

SUMMARY
Background
Apoptosis of vascular cells is considered to be a major determinant of atherosclerotic plaque vulnerability and potential rupture. Plasmin can be generated in atherosclerotic plaques and recent in vitro data suggest that plasminogen activation may trigger vascular smooth muscle cell (VSMC) apoptosis.
Objective
To determine whether plasminogen activation may induce aortic VSMC apoptosis ex vivo and in vivo.
Methods and results
Mice with single or combined deficiencies of ApoE and PAI-1 were used. Ex vivo incubation of plasminogen (1.3 μM) with isolated aortic tunica media from PAI-1-deficient mice induced plasminogen activation and VSMC apoptosis, which was inhibited by α2-antiplasmin. In vivo, levels of plasmin, active caspase 3 and VSMC apoptotic index were significantly higher in atherosclerotic aortas from mice with combined ApoE−/− and PAI-1−/− deficiencies than in those from littermates with single ApoE deficiency. A parallel decrease in VSMC density was also observed.
Conclusions
These data strongly suggest that, in vivo, plasminogen activation may contribute to VSMC apoptosis in atherosclerotic plaques.
Keywords: Animals, Antiplasmin, pharmacology, Aorta, drug effects, metabolism, pathology, Apolipoproteins E, genetics, Apoptosis, Atherosclerosis, metabolism, pathology, Disease Models, Animal, Mice, Mice, Inbred C57BL, Mice, Knockout, Muscle, Smooth, Vascular, drug effects, metabolism, pathology, Plasmin, metabolism, Plasminogen, metabolism, Plasminogen Activator Inhibitor 1, genetics, Tunica Media, drug effects, metabolism, pathology
Keywords: apoptosis, atherosclerosis, genetically altered mice, plasminogen, vascular smooth muscle cell
Apoptosis is considered to be a major determinant of atherosclerotic plaque vulnerability and potential rupture1–3. Little is known, however, about the mechanisms that trigger apoptosis of vascular cells in atherosclerotic plaques. In human atherosclerotic specimens, apoptotic vascular smooth muscle cells (VSMC) were observed in areas with loss of pericellular adhesion 3, indicating that pericellular proteolysis could be involved 4. Proteases capable of degrading pericellular matrix components are indeed secreted by resident vascular cells and infiltrating inflammatory cells.5 Components of the fibrinolytic system, including plasmin(ogen), plasminogen activators (either tissue-type, t-PA, predominantly expressed by VSMC6–8, or urokinase-type, u-PA, predominantly expressed by infiltrating macrophages), and plasminogen activator inhibitor-1 (PAI-1),9–11 are present in human atherosclerotic lesions. The activity of plasmin promotes cell migration, regulates growth factor activity (e.g. Transforming Growth Factor-β, TGF-β)12 and induces extracellular matrix proteolysis, either directly, via degradation of adhesive glycoproteins, such as fibronectin 13 or laminin 14 or indirectly, via activation of matrix metalloproteinases 15. Actually, it was recently demonstrated, using primary cultures of rat and human arterial VSMCs, that t-PA constitutively secreted by these cells can generate plasmin on the cell surface and induces thereby the proteolysis of extracellular matrix proteins, cell retraction and finally cell detachment leading to VSMC apoptosis8.
The fibrinolytic system plays a complex role in atherosclerosis, as assessed in experimental models using mice with targeted inactivation of its main components.16 Thus, lack of PAI-1 in transgenic mice with combined apolipoprotein E and PAI-1 deficiencies (ApoE−/−:PAI-1−/−) resulted in elevated plasmin levels, accompanied with extracellular matrix desorganization, increased accumulation of macrophages and a reduced density of myofibroblasts in advanced atherosclerotic lesions17. On the basis of the deleterious effect of plasmin on VSMC survival,8 we sought to investigate if plasminogen activation by VSMCs ex vivo on the one hand, and the increased plasmin generation within ApoE−/−:PAI-1−/− aortic plaques in vivo on the other hand could lead to VSMC apoptosis. We demonstrate that aortic VSMC apoptosis can be induced ex vivo by plasminogen activation. In vivo, VSMC apoptosis and plasmin activity were colocalized within atherosclerotic plaques. This mechanism may contribute to plaque destabilization and rupture.
METHODS
Reagents and animals
Ten weeks old mice with targeted inactivation of the gene encoding PAI-1 (PAI-1−/−) and wild-type (WT) mice of the same genetic background (75% C57B1/6 and 25% 129SV) and of either sex were obtained as described elsewhere 18.
Human or murine plasminogen, plasmin, and α2-antiplasmin were obtained as described 19,20. Primary monoclonal antibodies used are described below. Secondary antibodies used were goat anti-mouse-Alexa fluor® 568 (Molecular Probes, Eugene, Oregon, USA) and biotinylated rabbit anti-mouse Ig (Dako, Glostrup, Denmark).
