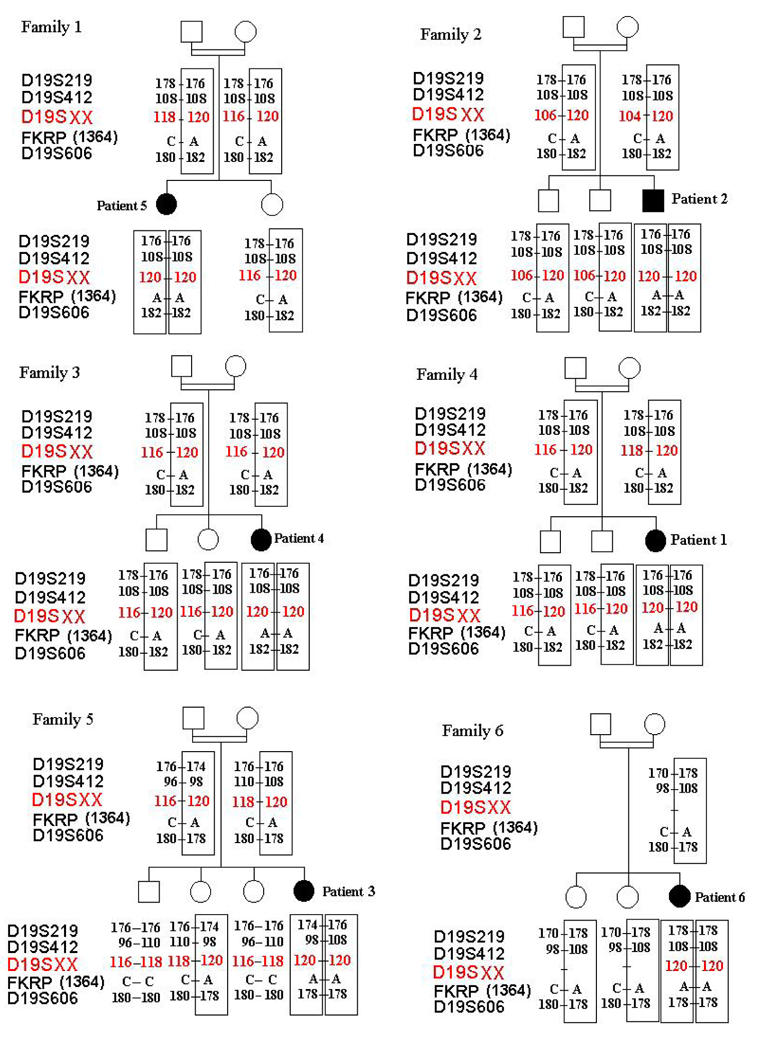
The pedigrees of the 6 Tunisian families showing the inheritance of the 1364C>A mutation. The haplotypes containing the mutation are boxed. We note the same founder haplotype for Families 1, 2, 3 and 4. Families 5 and 6 had the same mutation but not the same haplotype. With the fkrp 52 all families shared the same founder allele (120 bp) transmitted with the Tunisian mutation.
Figure 3.
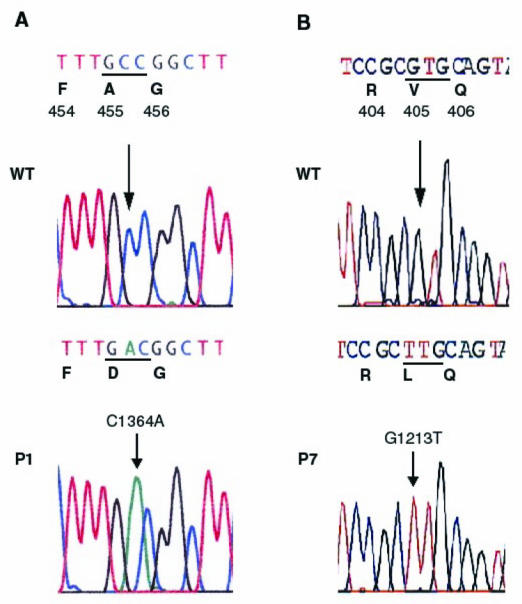
Sequence chromatograms from normal individual (wild type, WT) and affected individuals (P) are shown together with the expected amino-acid changes. Nucleotide variations are indicated by an arrow. A: 1364C>A inducing the A455D mutation in Patient 1 (P1), B: 1213G>T inducing the V405L mutation in Patient 7 (P7).
