

Abstract
Cyclooxygenase-derived prostaglandins modulate cardiovascular disease risk. We genotyped 2212 Atherosclerosis Risk in Communities study participants (1,023 incident coronary heart disease (CHD) cases; 270 incident ischemic stroke cases; 919 non-cases) with available DNA for polymorphisms in PTGS1 and PTGS2. Using a case–cohort design, associations between genotype and CHD or stroke risk were evaluated using proportional hazards regression. In Caucasians, the reduced function PTGS1 −1006A variant allele was significantly more common among stroke cases compared to non-cases (18.2 versus 10.6%, P = 0.027). In African Americans, the reduced function PTGS2 −765C variant allele was significantly more common in stroke cases (61.4 versus 49.4%, P = 0.032). No significant relationships with CHD risk were observed. However, aspirin utilization appeared to modify the relationship between the PTGS2 G-765C polymorphism and CHD risk (interaction P = 0.072). These findings suggest that genetic variation in PTGS1 and PTGS2 may be important risk factors for the development of cardiovascular disease events. Confirmation in independent populations is necessary.
Ischemic cardiovascular disease is a major public health problem. It is estimated that 1.2 million Americans will experience an acute coronary heart disease (CHD) event and 0.7 million will experience an acute stroke event this year.1 Prostaglandin endoperoxide H synthase-1 and -2 (more commonly described as cyclooxygenase (COX)-1 and COX-2, respectively)-derived prostaglandins are critical regulators of vascular, myocardial, and platelet function, and alterations in their expression and/or activity can significantly modify risk of cardiovascular disease events in humans.2–7 Recent evidence demonstrating associations between selective COX-2 inhibitor utilization and elevated risk of myocardial infarction,3,4 coupled with the well-known protective effects of low-dose aspirin,5 suggests that the relative contribution of COX-1 and COX-2-mediated prostaglandin synthesis is an integral mediator of cardiovascular homeostasis. Consequently, variation in the genes encoding COX-1 (PTGS1) and COX-2 (PTGS2) may be important modifiers of cardiovascular disease risk.
Numerous genetic polymorphisms in PTGS1 and PTGS2 have been identified and characterized.8–12 The G-765C polymorphism in the PTGS2 proximal promoter is associated with lower PTGS2 transcription.9,13,14 Moreover, due to the anti-inflammatory effects resulting from lower COX-2 expression, the variant −765C allele was associated with significantly lower risk of prevalent myocardial infarction or ischemic stroke events in a high-risk Italian population.15 We recently observed that certain non-synonymous polymorphisms in PTGS1, such as the rare G230S variant, have significantly lower COX-1 metabolic activity in vitro.12 However, potential associations between PTGS1 and PTGS2 polymorphisms and risk of incident cardiovascular disease events have not been rigorously evaluated. Our primary aim was to determine if genetic variation in PTGS1 and PTGS2 was associated with risk of incident CHD or ischemic stroke events in individuals enrolled in the biethnic Atherosclerosis Risk in Communities (ARIC) study. An important secondary aim was to determine if this risk was modified by aspirin utilization.
RESULTS
Study population
Significant baseline differences in various risk factors were observed between CHD and stroke cases and non-cases included in the cohort random sample, as described previously16 (Supplementary Materials). Incident CHD and stroke cases were significantly older and more likely to be male, smokers, diabetic, hypertensive, and have abnormal fasting lipid panels compared to non-cases. Stroke cases were also more likely to be African American and less likely to report aspirin use at baseline. Aspirin utilization at baseline was significantly lower in African Americans compared to Caucasians in the cohort random sample (11.4 versus 30.1%, P<0.001), CHD cases (14.2 versus 30.3%, P<0.001), and ischemic stroke cases (13.7 versus 23.8%, P = 0.026), respectively.
PTGS1/PTGS2 genotype
The location and observed race-specific allele frequencies of the six PTGS1 and two PTGS2 polymorphisms evaluated are listed in Table 1. The G-1006A and P17L polymorphisms in PTGS1 were in near-perfect linkage disequilibrium (LD) in Caucasians (D′ = 0.99, r2 = 0.96), but not African Americans (D′ = 0.46, r2 = 0.16) (Supplementary Materials). Significant LD was not observed between the other variants in either racial group (r2<0.02). The allelic distribution of the PTGS1 P17L polymorphism deviated significantly from Hardy–Weinberg equilibrium in Caucasians (P = 0.002); however, a disproportionate number of individuals carrying the −1006A variant allele had missing genotyping data for the P17L polymorphism. All other polymorphisms were in Hardy–Weinberg equilibrium in both Caucasians and African Americans (P>0.05).