Animal models
For ex vivo experiments, WT and PAI-1−/− mice were anesthetized and exsanguinated by perfusion at physiological pressure via cardiac puncture with a 0.9% NaCl solution. The aorta was dissected, the adventitia was extruded as described 21, and the aorta was cut first longitudinaly (to achieve de-endothelialization), and then transversely in 6 pieces of equal size. These sections were incubated in a humidified CO2 incubator at 37°C for 4 hours in Dulbecco’s modified Eagle’s Medium (without phenol red) containing 2 mM glutamine, 100 IU/ml penicillin and 0.1 mg/ml streptomycin, with and without 1.3 μM plasminogen, and in the absence or the presence of 1μM α2-antiplasmin. The aorta fragments were transferred to Jung tissue freezing medium™ (Leica Instruments, Nussloch, Germany) and snapfrozen in precooled 2-methyl butane.
For in vivo experiments, ApoE+/+:PAI-1−/− mice were intercrossed with ApoE−/−:PAI-1+/+ mice to generate breeding pairs with heterozygous deficiency of ApoE and PAI-1 (ApoE+/−:PAI-1+/−), which sired ApoE−/−:PAI-1−/− mice and ApoE−/−:PAI-1+/+ littermate offspring with a mixed genetic background of 87.5% C57B1/6 and 12.5% 129/SvJ, as described 17. Mice were kept on a regular chow diet for 5 weeks, and then fed a cholesterol/cholate rich diet for 25 weeks, as described 17. Aorta embedding procedures were previously described17.
For all surgical procedures, mice were anesthetized by intraperitoneal injection of Nembutal (60 mg/kg; Abbott Laboratories, North Chicago, IL, USA). All procedures were approved by the University Ethical Committee (P03112) and were performed in accordance with the guidelines of the International Society on Thrombosis and Haemostasis 22.
Protein assays
For immunoblotting of plasmin(ogen) and active caspase 3, equal protein amounts from whole protein extracts (from in vivo experiments) were electrophoresed on a 15% acrylamide gel, under reducing conditions. The nonspecific sites of the membranes were blocked with 10% non-fat dry milk in Tris Buffered Saline (TBS) containing 1% Tween 20 (TBST). The membranes were incubated with a rabbit anti-human/mouse active caspase 3 antibody (R&D systems) or a rabbit anti-murine plasmin(ogen)23 antibody overnight at 4°C in TBST containing 1% non-fat dry milk. Then, the membranes were washed and incubated with an anti-rabbit peroxidase-conjugated secondary antibody in TBST containing 1% non-fat dry milk. The membranes were washed with TBST, followed by detection with enhanced chemiluminiscence (ECL kit, Amersham).
Fibrinolytic activity was monitored by fibrin overlay (containing traces of plasminogen, with and without aprotinin) of non-fixed 8-μm arterial cryost at sections at 37°C for 24 hours 24.
Histological and immunohistochemical studies
Tissue sections (8 μm thick) were stained with hematoxylin-eosin under standard conditions. The following primary antibodies were used: for immunodetection of plasmin-α2-antiplasmin complexes, 7 AP (a mouse monoclonal antibody that recognizes neoantigen epitopes in the complexes but neither free plasmin nor α2-antiplasmin 25), for plasmin(ogen) a rabbit anti-murine plasminogen/plasmin 23, for active caspase 3 a rabbit anti-human/mouse active caspase 3 (R&D systems) and for VSMC detection a mouse anti-human smooth muscle actin (DAKO). Biotinylated secondary antibodies were applied in combination with the Vectastain system (ABC kit, Vector Laboratories Inc, Burlingame, CA, USA), using the appropriate negative controls.
VSMC were detected in paraffin embedded sections from in vivo experiments using a primary mouse monoclonal antibody against human α-actin and a secondary goat anti-mouse antibody labelled with Alexa fluor ® 568. A TUNEL reaction, using the appropriate negative controls (yielding FITC staining of apoptotic nuclei, with negligible background), and a nuclear counterstaining with DAPI were then performed. Cryosections from ex vivo experiments were also submitted to the TUNEL and DAPI reactions. Apoptotic VSMCs were defined as doubly (FITC and DAPI: ex vivo) or triply labelled cells (DAPI, FITC, and Alexa fluor 568: in vivo), and an apoptotic index was calculated using the formula: (number of TUNEL-positive VSMC/total VSMC)×100.
Images of the same microscopic (Zeiss Axioplan 2) fields were taken with each filter set (DAPI, FITC and Alexa 568), using a Zeiss AxioCam HRc digital camera with Zeiss Axiovision 3.0.6.38 SP4 Imaging Software, and were merged by using Adobe Photoshop software. Quantifications (areas and cell numbers) were performed by computer-assisted image analysis with the Zeiss KS300 Version 3.0 SP6 software. Briefly, the positive and negative cells within a defined area were counted automatically using a fixed threshold contrast. The operating system and the video-adaptor were Microsoft Windows 2000 SP1, and Matrox Meteor_II PCI frame grabber, respectively. Morphometric analyses were performed blinded for the genotype; for ex vivo experiments 18 areas in 6 sections (each 160 μm apart) randomly selected through the aortic segment were analyzed, and for in vivo experiments 18 areas randomly chosen in 3 sections (each 160 μm apart) were analyzed, the first one taken at the point where the cardiac valves were first visible.
Transmission electron microscopy was performed as described 26.
Statistical analysis
The statistics were performed with the Statview 5.0 software. Results are expressed as mean ± SEM or median (range). Comparisons were made by one-way analysis of variance with Scheffe’s F test, or Wilcoxon signed ranks, or Mann-Whitney U-test, as appropriate. Statistical significance was set at P < 0.05.
