

Abstract
Is monitoring initial response to treatment always helpful in clinical management of patients? Bell and colleagues have developed a framework for deciding whether surrogate outcomes should be used to monitor initial response to treatment in chronic disease.
Monitoring entails periodic measurement to guide management1 and is widely practised in clinical medicine to inform decisions throughout the course of a disease and to provide prognostic information to patients. It is helpful to divide monitoring into phases: pretreatment, initial response, maintenance, re-establish control, and post-treatment.1
Initial response monitoring uses repeat measurement soon after a new treatment is started to check that the response is within a range that maximises the benefits while minimising the harms. Table 1 summarises different types of initial response monitoring. We have limited our discussion to the use of surrogate outcomes for monitoring initial response to treatment. Surrogate outcomes are commonly used to monitor initial response in patients with chronic conditions. This type of initial response monitoring is common in clinical practice and can result in inappropriate decisions. We looked at two scenarios to develop a rational framework for deciding whether this form of initial monitoring should be done: Should change in blood pressure be monitored after addition of a diuretic to an angiotensin II receptor blocker in adults with essential hypertension? and Should change in cholesterol be monitored after giving patients with ischaemic heart disease a statin?
Table 1.
Rationale for monitoring initial response to drug treatment
| Aims and scenario | Target | Examples of monitoring |
|---|---|---|
| Maximise benefits | ||
| Therapy has rapid effect on patient centred outcome | Patient centred outcome | Observation of size and appearance of wound infection after empirically chosen antbiotics Monitoring for resolution of symptoms after treatment for depression |
| Therapy has slow effect on patient centred outcome | Surrogate outcome | Monitor cholesterol after statins Monitor blood pressure after antihypertensives |
| Minimise harms | ||
| Therapy has rapid effect on patient centred outcome | Patient centred outcome | Monitor for cough after angiotensin converting enzyme inhibitors |
| Therapy has slow effect on patient centred outcome | Surrogate outcome | Monitor drug levels after renal transplant patient on cyclosporin-A Monitor white cell count after clozapine in patient with psychosis |
| Maximise compliance | ||
| Compliance with therapy uncertain | Adherence | Use directly observed treatment strategy to ensure compliance with anti-tuberculosis treatment Count patient’s pills at follow-up visit to estimate how many have been taken if patient is suspected to be non-compliant Measure levels of anticonvulsant drugs to assess compliance with treatment after breakthrough seizure |
Rationale and pitfalls of monitoring initial response
Treatment for patients with chronic conditions (such as hypertension and raised cholesterol) is often monitored by using surrogate outcomes (such as blood pressure and cholesterol concentration). These outcomes are used to predict “hard” end points: the patient’s risk of a clinically important outcome (such as a stroke or myocardial infarction). These hard end points often occur many years after the patient was first diagnosed, are usually irreversible, and carry a substantial risk of mortality and so are unsuitable for monitoring purposes. The surrogate outcomes often respond to treatment, and by using treatment to alter the value of an intermediate outcome early on in the disease process, the clinician hopes to change the patient’s risk of developing later clinical outcomes.
A surrogate outcome should be considered for monitoring only if it is known to predict the effect of treatment on risk of the clinical outcome. Such evidence usually comes from population level meta-analyses of randomised controlled trials, where change in surrogate outcome is related to change in risk of clinical outcome for patients on active treatment relative to those on placebo. Although the population average treatment effect on a surrogate outcome might predict the population average treatment effect on the risk of a clinical outcome, the surrogate outcome might not be useful for monitoring the effect of treatment in an individual, in whom other factors besides treatment can cause change in the surrogate outcome. Failure to recognise such variation might lead to inappropriate changes in treatment or delays in necessary action.2
This problem is illustrated by a study that examined measurements of bone mineral density in postmenopausal women after bisphosphonate treatment.3 4 Most women who seemed to lose density in the first year of treatment were found to have gained density by the next measurement. The benefit of bisphosphonate in reducing risk of fracture applied both to women who lost and gained bone density in the first year. Because of the potential for bone density measurements to mislead the clinicians about the adequacy of biphosphonate, the authors concluded that it may be preferable not to monitor after the start of treatment. This emphasises the fact that while there is often considerable variation in the surrogate outcome observed for any group of patients on treatment, much of this may be explained by short term variability and measurement error. Variation that is actually caused by differences in the effect of treatment will usually be much less and may even be zero.