Table 1.
Selected PTGS1 and PTGS2 polymorphisms
| CRS minor allele frequency
|
||||||
|---|---|---|---|---|---|---|
| Polymorphism | Nucleotidea | Location | Amino acid | Caucasian (N=698) | African American (N=367) | Functionb |
| PTGS1 | ||||||
| G>Ac | −1006 | 5′-UTR | — | 0.059 | 0.116 | Unknown |
| C>T (rs1236913) | 250 | Exon 2 | R8W | 0.070 | 0.012 | Normal |
| C>T (rs3842787) | 278 | Exon 2 | P17L | 0.061 | 0.145 | Normal |
| G>A (rs3842789) | 7010 | Exon 3 | R53H | 0 | 0.002 | Dec. activity |
| G>A (rs3842795) | 10721 | Exon 7 | G230S | 0 | 0.005 | Dec. activity |
| C>A (rs5789) | 10742 | Exon 7 | L237M | 0.033 | 0.005 | Dec. activity |
| PTGS2 | ||||||
| G>C (rs20417) | −765 | Promoter | — | 0.164 | 0.301 | Dec. transcription |
| T>C (rs5273) | 5792 | Exon 10 | V511A | 0 | 0.045 | Normal |
CRS, cohort random sample; UTR, untranslated region.
Nucleotide position relative to gene transcriptional start site (GenBank accession numbers: PTGS1 AF440204; PTGS2 AY229989).
No rs number is currently available for the G-1006A polymorphism in PTGS1. Note: it is in perfect linkage disequilibrium with the A-707G (rs10306114) and A-918G (rs10306110) polymorphisms in both European/Caucasian and African individuals.12
PTGS1 polymorphisms
In Caucasians, the variant −1006A allele was significantly more common among ischemic stroke cases compared to cohort random sample non-cases (18.2 versus 10.6%, respectively, P = 0.027) (Table 2). Presence of at least one −1006A allele was associated with significantly higher risk of incident ischemic stroke events relative to −1006G homozygotes after adjusting for age, gender, and study center (model 1, hazard rate ratio (HRR) 1.78, 95% confidence interval (CI) 1.08–2.92, P = 0.024, q = 0.080) (Table 3). A similar association was observed after also adjusting for cigarette smoking, diabetes, and hypertension status (model 2, HRR 1.70, 95% CI 0.99–2.93, P = 0.056, q = 0.120); however, this association was not statistically significant. This relationship was not modified by gender (interaction P = 0.252). A similar relationship was also observed with the reconstructed haplotype tagged by both the variant −1006A and P17L alleles (data not shown). No association between the −1006A allele and risk of ischemic stroke in African Americans, or risk of CHD in either Caucasians or African Americans, was observed (Tables 2 and 3).
Table 2.
PTGS1 and PTGS2 polymorphism frequency by incident ischemic stroke or CHD case status
| Caucasian
|
African American
|
|||||
|---|---|---|---|---|---|---|
| Genotypea | Non-cases | Cases | P-value | Non-cases | Cases | P-value |
| Stroke | n=671b | n=151b | n=326b | n=119b | ||
| G-1006A (PTGS1) | ||||||
| G/G | 583 (89.4%) | 121 (81.8%) | 246 (76.9%) | 84 (73.0%) | ||