RESULTS
Aortic VSMC apoptosis parallels plasminogen activation ex vivo
In order to isolate the effect of plasminogen activation on VSMC apoptosis from other cellular components of the vascular wall, we performed ex vivo experiments on isolated aortic tunica media from WT and PAI-1−/− mice. The tunica media were incubated with plasminogen at 1.3 μM for 4 h, with or without 1 μM α2-antiplasmin. Plasmin generation developed faster in PAI-1−/− than in WT aortas, as assessed by in situ fibrin zymography (Fig. 1A). VSMC apoptotic levels, as assessed by TUNEL, was not significantly different between both genotypes in the absence of plasminogen (n=9, P=0.1). Following plasminogen activation, PAI-1−/− aortas displayed a fivefold increase in VSMC apoptosis (n= 9, P= 0.03) that was inhibited by α2-antiplasmin, thus suggesting a plasmin-dependent effect (Fig. 1B–C). In contrast, apoptotic levels in WT aortas remained unchanged following the incubation with plasminogen (n=9, P>0.99; Fig. 1 B–C).
Figure 1. Plasminogen activation within the aortic wall leads to ex vivo VSMC apoptosis.
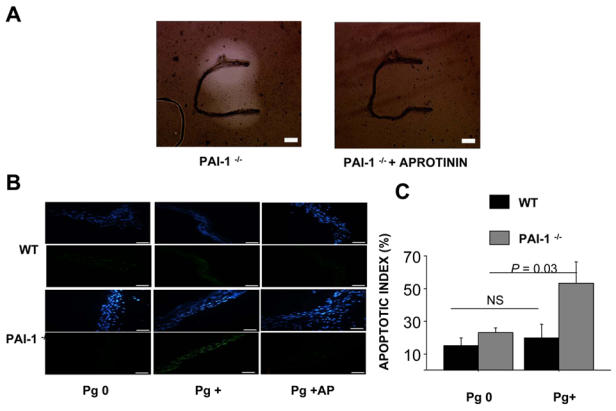
A. In situ fibrin zymography for 24 hours on de-endothelialized aortic tunica media from a PAI-1−/− mouse in the absence (left panel) or in the presence of 10 k.I.U/ml aprotinin (right panel). The scale bar corresponds to 100 μm.
B, C. De-endothelialized aortic tunica media from WT and PAI-1−/− mice were incubated for 4 hours without (Pg 0) or with 1.3 μM plasminogen (Pg+), without and with 1 μM α2-antiplasmin (Pg+AP). n= 5 to 9 in each condition tested.
B. Representative WT (upper panels) and PAI-1−/− (lower panels) mouse de-endothelialized aortas, stained with DAPI (blue, for DNA detection) after the TUNEL reaction (green, for DNA fragmentation). No increased apoptosis was detectable in WT aortas after 4 hours of plasminogen incubation, whereas PAI-1−/− aortas displayed increased apoptotic levels (Pg+), inhibited in the presence of α2-antiplasmin (Pg + AP). The scale bar corresponds to 20 μm.
C. VSMC apoptotic index (%) in aortas of WT and PAI-1−/− mice.
Increased plasmin generation and VSMC apoptosis in atherosclerotic plaques from ApoE−/−:PAI-1−/− mice
To investigate the involvement of the plasminogen activation system in apoptosis of vascular cells in vivo and its relevance for atherosclerosis, we analyzed atherosclerotic and non-atherosclerotic aorta fragments from ApoE−/−:PAI-1+/+ and ApoE−/−:PAI-1−/− mice. In both genotypes, plasmin activity was increased in extracts from aortic areas with plaques, as compared to areas without plaques, being six-fold more abundant (n=4, P<0.05) in plaques from ApoE−/−:PAI-1−/− mice as compared to plaques from ApoE−/−:PAI-1+/+ mice (Fig. 2A, upper panel). Activated caspase-3 (Fig. 2A, lower panel) was barely detectable in areas without plaques, in either genotype (n=4, P= 0.96) but was concomitantly increased six-fold in plaques from ApoE−/−:PAI-1−/− mice (plaque vs non plaque, n=4, P= 0.003) but not in ApoE−/−:PAI-1+/+ mice (n=4, P= 0.24). Similarly, caspase-3 activity levels were higher in plaques from ApoE−/−:PAI-1−/− as compared to plaques from ApoE−/−:PAI-1+/+ mice (n=4, P= 0.04). In short, active caspase-3 levels paralleled plasmin levels in atherosclerotic plaques. Tissue section analysis confirmed that plasminogen activation as assessed by plasmin-α2-antiplasmin complexes (data not shown), plasmin detection (Fig. 2B) and apoptosis (activated caspase 3 in Fig. 2C, TUNEL in Fig. 2D and electron microscopy analysis in Fig. 2E), were present within the aortic intima only in areas with plaques, in both genotypes. Apoptosis within atherosclerotic plaques was a patchy phenomenon, but apoptotic cells were mainly located in the fibrous cap and were predominantly foam-cells.
Figure 2. Differential expression patterns of plasmin and active caspase 3 in aorta compartments of ApoE−/− :PAI-1+/+ and ApoE−/− :PAI-1−/− mice.