Figure 4.
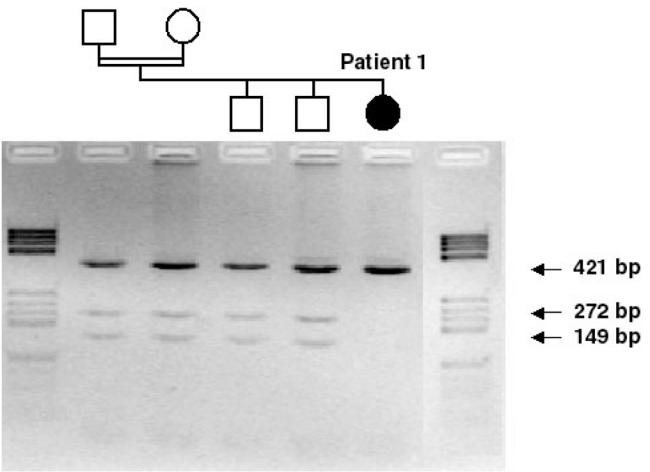
Restriction enzyme analysis showing the inheritance of the 1364C>A mutation in family 4. A 421bp PCR fragment was digested with Nae I. Normal control DNA is cut into two fragments of 272 bp and 149 bp, whereas products containing the mutation remained uncut at 421 bp in Patient 1, and only one of the two alleles was cut for parents and the two brothers who are heterozygous carriers of the A455D mutation.
Since the 6 families originated from Tunisia, we searched for a founder origin of the mutation. The known markers flanking FKRP, D19S219, D19S412 and D19S606, were physically mapped according to the Human Genome Browser and Ensembl Genome Server maps (http://genome.cse.ucsc.edu/goldenPath/hgTracks.html and http://www.ensembl.org). They are located at 1255 kb and 238 kb proximal, and 722 kb distal to FKRP, respectively. Among the families tested, Families 1 to 4 belonging to the same region in the South of Tunisia shared the same alleles (Figure 5) while the two others did not. We searched for new (CA)n microsatellite markers closer to FKRP. One was identified, fkrp 52, with alleles varying from 92 to 122 bp, 42 kb proximal to FKRP. A rare 120 bp allele containing 26 CA repeats was shared by the 6 families suggesting that the mutation has been transmitted by a common ancestor (Figure 5).
DISCUSSION
In this study, we report two new homozygous missense FKRP mutations in 7 patients affected with MDC1C associated with mental retardation and CNS abnormalities. Six of them originating from Tunisia share the same mutation, which strongly suggests that this particular FKRP mutation in the homozygous state invariably leads to the CNS involvement.
Mutations in the FKRP gene previously reported account for a broad spectrum of patients with muscular dystrophy, from a severe congenital form (MDC1C) (11) to a much milder limb-girdle muscular dystrophy (LGMD2I) (16). There exist intermediate phenotypes which look like Duchenne muscular dystrophy. The intermediate and congenital forms may share an identical end-stage picture characterized by a paralytic atrophic-hypertrophic phenotype including extreme diffuse weakness, tongue hypertrophy and need of continuous mechanical ventilation (11, 12). Genotype-phenotype correlation has been evidenced by the identification of “mild” and “severe” mutations in cases with LGMD2I and MDC1C respectively, and which lead to an intermediate phenotype when combined (17, 18, 19). In spite of differences in the degree of severity, a common phenotypical pattern including some features such as high CK levels, muscle hypertrophy, a progressive course and reduced expression of laminin α2 chain and α-dystroglycan in muscle characterizes this entity.