Because of the potential for misinterpretation, we should avoid monitoring initial response to treatment unless we think it will usefully inform clinical decision making. We have developed a method to determine whether monitoring might be useful by examining outcome variability in placebo controlled randomised trials.
Estimating variability in treatment effects between individuals from placebo controlled randomised trials
Table 2 summarises four important sources of temporal variation in surrogate outcomes used in randomised controlled trials.5 The combined effects result in observed variability in the surrogate (monitored) outcome over time. Variation caused by differences between treatments (T) occurs when there is a difference in the mean effect of treatment and placebo and is often reported in trials. Variation caused by “true” differences between patients in the surrogate outcome over time (B) may arise because of differences in severity of disease, rate of progression, or other prognostic factors such as demographic factors and coexistent disease. Variation caused by “noise” within patients (W) occurs when measurements repeated on the same individual vary because of short term biological fluctuations in the surrogate outcome as well as technical measurement error. Variation caused by differences between patients in treatment effect (R) occurs when the effect of treatment is different for each patient.
Table 2.
Sources of variation in measured outcomes in randomised controlled trials
| Label | Source | Description |
|---|---|---|
| T | Between treatments | Variation caused by average effect of treatment |
| B | Between patients | Variation caused by differences between patients unrelated to treatment |
| W | Within patients | Variation caused by measurement variability |
| R | Between patients in treatment effect | Variation caused by differences between patients in effect of treatment |
Individuals’ measurements of change in the placebo arm vary around the group’s mean change. The amount of variation around this mean reflects the combined effects of B and W. Measurements in the treatment arm vary around a different mean change. The amount of variation around this mean reflects the combined effects of B, W, and R. Comparison of the mean change in outcome between treatment and placebo groups allows estimation of T (mean treatment effect). Comparison of the variances of change in outcome in the treatment and placebo groups allows estimation of R (variation in treatment effect).
Figure 1 depicts hypothetical distributions of change in measurement of a surrogate outcome in a randomised controlled trial. The target level of change in this figure reflects the difference between an individual’s true baseline level (the average of many measurements before treatment) and an externally set treatment target (such as a 1 mmol reductionin cholesterol concentration). In this particular case the target level of change is 1.5 units of the surrogate outcome.
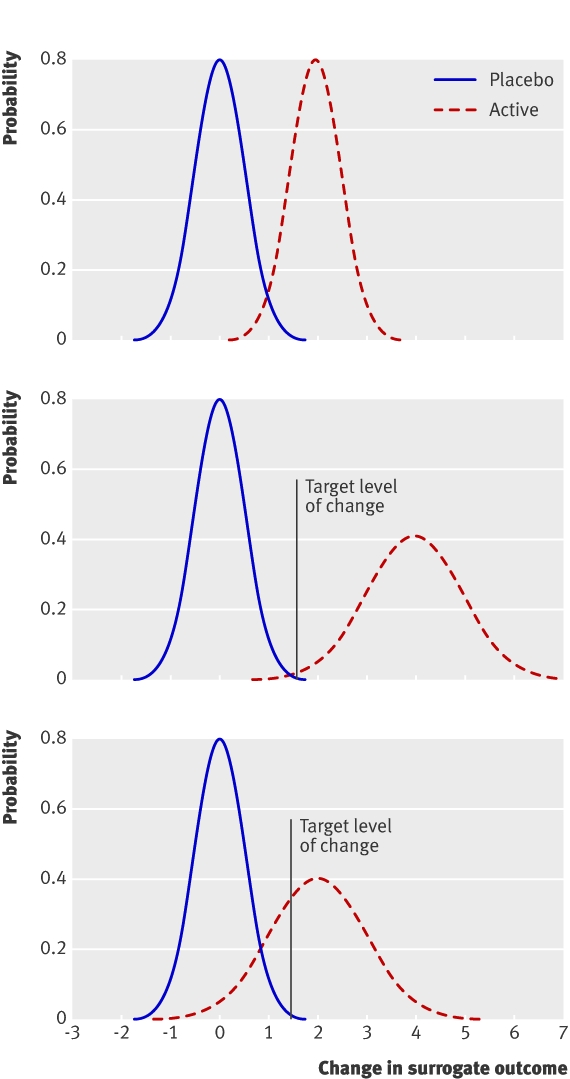
Fig 1 Hypothetical distributions of change in surrogate outcome in randomised controlled trial. Top: no variation in treatment; no need to monitor initial response. Middle: variation in effect of treatment, target levels guaranteed; no need to monitor initial response. Bottom: variation in treatment effect, target levels not guaranteed; initial monitoring of response justified
When the variance in the active arm is the same as that in the placebo arm there is no evidence of variation in treatment effect (fig 1, top). All of the variation in surrogate outcome could be explained by other sources (B and W). In this scenario there is no reason for monitoring and the effect of treatment for the individual should be predicted from that seen on average for the population.