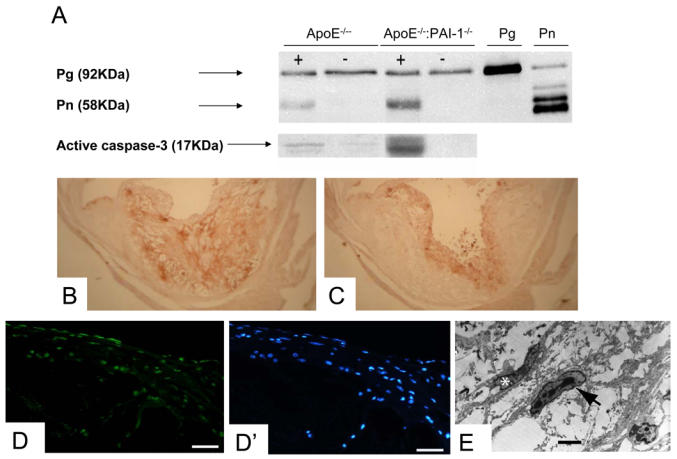
A. Plasminogen (Pg), plasmin (Pn) (upper gel) and active caspase 3 (lower gel) were detected by Western blot under reducing conditions in the same extracts from aortic areas with (+) and without (−) atherosclerotic plaques of ApoE−/− :PAI-1+/+(ApoE−/−) and ApoE−/− :PAI-1−/− mice.
B,C. Light microscopic analysis of an ascending aorta section in a representative ApoE−/−:PAI-1−/− mouse, after immunostaining for plasmin(ogen) (B) or active caspase 3 (C). Both were detected within atherosclerotic plaques but not in the tunica media nor in areas without atherosclerotic plaques.
D (TUNEL reaction) and D′ (staining with DAPI) focused on a fibrous cap within an atherosclerotic plaque in a representative ApoE−/−:PAI-1−/− mouse. Magnification bar: 20 μm.
E. Transmission electron micrograph of the aortic intima of a representative ApoE−/− :PAI-1−/− mouse showing the presence of a VSMC with chromatin condensation and fragmentation (arrow), which represent distinctive ultrastructural features of apoptotic cells. A VSMC with normal nucleus is marked with asterisk. Magnification bar: 10 μm.
We used the TUNEL to quantitatively compare the VSMC index in plaques from both genotypes. Since about 25% of the examined areas did not contain α-actin positive cells, to overcome a potential bias, we calculated the VSMC apoptotic index as a ratio (apoptotic VSMC/total VSMC)×100, each parameter being the average of the 18 values collected from the 18 areas (from 3 sections, 100μm apart) evaluated per mouse. ApoE−/−:PAI-1−/− plaques had a significantly lower VSMC density (13±1% vs. 23±2%, P<0.05, n=5) but the total cellular density was not significantly different between both genotypes. Although the overall apoptotic cell density was not significantly increased in ApoE−/−:PAI-1−/− mice plaques, the VSMC apoptotic index was significantly higher within atherosclerotic plaques from ApoE−/−:PAI-1−/−(1.5% (0–9.5), n=8) as compared to ApoE−/−:PAI-1−/− mice (0% (0–0.5), n=7, P<0.03). Averaging in each mouse the indices calculated in each individual area (i.e omitting areas without α-actin positive cells, where the apoptotic index could not be calculated) yielded similar trends (not shown).
DISCUSSION
Cell death by apoptosis is considered to be a major determinant of atherosclerotic plaque vulnerability 1–3. VSMCs can stabilize atherosclerotic plaques by maintaining the tensile strength of the fibrous cap via the synthesis of collagen isoforms and protease inhibitors. VSMC apoptosis may therefore potentially contribute to the instability and rupture of plaques. Although apoptosis has been detected in human plaques, the mechanisms that trigger this apoptosis and its contribution to VSMC loss, remain unclear.
It has been reported that VSMCs in atherosclerotic lesions may undergo apoptosis in response to effectors secreted by infiltrating inflammatory cells. Macrophages, for instance, may contribute to VSMC apoptosis by direct cell-cell contact, Fas-L/Fas signaling, nitric oxide and TNF-α production27, 28. However, VSMC autocrine destruction has not been demonstrated as yet, although a study suggested that self autodestruction may occur via Fas-mediated apoptosis by relocating Fas-L to the VSMC surface.29 t-PA, an other glycoprotein constitutively expressed by VSMCs30, can be located at the cell surface where it transforms plasminogen into plasmin, which triggers proteolysis-induced cell detachment and apoptosis.8 The hypothesis that this sequence of reactions may be participating in the apoptosis of vascular cells on atherosclerotic plaques in vivo, was evaluated in this study. The well characterized ApoE−/− atherosclerosis prone mouse model mimics several features of human atherosclerosis31, 32, including the lack of VSMC apoptosis in the normal media 1,2,35. Moreover, apoptosis is a heterogeneous and patchy phenomenon in this murine model, as also observed in humans 2,36. The combined deficiency of PAI-1 with ApoE−/− allows a better assessment of the effects of plasminogen activation on atherosclerosis progression in vivo while discarding possible direct effects of PAI-1 on apoptosis33 34. Indeed, in both genotypes VSMC apoptosis was undetectable in aortic areas without atherosclerotic plaques.