The patients of our series presented with a typical MDC1C phenotype, but in addition they had severe psycho-motor retardation and developed a microcephaly which was not observed at birth. They showed late and unsteady head control, inability to stand or walk, and marked mental retardation. Very high CK levels were constant, and calf hypertrophy was noted in four of the patients. The oldest patient developed at the end of the first decade the typical paralytic atrophic-hypertrophic FKRP phenotype. This was not observed in the other six patients probably because they were too young to manifest this progression which is not usually observed in the first years of life (12). Regarding neuroradiological findings, all the patients in our series showed white matter changes and/or cerebellar structural abnormalities. We have previously reported the presence of minor signs of central involvement such as speech delay, brain atrophy and mild white matter lesions in some MDC1C patients, but such findings were thought to be non-specific or secondary to hypoxic complications related to the respiratory insufficiency in advanced stages of the disease (12). Therefore, MDC1C was initially classified as a CMD without neurological involvement, in contradistinction to the CMD-syndromes with structural brain abnormalities such as MEB, FCMD and WWS. Brain abnormalities such as agyria, pachygyria or polymicrogyria, hypomyelination of the white matter, cerebellar cysts or ponto-cerebellar hypoplasia are frequent findings in these three conditions (8, 9, 10). In our series, brain MRI was available for six of the seven patients and revealed cerebellar cysts in all of them, and hypoplasia of the vermis in five. MRI has not been performed in Patient 5 and, therefore, we cannot exclude the presence of posterior fossa abnormalities in this patient. Our patients did not show any gyral malformation in contrast to what may be observed in MEB, FCMD or WWS. White matter abnormalities were observed in five of our patients, with significant regression in one of them, detected by serial MRIs at 3 and 6 years of age. Such transient infra-myelination has also been described in MEB and FCMD (19, 20) while in primary laminin α2 chain deficient CMD, brain white matter abnormalities do not regress with time. It is also noteworthy that one patient presented with strabismus and another with ophthalmoplegia in the second decade of life.
Very recently, different FKRP homozygote mutations were identified in three Turkish patients, two with mental retardation, white matter changes and cerebellar cysts (13), and another with a typical Walker-Warburg syndrome (Beltran-Valero de Bernabé et al, 2003, personal communication). This finding raised the question of the role of specific homozygous FKRP mutations in the development of neurological disturbances. Our study revealing two new homozygous missense FKRP mutations, A455D and V405L, in seven additional patients of similar phenotype clearly supports this hypothesis. Fukutin related protein has been shown to be localized in the Golgi apparatus (21). It is involved in the glycosylation processing of α-dystroglycan, a heavily glycosylated membrane protein that forms a link between the actin associated cytoskeleton and the extracellular matrix via the laminin α2 chain (22). Hypoglycosylation of α-dystroglycan abolishes binding not only to laminin, but also to neurexin and agrin, and such abnormal interactions underlie the pathogenic mechanism of muscular dystrophy with brain abnormalities (23, 24). The mutations identified in our patients, V405L and A455D, are localized in the C-terminal domain (catalytic domain) of the fukutin related protein, where other severe MDC1C mutations (P448L, Y465S) without CNS involvement have also been identified. These different mutations may affect in various degrees the conformation or the catalytic activity of the protein and/or recognition sites for α-dystroglycan and other potential substrates (16). In contrast, the two mutations reported by Topaloglu et al, P315T and S221A, which cause a similar phenotype with CNS involvement are located outside the catalytic domain in the stem region. This region is believed to be involved in protein-protein interaction (13).
In summary, the report describes two new missense mutations in the FKRP gene in seven children with MDC1C, mental retardation and central nervous system abnormalities. While brain white matter changes may regress with time, structural cerebellar abnormalities were present early in life and did not seem to progress. Our results support the existence of particular homozygous FKRP mutations associated with severe neurological involvement, and relate MDC1C to other congenital muscular dystrophies with abnormal protein glycosylation and affecting brain function.
Acknowledgments
We thank the patients and their families for their participation. This work was supported by funds from the Secrétariat d’Etat à la Recherche Scientifique et à la Technologic (Tunisia), the Institut National de la Santé et de la Recherche Médicale (INSERM), Association Française contre les Myopathies (AFM), and the European Commission (contract N° QLG1-CT1999-00870).