| G/A+A/A | 71 (10.6%) | 27 (18.2%) | 0.027 | 70 (23.1%) | 31 (27.0%) | 0.436 |
| R8W (PTGS1) | ||||||
| C/C | 571 (86.7%) | 131 (87.9%) | 308 (97.1%) | 112 (97.4%) | ||
| C/T+T/T | 85 (13.3%) | 18 (12.1%) | 0.694 | 8 (2.9%) | 3 (2.6%) | NA |
| P17L (PTGS1) | ||||||
| C/C | 564 (89.3%) | 123 (84.3%) | 229 (72.1%) | 81 (71.7%) | ||
| C/T+T/T | 69 (10.7%) | 23 (15.7%) | 0.125c | 90 (27.9%) | 32 (28.3%) | 0.758 |
| G230S (PTGS1) | ||||||
| G/G | 654 (100%) | 149 (100%) | 311 (98.8%) | 112 (97.4%) | ||
| G/A+A/A | 0 (0%) | 0 (0%) | NA | 3 (1.2%) | 3 (2.6%) | NA |
| L237M (PTGS1) | ||||||
| C/C | 604 (93.6%) | 143 (94.7%) | 312 (99.2%) | 114 (99.1%) | ||
| C/A+A/A | 43 (6.4%) | 8 (5.3%) | 0.613 | 3 (0.8%) | 1 (0.9%) | NA |
| G-765C (PTGS2) | ||||||
| G/G | 457 (70.7%) | 111 (74.5%) | 154 (50.6%) | 44 (38.6%) | ||
| G/C+C/C | 195 (29.3%) | 38 (25.5%) | 0.353 | 149 (49.4%) | 70 (61.4%) | 0.032 |
| V511A (PTGS2) | ||||||
| T/T | 635 (100%) | 149 (100%) | 290 (91.6%) | 104 (92.0%) | ||
| T/C+C/C | 0 (0%) | 0 (0%) | NA | 28 (8.4%) | 9 (8.0%) | 0.877 |
| CHD | n=626b | n=792b | n=315b | n=231b | ||
| G-1006A (PTGS1) | ||||||
| G/G | 542 (89.2%) | 666 (86.6%) | 234 (75.9%) | 168 (76.4%) | ||
| G/A+A/A | 68 (10.8%) | 103 (13.4%) | 0.164 | 72 (24.1%) | 52 (23.6%) | 0.906 |
| R8W (PTGS1) | ||||||
| C/C | 533 (86.7%) | 669 (86.6%) | 298 (97.0%) | 218 (98.2%) | ||
| C/T+T/T | 79 (13.3%) | 104 (13.5%) | 0.952 | 8 (3.0%) | 4 (1.8%) | NA |
| P17L (PTGS1) | ||||||
| C/C | 524 (89.1%) | 671 (87.9%) | 219 (71.5%) | 159 (72.0%) | ||
| C/T+T/T | 66 (10.9%) | 92 (12.1%) | 0.534c | 90 (28.5%) | 62 (28.0%) | 0.913 |
| G230S (PTGS1) | ||||||
| G/G | 610 (100%) | 773 (100%) | 301 (98.8%) | 215 (98.2%) | ||
| G/A+A/A | 0 (0%) | 0 (0%) | NA | 3 (1.2%) | 4 (1.8%) | NA |
| L237M (PTGS1) | ||||||
| C/C | 563 (93.7%) | 733 (94.0%) | 301 (99.2%) | 220 (99.6%) | ||
| C/A+A/A | 39 (6.3%) | 47 (6.0%) | 0.851 | 3 (0.8%) | 1 (0.4%) | 0.659 |
| G-765C (PTGS2) | ||||||
| G/G | 425 (71.0%) | 546 (70.4%) | 147 (49.7%) | 113 (50.5%) | ||
| G/C+C/C | 182 (29.0%) | 230 (29.6%) | 0.808 | 144 (50.3%) | 111 (49.6%) | 0.873 |
| V511A (PTGS2) | ||||||
| T/T | 591 (100%) | 775 (100%) | 280 (91.9%) | 202 (90.2%) | ||
| T/C+C/C | 0 (0%) | 0 (0%) | NA | 27 (8.1%) | 22 (9.8%) | 0.514 |
CHD, coronary heart disease; NA, not available.
Genotype data presented as absolute (percent) genotype frequency. Frequencies are weighted according to the sampling fraction. The R53H variant allele in PTGS1 (rs3842789) was identified in only one African-American non-case.
Number of cases and non-cases with available DNA for genotyping (n=131/2,275 did not have DNA available).
Distribution of the P17L polymorphism in PTGS1 significantly deviated from Hardy–Weinberg equilibrium in Caucasians (P=0.002).
Table 3.