Corroborating previous in vitro studies8, our ex vivo experiments revealed that, in the presence of physiological circulating plasminogen concentrations, VSMC apoptosis was associated with plasmin generation in PAI-1−/− aortic tunica media. In the absence of plasminogen, similar apoptotic rates were observed in WT and PAI-1−/− VSMCs, suggesting that PAI-1 is not a pro-apoptotic factor in this model. Furthermore, we recently showed that fibroblasts in two or three dimensional culture systems became resistant to plasminogen activation-induced cell detachment and apoptosis when transfected either with PAI-1 or protease-nexin-1; these serpins inhibited plasminogen activation at the cell surface and subsequent pericellular proteolysis35. These findings are in agreement with previous data indicating that addition of PAI-1 inhibited plasminogen activation-induced gel contraction and capillary regression, whereas anti-PAI-1 antibodies potentiated these processes36.
Expression of the main components of the plasminogen/plasmin system has been documented in the atherosclerotic arterial wall of mice and humans 9, 17. To test whether this system may be involved in vivo in VSMC apoptosis within plaques, we have compared VSMC apoptosis in ApoE−/− :PAI-1+/+ and ApoE−/− :PAI-1−/− mice. Our data show that PAI-1 deficiency is associated with strongly enhanced apoptosis within advanced atherosclerotic plaques in ApoE deficient mice, as assessed by active caspase 3 generation. Election microscopy revealed that most apoptotic cells were foam cells, probably of macrophage and VSMC origin. Our TUNEL results suggest that among the plaque cells, VSMCs maybe particularly sensitive to plasmin-induced apoptosis, since the overall and non-VSMC apoptotic rates were not significantly different between both genotypes. In contrast, VSMCs were almost only detected within plaques from ApoE−/− :PAI-1−/− mice. Moreover, we observed an associated decrease in VSMC cell density in ApoE−/− :PAI-1−/− mice, whereas the overall cell density did not significantly differ between both genotypes. A similar decrease in VSMC content and increase in caspase activity, was also observed in the inflammatory region of human carotid atherosclerotic plaques as reported recently37.
The apoptosis observed in ApoE−/− :PAI-1−/− plaques appears to be dependent on plasminogen activation, as suggested by the six-fold higher plasmin activity detected in atherosclerotic extracts of ApoE−/− :PAI-1−/− as compared to ApoE−/− :PAI-1+/+ mice, and by the colocalization of plasmin and active caspase 3 in situ. An extracellular pathway leading to plasmin induced cell detachment and apoptosis of VSMCs has recently been described.8 A similar plasmin(ogen)-dependent cell detachment mechanism has also been described for retinal ganglion cells 38, endothelial cells 36, 39, fibroblasts 35, and cerebrovascular smooth muscle cells 7, thus suggesting that autocrine production of plasminogen activators by VSMCs and other cell types may induce apoptotic autodestruction by cell surface-formed plasmin. As in vitro studies have revealed that plaque-infiltrating inflammatory cells may degrade pericellular matrix components 4, it is possible that in our atherosclerotic plaque mouse model, paracrine expression of urokinase by macrophages may also contribute to vascular cell detachment and apoptosis. Of note, macrophages overexpressing urokinase show accelerated atherosclerosis in ApoE−/− mice and had elevated lesion proteolytic activity that may causes plaque rupture40. Other proteases (elastase, chymase, granzyme B, matrix metalloproteinases) secreted by macrophages, T-lymphocytes and mast cells, may also trigger apoptosis by breaking-down cell-extracellular matrix interactions 4.
Plasmin may also activate growth factors such as latent TGF-β, which may induce VSMC apoptosis 41. Interestingly, we found, in ApoE−/− :PAI-1−/− mice, more pronounced extracellular matrix disorganisation, proMMP activation, and activation of latent TGF-β, as compared to ApoE−/− :PAI-1+/+ mice 17.
Detection of cell apoptosis by TUNEL may have some potential limitations. Firstly, it is known that the TUNEL technique may overestimate the apoptotic rate, especially in highly proliferative cells 42. However, the proliferation rate of VSMC is known to be low in advanced atherosclerotic plaques 2, 43, 44. Comparing the two ApoE−/− :PAI-1 genotypes, we observed no significant background, and our VSMC apoptotic rates were comparable to those observed in other studies using stringent criteria to define apoptosis 42. Secondly, VSMCs within atherosclerotic plaques may lose α-actin immunoreactivity, thus compromising identification of VSMC-derived foam cells and determination of the apoptotic rate 1, 42, 44. However, active caspase 3 detection within atherosclerotic extracts corroborated our TUNEL data and, most importantly, it parallelled plasmin activity.
In summary, we have found that ex vivo plasmin generation may lead to apoptosis of murine VSMCs. In vivo, plasmin activity in atherosclerotic plaques was associated with VSMC apoptosis in ApoE deficient mice, suggesting that plasmin may induce VSMC apoptosis during atherogenesis. Bot et al. 45 reported that the serine protease inhibitor Serp-1 (which inhibits plasmin and plasminogen activators) impaired atherosclerotic lesion formation and stabilized plaques in ApoE−/− mice. Therapeutic strategies aimed at preventing pericellular plasminogen activation might thus be beneficial to stabilize atherosclerotic plaques.