Greater variance in the active arm compared with the placebo suggests variation in treatment effect (fig 1, middle and bottom). The variation in surrogate outcome exceeds that expected from other sources. When the average effect of treatment is sufficiently large, all patients meet the target level with treatment (fig 1, middle). In this case, even though the treatment effect does vary between individuals, all patients can be expected to meet the target level and there is no need for response monitoring. When the average effect of treatment is not sufficiently large, not all patients will meet the target level with treatment (fig 1, bottom). In this case, as the treatment effect for the individual cannot be predicted and we cannot guarantee that the patient will meet the target level, monitoring might be justified.
We used data from two large placebo controlled trials to illustrate this concept. Both trials showed a significant and clinically meaningful difference in mean levels of surrogate outcome between the treatment and placebo groups, providing evidence of treatment effect at the population level (T). We compared variances of change in surrogate outcome between treatment groups to make inferences about treatment effect at the individual level (that is, to examine the importance of R). The method used assumes that data are normally distributed with no relation between variability of measurement and level and that there is no substantial correlation between change in surrogate outcome caused by treatment and background change. Further discussion of these assumptions and details of the statistical methods used are on bmj.com.
Blood pressure and lipid lowering
Should blood pressure be monitored after addition of a diuretic to angiotensin II receptor antagonist treatment for patients with essential hypertension?
A study compared the effects of hydrochlorothiazide (12.5 mg and 25 mg daily) and placebo in 535 adults with essential hypertension who were already taking 20 mg of olmesartan.6 The summary estimates for change in blood pressure at two months were based on measurements made on 534 patients (data were not available for one patient allocated to hydrochlorothiazide 25 mg). We calculated estimates of variances in change in blood pressure and compared the two variances. We assumed that the variability of measurement was unrelated to level of blood pressure, the change in blood pressure was normally distributed for all treatment groups, and the treatment induced change in blood pressure was independent of the background change in measurement over the two months of the study.
Figure 2 shows the distributions of change in systolic blood pressure. The mean decreases in blood pressure were 4 mm Hg (SD of change 12.5 mm Hg) in the placebo group, 7.8 mm Hg (12.8 mm Hg) in the 12.5 mg hydrochlorothiazide group, and 11.4 mm Hg (12.2 mm Hg) in the 25 mg hydrochlorothiazide group. The standard deviations of change for the two hydrochlorothiazide groups were about the same as that of the placebo group.