References
- Dubowitz V, Fardeau M. Proceedings of the 27th ENMC sponsored workshop on congenital muscular dystrophy, 22–24 April 1994, The Netherlands. Neuromuscul Disord. 1995;5:253–258. doi: 10.1016/0960-8966(95)90011-x. [DOI] [PubMed] [Google Scholar]
- Tome FM, Evangelista T, Leclerc A, Sunada Y, Manole E, Estournet B, Barois A, Campbell KP, Fardeau M. Congenital muscular dystrophy with merosin deficiency. CR Acad Sci [III] 1994;317:351–357. [PubMed] [Google Scholar]
- Helbling-Leclerc A, Zhang X, Topaloglu H, Cruaud C, Tesson F, Weissenbach J, Tome FM, Schwartz K, Fardeau M, Tryggvason K, et al. Mutations in the laminin alpha2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat Genet. 1995;11:216–218. doi: 10.1038/ng1095-216. [DOI] [PubMed] [Google Scholar]
- Allamand V, Guicheney P. Merosin-deficient congenital muscular dystrophy, autosomal recessive (MDC1A, MIM#156225, LAMA2 gene coding for alpha2 chain of laminin) Eur J Hum Genet. 2002;10:91–94. doi: 10.1038/sj.ejhg.5200743. [DOI] [PubMed] [Google Scholar]
- Philpot J, Cowan F, Pennock J, Sewry C, Dubowitz V, Bydder G, Muntoni F. Merosin-deficient congenital muscular dystrophy: the spectrum of brain involvement on magnetic resonance imaging. Neuromuscul Disord. 1999;9:81–85. doi: 10.1016/s0960-8966(98)00110-2. [DOI] [PubMed] [Google Scholar]
- Pini A, Merlini L, Tome FM, Chevallay M, Gobbi G. Merosin-negative congenital muscular dystrophy, occipital epilepsy with periodic spasms and focal cortical dysplasia. Report of three Italian cases in two families. Brain Dev. 1996;18:316–322. doi: 10.1016/0387-7604(96)00028-9. [DOI] [PubMed] [Google Scholar]
- Sunada Y, Edgar TS, Lotz BP, Rust RS, Campbell KP. Merosin-negative congenital muscular dystrophy associated with extensive brain abnormalities. Neurology. 1995;45:2084–2089. doi: 10.1212/wnl.45.11.2084. [DOI] [PubMed] [Google Scholar]
- Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, Mizuno M, Inazu T, Mitsuhashi H, Takahashi S, Takeuchi M, Herrmann R, Straub V, Talim B, Voit T, Topaloglu H, Toda T, Endo T. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell. 2001;1:717–724. doi: 10.1016/s1534-5807(01)00070-3. [DOI] [PubMed] [Google Scholar]
- Beltran-Valero de Bernabe D, Currier S, Steinbrecher A, Celli J, van Beusekom E, van der Zwaag B, Kayserili H, Merlini L, Chitayat D, Dobyns WB, Cormand B, Lehesjoki AE, Cruces J, Voit T, Walsh CA, Bokhoven H, van Brunner HG. Mutations in O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet. 2002;71:1033–1043. doi: 10.1086/342975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Nakahori Y, Miyake M, Matsumura K, Kondo-Iida E, Nomura Y, Segawa M, Yoshioka M, Saito K, Osawa M, Hamano K, Sakakihara Y, Nonaka I, Nakagome Y, Kanazawa I, Nakamura Y, Tokunaga K, Toda T. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388–392. doi: 10.1038/28653. [DOI] [PubMed] [Google Scholar]
- Brockington M, Blake DJ, Prandini P, Brown SC, Torelli S, Benson MA, Ponting CP, Estournet B, Romero NB, Mercuri E, Voit T, Sewry CA, Guicheney P, Muntoni F. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet. 2001;69:1198–1209. doi: 10.1086/324412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quijano-Roy S, Galan L, Ferreiro A, Cheliout-Heraut F, Gray F, Fardeau M, Barois A, Guicheney P, Romero NB, Estournet B. Severe progressive form of congenital muscular dystrophy with calf pseudohypertrophy, macroglossia and respiratory insufficiency. Neuromuscl Disord. 2002;12:466–475. doi: 10.1016/s0960-8966(01)00331-5. [DOI] [PubMed] [Google Scholar]