Acknowledgments
Patrick Rossignol received a grant from the Institut National de la Santé et de la Recherche Médicale (INSERM, France), and was on leave of absence from INSERM U698. P.R., J.-L.M.V., F.L., E. AC., H.R.L. were granted by the Leducq Foundation (Transatlantic Networks).
References
- 1.Geng YJ, Libby P. Evidence for apoptosis in advanced human atheroma. Colocalization with interleukin-1 beta-converting enzyme. Am J Pathol. 1995;147:251–266. [PMC free article] [PubMed] [Google Scholar]
- 2.Han DK, Haudenschild CC, Hong MK, Tinkle BT, Leon MB, Liau G. Evidence for apoptosis in human atherogenesis and in a rat vascular injury model. Am J Pathol. 1995;147:267–277. [PMC free article] [PubMed] [Google Scholar]
- 3.Bauriedel G, Hutter R, Welsch U, Bach R, Sievert H, Luderitz B. Role of smooth muscle cell death in advanced coronary primary lesions: implications for plaque instability. Cardiovasc Res. 1999;41:480–488. doi: 10.1016/s0008-6363(98)00318-6. [DOI] [PubMed] [Google Scholar]
- 4.Lindstedt KA, Leskinen MJ, Kovanen PT. Proteolysis of the Pericellular Matrix. A Novel Element Determining Cell Survival and Death in the Pathogenesis of Plaque Erosion and Rupture. Arterioscler Thromb Vasc Biol. 2004;24:1–9. doi: 10.1161/01.ATV.0000135322.78008.55. [DOI] [PubMed] [Google Scholar]
- 5.Garcia-Touchard A, Henry TD, Sangiorgi G, Spagnoli LG, Mauriello A, Conover C, Schwartz RS. Extracellular proteases in atherosclerosis and restenosis. Arterioscler Thromb Vasc Biol. 2005;25:1119–1127. doi: 10.1161/01.ATV.0000164311.48592.da. [DOI] [PubMed] [Google Scholar]
- 6.Houard X, Monnot C, Dive V, Corvol P, Pagano M. Vascular smooth muscle cells efficiently activate a new proteinase cascade involving plasminogen and fibronectin. J Cell Biochem. 2003;88:1188–1201. doi: 10.1002/jcb.10460. [DOI] [PubMed] [Google Scholar]
- 7.Davis J, Wagner MR, Zhang W, Xu F, Van Nostrand WE. Amyloid beta-protein stimulates the expression of urokinase-type plasminogen activator (uPA) and its receptor (uPAR) in human cerebrovascular smooth muscle cells. J Biol Chem. 2003;278:19054–19061. doi: 10.1074/jbc.M301398200. [DOI] [PubMed] [Google Scholar]
- 8.Meilhac O, Ho-Tin-Noe B, Houard X, Philippe M, Michel JB, Angles-Cano E. Pericellular plasmin induces smooth muscle cell anoikis. Faseb J. 2003;17:1301–1303. doi: 10.1096/fj.02-0687fje. [DOI] [PubMed] [Google Scholar]
- 9.Raghunath PN, Tomaszewski JE, Brady ST, Caron RJ, Okada SS, Barnathan ES. Plasminogen activator system in human coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 1995;15:1432–1443. doi: 10.1161/01.atv.15.9.1432. [DOI] [PubMed] [Google Scholar]
- 10.Fontaine V, Jacob MP, Houard X, Rossignol P, Plissonnier D, Angles-Cano E, Michel JB. Involvement of the mural thrombus as a site of protease release and activation in human aortic aneurysms. Am J Pathol. 2002;161:1701–1710. doi: 10.1016/S0002-9440(10)64447-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stoop AA, Lupu F, Pannekoek H. Colocalization of thrombin, PAI-1, and vitronectin in the atherosclerotic vessel wall: A potential regulatory mechanism of thrombin activity by PAI-1/vitronectin complexes. Arterioscler Thromb Vasc Biol. 2000;20:1143–1149. doi: 10.1161/01.atv.20.4.1143. [DOI] [PubMed] [Google Scholar]
- 12.Grainger DJ, Kemp PR, Liu AC, Lawn RM, Metcalfe JC. Activation of transforming growth factor- beta is inhibited in transgenic apolipoprotein(a) mice. Nature. 1994;370:460–462. doi: 10.1038/370460a0. [DOI] [PubMed] [Google Scholar]
- 13.Bonnefoy A, Legrand C. Proteolysis of subendothelial adhesive glycoproteins (fibronectin, thrombospondin, and von Willebrand factor) by plasmin, leukocyte cathepsin G, and elastase. Thromb Res. 2000;98:323–332. doi: 10.1016/s0049-3848(99)00242-x. [DOI] [PubMed] [Google Scholar]
- 14.Liotta LA, Goldfarb RH, Terranova VP. Cleavage of laminin by thrombin and plasmin: alpha thrombin selectively cleaves the beta chain of laminin. Thromb Res. 1981;21:663–673. doi: 10.1016/0049-3848(81)90268-1. [DOI] [PubMed] [Google Scholar]
- 15.Lijnen HR. Plasmin and matrix metalloproteinases in vascular remodeling. Thromb Haemost. 2001;86:324–333. [PubMed] [Google Scholar]