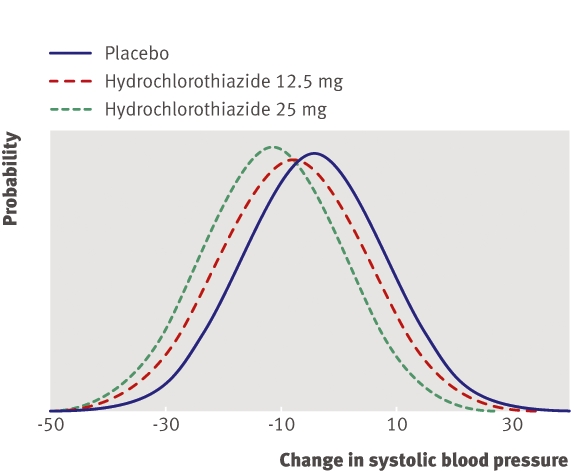
Fig 2 Change in systolic blood pressure eight weeks after addition of hydrochlorothiazide (12.5 mg or 25 mg) to olmesartan 20 mg
There was no evidence of variability between patients in the effect of 12.5 mg hydrochlorothiazide (F183,173=1.06, P=0.36). The mean effect of treatment was estimated as a decrease in blood pressure of 3.8 mm Hg. We can conclude that initial response monitoring is not needed. Clinicians should expect a decrease in blood pressure of about 3.8 mm Hg. Additional treatment will be needed if a decrease >4 mm Hg is required to meet the target blood pressure.
There was also no evidence of variability in effect if the dose was increased from 12.5 mg to 25 mg (F183,175=1.10, P=0.25). If the dose is increased from 12.5 mg to 25 mg, monitoring is not needed and the clinician can expect blood pressure to further decrease by about 3.6 mm Hg (if treatment starts at 25 mg without up-titration, blood pressure can be expected to decrease by about 7.4 mm Hg). Again, additional treatment will be needed when a greater decrease than this is required to meet the target blood pressure.
We found no evidence of variability of treatment effect on diastolic blood pressure at either dose of hydrochlorothiazide. With 12.5 mg, clinicians should expect a decrease in diastolic blood pressure of 1.9 mm Hg, and if the dose is 25 mg, they should expect a decrease of 3.7 mm Hg.
Is monitoring of cholesterol concentration needed after patients with known ischaemic heart disease start taking 40 mg pravastatin?
Another study compared the effects of 40 mg pravastatin and placebo in 9014 patients with known ischaemic heart disease.7 The summary estimates for change in cholesterol concentration at six months are based on measurements on 8625 patients (data were not available for 195 patients allocated to placebo and 194 patients allocated to pravastatin). (These data were not included in earlier papers for this trial but have recently been published8). We calculated estimates of variances in change in cholesterol concentration using the standard deviations of changes and compared the two variances. There was no relation between variability of measurement and cholesterol concentration, and the data were approximately normally distributed. The change induced by treatment was thought to be independent of background change in measurement over the six months studied.
Figure 3 shows the distributions of change in cholesterol concentration after six months. The placebo group had a mean increase of 0.02 mmol/l (SD of change 0.65 mmol/l) and the pravastatin group had a mean decrease of 1.16 mmol/l (0.75 mmol/l). The standard deviation of change in the pravastatin group was about 15% higher than that in the placebo group.
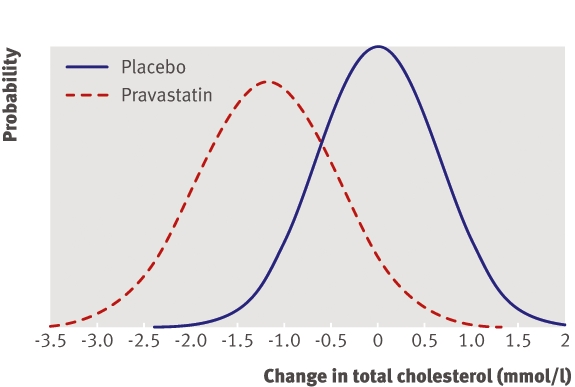
Fig 3 Change in cholesterol concentration six months after start of treatment with 40 mg pravastatin
Comparisons of variances show strong evidence of variability in the effect of pravastatin (F4317,4306=1.33, P<0.001). The mean effect of treatment was estimated as a decrease in cholesterol concentration of 1.18 mmol/l. The distribution of treatment effect for 95% of patients was estimated to range from a decrease of 0.45 mmol/l to a decrease of 1.91 mmol/l (note that this distribution is considerably narrower than the distribution of change actually observed in the pravastatin group, which includes non-treatment sources of variation as well as variation in treatment effect).