- Topaloglu H, Brockington M, Yuva Y, Talim B, Haliloglu G, Blake D, Torelli S, Brown SC, Muntoni F. FKRP gene mutations cause congenital muscular dystrophy, mental retardation and cerebellar cysts. Neurology. 2003;60:988–992. doi: 10.1212/01.wnl.0000052996.14099.dc. [DOI] [PubMed] [Google Scholar]
- Kawazaki ES. Sample preparation from blood, cells, and other fluids. PCR protocols. In: Innis M, Gelffand D, Snisky G, White T, editors. A guide to methods and application. Academic Press; San Diego: 1990. pp. 146–52. [Google Scholar]
- Triki C, Louhichi N, Meziou M, Choyakh F, Kechaou MS, Jlidi R, Mhiri C, Fakhfakh F, Ayadi H. Merosin-deficient congenital muscular dystrophy with mental retardation and cerebellar cysts in three Tunisian patients unlinked to the LAMA2, FCMD, MEB and CMD1B. Neuromuscul Disord. 2003;13:4–12. doi: 10.1016/s0960-8966(02)00188-8. [DOI] [PubMed] [Google Scholar]
- Brockington M, Yuva Y, Prandini P, Brown SC, Torelli S, Benson MA, Herrmann R, Anderson LV, Bashir R, Burgunder JM, Fallet S, Romero N, Fardeau M, Straub V, Storey G, Pollitt C, Richard I, Sewry CA, Bushby K, Voit T, Blake DJ, Muntoni F. Mutations in the ukutin related-protein gene (FKRP) identifies limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet. 2001;10:2851–285. doi: 10.1093/hmg/10.25.2851. [DOI] [PubMed] [Google Scholar]
- Mercuri E, Brockington M, Straub V, Quijano-Roy S, Yuva Y, Herrmann R, Brown SC, Torelli S, Dubowitz V, Blake DJ, Romero NB, Estournet B, Sewry CA, Guicheney P, Voit T, Muntoni F. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann Neurol. 2003;53:537–542. doi: 10.1002/ana.10559. [DOI] [PubMed] [Google Scholar]
- Quijano-Roy S, Romero NB, Louhichi N, Brockington M, Many H, Yeliz Y, Richard P, Estournet B, Muntoni F, Fardeau F, Barois A, Guicheney P. FKRP (fukutin related protein) gene mutations associated with intermediate phenotype between CMD type 1C and LGMD2I (abstract) Neuromuscul Disord. 2002;12:743. [Google Scholar]
- Muntoni F, Valero de Bernabe B, Bittner R, Blake D, Bokhoven H, Brockington M, Brown S, Bushby K, Fiszman M, Gruenewald S, Merlini L, Quijano-Roy S, Romero N, Sabatelli P, Sewry CA, Straub V, Talim H, Topaloglu H, Voit T, Yurchenco PD, Urtizberea JA, Wewer U, Guicheney P, Naarden The Netherlands: (8th Workshop of the International Consortium on CMD; 3rd Workshop of the MYO-CLUSTER project GENRE) Neuromuscul Disord. 2003;13:579–88. doi: 10.1016/s0960-8966(03)00072-5. [DOI] [PubMed] [Google Scholar]
- Osawa M, Sumida S, Suzuki N, Arai Y, Ikenaka H, Murasugi H, Shishikura K, Suzuki H, Saito K, Fukuyama Y. Fukuyama type congenital progressive muscular dystrophy. In: Fukujama Y, Osawa M, Saito K, editors. Congenital muscular dystrophies. Elsevier Science; 1997. pp. 31–68. [Google Scholar]
- Esapa CT, Benson MA, Schroder JE, Martin-Rendon E, Brockington M, Brown SC, Muntoni F, Kroger S, Blake DJ. Functional requirements for fukutin-related protein in the Golgi apparatus. Hum Mol Genet. 2002;11:3319–3331. doi: 10.1093/hmg/11.26.3319. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmenbrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS, Dollar J, Nishino I, Kelley RI, Somer H, Straub V, Mathews KD, Moore SA, Campbell KP. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–422. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, Hoshi T, Campbell KP. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418:422–425. doi: 10.1038/nature00838. [DOI] [PubMed] [Google Scholar]