- 16.Carmeliet P, Moons L, Dewerchin M, Mackman N, Luther T, Breier G, Ploplis VA, Müller M, Nagy A, Plow E, Gerard R, Edgington T, Risau W, Collen D. Insights in vessel development and vascular disorders using targeted inactivation and transfer of vascular endothelial growth factor, the tissue factor receptor, and the plasminogen system. Ann N Y Acad Sci. 1997;811:191–206. doi: 10.1111/j.1749-6632.1997.tb52002.x. [DOI] [PubMed] [Google Scholar]
- 17.Luttun A, Lupu F, Storkebaum E, Hoylaerts MF, Moons L, Crawley J, Bono F, Poole AR, Tipping P, Herbert JM, Collen D, Carmeliet P. Lack of plasminogen activator inhibitor-1 promotes growth and abnormal matrix remodeling of advanced atherosclerotic plaques in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2002;22:499–505. doi: 10.1161/hq0302.104529. [DOI] [PubMed] [Google Scholar]
- 18.Carmeliet P, Kieckens L, Schoonjans L, Ream B, van Nuffelen A, Prendergast G, Cole M, Bronson R, Collen D, Mulligan RC. Plasminogen activator inhibitor-1 gene-deficient mice. I. Generation by homologous recombination and characterization. J Clin Invest. 1993;92:2746–2755. doi: 10.1172/JCI116892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lijnen HR, van Hoef B, Beelen V, Collen D. Characterization of the murine plasma fibrinolytic system. Eur J Biochem. 1994;224:863–871. doi: 10.1111/j.1432-1033.1994.00863.x. [DOI] [PubMed] [Google Scholar]
- 20.Lijnen HR, Van Hoef B, Collen D. Characterization of the murine plasminogen/urokinase-type plasminogen activator system. Eur J Biochem. 1996;241:840–848. doi: 10.1111/j.1432-1033.1996.00840.x. [DOI] [PubMed] [Google Scholar]
- 21.Battle T, Arnal JF, Challah M, Michel JB. Selective isolation of rat aortic wall layers and their cell types in culture-application to converting enzyme activity measurement. Tissue Cell. 1994;26:943–955. doi: 10.1016/0040-8166(94)90043-4. [DOI] [PubMed] [Google Scholar]
- 22.Giles AR. Guidelines for the use of animals in biomedical research. Thromb Haemost. 1987;58:1078–1084. [PubMed] [Google Scholar]
- 23.Lijnen HR, Okada K, Matsuo O, Collen D, Dewerchin M. Alpha2-antiplasmin gene deficiency in mice is associated with enhanced fibrinolytic potential without overt bleeding. Blood. 1999;93:2274–2281. [PubMed] [Google Scholar]
- 24.Lijnen HR, Van Hoef B, Dewerchin M, Collen D. Alpha(2)-antiplasmin gene deficiency in mice does not affect neointima formation after vascular injury. Arterioscler Thromb Vasc Biol. 2000;20:1488–1492. doi: 10.1161/01.atv.20.6.1488. [DOI] [PubMed] [Google Scholar]
- 25.Wimazal F, Sperr WR, Horny HP, Carroll V, Binder BR, Fonatsch C, Walchshofer S, Fodinger M, Schwarzinger I, Samorapoompichit P, Chott A, Dvorak AM, Lechner K, Valent P. Hyperfibrinolysis in a case of myelodysplastic syndrome with leukemic spread of mast cells. Am J Hematol. 1999;61:66–77. doi: 10.1002/(sici)1096-8652(199905)61:1<66::aid-ajh12>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 26.Chen K, Wight TN. Proteoglycans in arterial smooth muscle cell cultures: an ultrastructural histochemical analysis. J Histochem Cytochem. 1984;32:347–357. doi: 10.1177/32.4.6200530. [DOI] [PubMed] [Google Scholar]
- 27.Boyle JJ, Bowyer DE, Weissberg PL, Bennett MR. Human blood-derived macrophages induce apoptosis in human plaque-derived vascular smooth muscle cells by Fas-ligand/Fas interactions. Arterioscler Thromb Vasc Biol. 2001;21:1402–1407. doi: 10.1161/hq0901.094279. [DOI] [PubMed] [Google Scholar]
- 28.Boyle JJ, Weissberg PL, Bennett MR. Human macrophage-induced vascular smooth muscle cell apoptosis requires NO enhancement of Fas/Fas-L interactions. Arterioscler Thromb Vasc Biol. 2002;22:1624–1630. doi: 10.1161/01.atv.0000033517.48444.1a. [DOI] [PubMed] [Google Scholar]
- 29.Chan SW, Hegyi L, Scott S, Gary NR, Weissberg PL, Bennett MR. Sensitivity to Fas-mediated apoptosis is determined below receptor level in human vascular smooth muscle cells. Circ Res. 2000;86:1038–1046. doi: 10.1161/01.res.86.10.1038. [DOI] [PubMed] [Google Scholar]
- 30.Razzaq TM, Bass R, Vines DJ, Werner F, Whawell SA, Ellis V. Functional regulation of tissue plasminogen activator on the surface of vascular smooth muscle cells by the type-II transmembrane protein p63 (CKAP4) J Biol Chem. 2003;278:42679–42685. doi: 10.1074/jbc.M305695200. [DOI] [PubMed] [Google Scholar]