The clinical relevance of this level of variability will depend on the patient’s baseline cholesterol concentration. For patients with a baseline concentration <5.45 mmol/l we can reasonably assume that the recommended target level of 5 mmol/l9 will be reached with 40 mg pravastatin and initial response monitoring will not be necessary. For patients with baseline concentration >6.91 mmol/l we can reasonably assume additional treatment will be needed to meet target levels and initial response monitoring is not necessary. It is only in patients with baseline concentration ≥5.45 mmol/l and ≤6.91 mmol/l that we are uncertain as to whether the target will be met and initial response monitoring may be necessary.
A framework for choosing whether to monitor initial response to a new drug
Monitoring initial response is necessary only when there is evidence of variation in treatment effect between patients. Figure 4 provides a general framework for monitoring decisions for individual patients. As a preliminary step clinicians need to decide whether to consider the trial data as a whole or in subgroups. Subgroups can be defined on the basis of demographics or clinicopathological features such as baseline level of disease activity. Guidelines for deciding if an apparent subgroup difference is real assess whether the magnitude of the difference is clinically important and statistically significant, whether the subgroup analysis was one of a small number that were prespecified, whether the comparisons were made within studies and the difference consistent across studies, and whether other indirect evidence is supportive.10
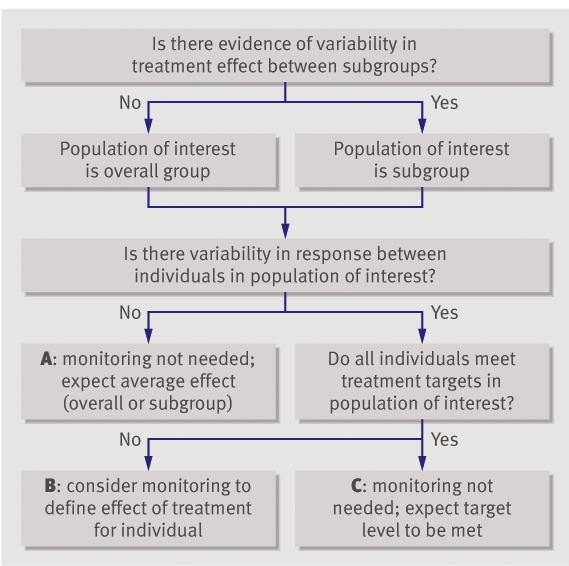
Fig 4 Algorithm to decide whether to monitor initial response
The next step is to examine the population of interest for evidence of variation in treatment effect. Monitoring is unlikely to be helpful when there is no evidence of variation in treatment effect (A in fig 4). The effect of treatment for the individual can be predicted from the average effect for the population (either as a whole or subgroup). When there is evidence of variation in treatment effect but all patients meet a predetermined target level, again there will be no need for monitoring (C in fig 4). It is only when there is evidence of variation in treatment effect and uncertainty that target levels will be met with treatment that monitoring is potentially helpful (B in fig 4).
What to do next
There is a long history of monitoring in clinical medicine aimed at ensuring treatment is tailored to the individual. Where monitoring achieves this the patient is likely to benefit, but current monitoring schedules are rarely based on empirical research and may be harmful. Many monitoring decisions in clinical practice could be informed by available trial data. When variability in treatment effect suggests that monitoring might be of benefit, the next stage will be to design and carry out randomised controlled trials to determine optimum monitoring strategies.
Readers of trials that estimate the variability of effect for other treatments should look for summary statistics for surrogate outcomes. They need information on the mean and standard deviation of the outcome (or other parameters that enable calculation of these such as standard error of the mean or 95% confidence limits of the mean together with number of patients). Graphical displays of outcome distributions (such as histograms) allow readers to assess normality. If the outcome is not approximately normally distributed, readers should look for results of transformed data. Readers should also look for Bland-Altman plots of individuals’ measurements in the study population to see if measurement variability varies with level. The inferences made on the basis of trial data will often be context specific. This means that readers need to look for trial information on not only the specific treatment being started but also the specific population of patients, dose of treatment, and surrogate outcome. Finally, because there may be substantial sampling error in variance estimates of trials with only a small number of patients, inferences on the variability of treatment effect should be made from trials of a large size.