- 31.Reddick RL, Zhang SH, Maeda N. Atherosclerosis in mice lacking apo E. Evaluation of lesional development and progression. Arterioscler Thromb. 1994;14:141–147. doi: 10.1161/01.atv.14.1.141. [DOI] [PubMed] [Google Scholar]
- 32.Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- 33.Al-Fakhri N, Chavakis T, Schmidt-Woll T, Huang B, Cherian SM, Bobryshev YV, Lord RS, Katz N, Preissner KT. Induction of apoptosis in vascular cells by plasminogen activator inhibitor-1 and high molecular weight kininogen correlates with their anti-adhesive properties. Biol Chem. 2003;384:423–435. doi: 10.1515/BC.2003.048. [DOI] [PubMed] [Google Scholar]
- 34.Kwaan HC, Wang J, Svoboda K, Declerck PJ. Plasminogen activator inhibitor 1 may promote tumour growth through inhibition of apoptosis. Br J Cancer. 2000;82:1702–1708. doi: 10.1054/bjoc.2000.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rossignol P, Ho-Tin-Noe B, Vranckx R, Bouton MC, Meilhac O, Lijnen HR, Guillin MC, Michel JB, Angles-Cano E. Protease-nexin-1 inhibits plasminogen activation-induced apoptosis of adherent cells. J Biol Chem. 2004;279:10346–10356. doi: 10.1074/jbc.M310964200. [DOI] [PubMed] [Google Scholar]
- 36.Davis GE, Pintar Allen KA, Salazar R, Maxwell SA. Matrix metalloproteinase-1 and -9 activation by plasmin regulates a novel endothelial cell-mediated mechanism of collagen gel contraction and capillary tube regression in three-dimensional collagen matrices. J Cell Sci. 2001;114:917–930. doi: 10.1242/jcs.114.5.917. [DOI] [PubMed] [Google Scholar]
- 37.Martin-Ventura JL, Blanco-Colio LM, Munoz-Garcia B, Gomez-Hernandez A, Arribas A, Ortega L, Tunon J, Egido J. NF-kappaB activation and Fas ligand overexpression in blood and plaques of patients with carotid atherosclerosis: potential implication in plaque instability. Stroke. 2004;35:458–463. doi: 10.1161/01.STR.0000114876.51656.7A. [DOI] [PubMed] [Google Scholar]
- 38.Zhang X, Chaudhry A, Chintala SK. Inhibition of plasminogen activation protects against ganglion cell loss in a mouse model of retinal damage. Mol Vis. 2003;9:238–248. [PubMed] [Google Scholar]
- 39.Reijerkerk A, Mosnier LO, Kranenburg O, Bouma BN, Carmeliet P, Drixler T, Meijers JC, Voest EE, Gebbink MF. Amyloid endostatin induces endothelial cell detachment by stimulation of the plasminogen activation system. Mol Cancer Res. 2003;1:561–568. [PubMed] [Google Scholar]
- 40.Cozen AE, Moriwaki H, Kremen M, DeYoung MB, Dichek HL, Slezicki KI, Young SG, Veniant M, Dichek DA. Macrophage-targeted overexpression of urokinase causes accelerated atherosclerosis, coronary artery occlusions, and premature death. Circulation. 2004;109:2129–2135. doi: 10.1161/01.CIR.0000127369.24127.03. Epub 2004 Apr 2119. [DOI] [PubMed] [Google Scholar]
- 41.Hishikawa K, Nakaki T, Fujii T. Transforming growth factor-beta induces apoptosis via connective tissue growth factor in human aortic smooth muscle cells. Eur J Pharmacol. 1999;385:287–290. doi: 10.1016/s0014-2999(99)00763-3. [DOI] [PubMed] [Google Scholar]
- 42.Kockx MM, De Meyer GR, Muhring J, Jacob W, Bult H, Herman AG. Apoptosis and related proteins in different stages of human atherosclerotic plaques. Circulation. 1998;97:2307–2315. doi: 10.1161/01.cir.97.23.2307. [DOI] [PubMed] [Google Scholar]
- 43.Bennett MR, Evan GI, Schwartz SM. Apoptosis of human vascular smooth muscle cells derived from normal vessels and coronary atherosclerotic plaques. J Clin Invest. 1995;95:2266–2274. doi: 10.1172/JCI117917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lutgens E, de Muinck ED, Kitslaar PJ, Tordoir JH, Wellens HJ, Daemen MJ. Biphasic pattern of cell turnover characterizes the progression from fatty streaks to ruptured human atherosclerotic plaques. Cardiovasc Res. 1999;41:473–479. doi: 10.1016/s0008-6363(98)00311-3. [DOI] [PubMed] [Google Scholar]
- 45.Bot I, von der Thusen JH, Dormers MM, Lucas A, Fekkes ML, de Jager SC, Kuiper J, Daemen MJ, van Berkel TJ, Heeneman S, Biessen EA. Serine protease inhibitor Serp-1 strongly impairs atherosclerotic lesion formation and induces a stable plaque phenotype in ApoE−/−mice. Circ Res. 2003;93:464–471. doi: 10.1161/01.RES.0000090993.01633.D4. [DOI] [PubMed] [Google Scholar]