Decisions on whether to monitor patients for initial response to treatment are potentially informed by knowledge of outcome variability in trials, but this is not often provided. Updating the commonly used CONSORT statement11 to include comprehensive reporting on outcome variability would be an effective way to remedy this.
Summary points
Clinicians routinely monitor individual patients after they start a new treatment; sometimes this may be unnecessary and even potentially harmful
Monitoring is unlikely to be of value when there is no evidence of variation in the response to treatment or when there is a high probability that therapeutic targets will be met
Data from placebo controlled randomised trials can be used to decide on the need for monitoring initial response in different clinical scenarios
Updating the CONSORT statement to include the detailed reporting of outcome variability will allow clinicians to make informed decisions on the need to monitor initial response to treatment
Supplementary Material
We thank P Glasziou, N Cross, and M Turner for comments on previous drafts.
Contributors and sources: All authors are affiliated with the screening and diagnostic test evaluation programme, University of Sydney. KJLB is a PhD candidate researching the use of surrogate outcomes to monitor initial response to treatment in chronic disease. LI is an epidemiologist with particular expertise in the evaluation of medical tests. JCC has a personal chair in clinical epidemiology with a specialty interest in kidney disease and child health. PM is a biostatistician who has published applied and methodological papers on diagnostic testing. KJLB and LI conceived the study, and all authors contributed to the ideas and writing. KJLB is guarantor.
Funding: Australian National Health and Medical Research Council Program Grants (No 402764).
Competing interests: None declared.
Ethical approval: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Glasziou PP, Irwig L, Mant D. Monitoring in chronic disease: a rational approach. BMJ 2005;330:644-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morton V, Torgenson D. Regression to the mean: treatment effect without the intervention. J Eval Clin Pract 2004;11:59. [DOI] [PubMed] [Google Scholar]
- 3.Cummings S, Palermo L, Browner W, Marcus R, Wallace R, Pearson J, et al. Monitoring osteoporosis therapy with bone densitometry: misleading changes and regression to the mean. JAMA 2000;283:1318-21. [DOI] [PubMed] [Google Scholar]
- 4.Cummings S, Karpf D, Harris F, Genant H, Ensrud K, LaCroix A, et al. Improvement in spine bone density and reduction in risk of vertebral fractures during treatment with antiresorptive drugs. Am J Med 2002;112:281-9. [DOI] [PubMed] [Google Scholar]
- 5.Senn S. Individual therapy: new dawn or false dawn? Drug Inf J 2001;35:1479-94. [Google Scholar]
- 6.Sellin L, Stegbauer J, Laeis P, Rump L. Adding hydrochlorothiazide to olmesartan dose dependently improves 24-h blood pressure and response rates in mild-moderate hypertension. J Hypertens 2005;23:2083-92. [DOI] [PubMed] [Google Scholar]
- 7.LIPID Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med 1998;339:1349-57. [DOI] [PubMed] [Google Scholar]
- 8.Glasziou P, Irwig L, Heritier S. Monitoring cholesterol-modifying interventions. In: Glasziou P, Aronson J, Irwig l, eds. Evidence-based medical monitoring. From principles to practice Oxford: Blackwell Publishing, 2007:245-53.
- 9.NCEP. Implications of recent clinical trials for the national cholesterol education program adult treatment panel III guidelines. Circulation 2004;110:227-39. [DOI] [PubMed] [Google Scholar]
- 10.Oxman A, Guyatt G. A Consumer’s guide to subgroup analyses. Ann Intern Med 1992;116:78-84. [DOI] [PubMed] [Google Scholar]
- 11.Moher D, Schulz KF, Altman DG. The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomised trials. Lancet 2001;357:1191. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.