HRR between PTGS1 and PTGS2 polymorphisms and risk of incident ischemic stroke or CHD events
| Caucasian
|
African American
|
|||||
|---|---|---|---|---|---|---|
| Polymorphism | HRR | 95% CI | P-value | HRR | 95% CI | P-value |
| Stroke | ||||||
| G-1006A (PTGS1) | G/A+A/A versus G/G | G/A+A/A versus G/G | ||||
| Model 1a | 1.78 | 1.08–2.92 | 0.024 | 1.46 | 0.88–2.40 | 0.137 |
| Model 2b | 1.70 | 0.99–2.93 | 0.056 | 1.16 | 0.64–2.10 | 0.616 |
| G-765C (PTGS2) | G/C+C/C versus G/G | G/C+C/C versus G/G | ||||
| Model 1a | 0.78 | 0.51–1.18 | 0.236 | 1.54 | 0.98–2.41 | 0.059 |
| Model 2b | 0.79 | 0.50–1.25 | 0.314 | 1.76 | 1.05–2.94 | 0.031 |
| CHD | ||||||
| G-1006A (PTGS1) | G/A+A/A versus G/G | G/A+A/A versus G/G | ||||
| Model 1a | 1.13 | 0.80–1.60 | 0.487 | 1.15 | 0.76–1.75 | 0.509 |
| Model 2c | 1.28 | 0.87–1.89 | 0.206 | 1.25 | 0.77–2.04 | 0.367 |
| G-765C (PTGS2) | G/C+C/C versus G/G | G/C+C/C versus G/G | ||||
| Model 1a | 1.01 | 0.79–1.29 | 0.929 | 1.00 | 0.70–1.41 | 0.991 |
| Model 2c | 1.08 | 0.83–1.42 | 0.567 | 1.03 | 0.69–1.52 | 0.894 |
CI, confidence interval; HRR, hazard rate ratio.
Adjusted for age, gender, and study center.
Adjusted for age, gender, study center, current smoker, diabetes, and hypertension.
Adjusted for age, gender, study center, current smoker, diabetes, hypertension, high-density lipoprotein cholesterol, total cholesterol, and body mass index.
In African Americans, the rare G230S variant allele appeared to be more common in ischemic stroke cases (2.6%) and CHD cases (1.8%) compared to non-cases (1.2%) (Table 2). One individual carrying the G230S variant allele experienced a stroke and CHD event on separate occasions. When considering the composite end point, the G230S variant allele was associated with higher risk of an incident ischemic stroke or CHD event after covariate adjustment (model 2, HRR 8.37, 95% CI 1.53–45.8, P = 0.014). No relationship between the R8W, P17L, R53H, or L237M variant alleles and risk of either end point was observed (Table 2).
PTGS2 polymorphisms
In African Americans, the variant −765C allele was significantly more common among ischemic stroke cases compared to cohort random sample non-cases (61.4 versus 49.4%, respectively, P = 0.032) (Table 2). Presence of at least one −765C allele was associated with significantly higher risk of incident ischemic stroke events relative to −765G homozygotes after covariate adjustment (model 2, HRR 1.76, 95% CI 1.05–2.94, P = 0.031, q = 0.146) (Table 3). This relationship was not modified by gender (interaction P = 0.645). The −765C allele was not significantly associated with risk of ischemic stroke in Caucasians, or risk of CHD in either Caucasians or African Americans (Tables 2 and 3). No relationship between the V511A variant allele and risk of either end point was observed (Table 2).
Aspirin utilization
In Caucasians, aspirin utilization at baseline appeared to modify the relationship between the G-765C polymorphism in PTGS2 and risk of incident CHD events (interaction P = 0.072, q = 0.137) (Table 4); however, the interaction was not statistically significant. When stratified by aspirin use at baseline, the variant −765C allele demonstrated a nonsignificant association with higher risk of CHD in aspirin non-users (model 2, HRR 1.36, 95% CI 0.97–1.90, P = 0.075) and lower risk of CHD in those reporting aspirin use (model 2, HRR 0.60, 95% CI 0.36–1.02, P = 0.058). Moreover, similar results were obtained after restriction to only those reporting aspirin use or no aspirin use at both their baseline and first follow-up visit (interaction P = 0.047, q = 0.137) (Table 4). Aspirin utilization at baseline did not modify the relationship between the G-765C polymorphism and risk of ischemic stroke (interaction P = 0.639), or the relationship between the G-1006A polymorphism in PTGS1 and risk of either ischemic stroke (interaction P = 0.254) or CHD (interaction P = 0.965).
Table 4.
G-765C polymorphism in PTGS2, aspirin utilization, and risk of incident CHD events in Caucasians
| Non-cases G/C+C/C | CHD cases G/C+C/C | P-value | HRRaG/C+C/C versus G/G | 95% CI | P-value | |
|---|---|---|---|---|---|---|
| All patients | 182 (29.0%) | 230 (29.6%) | 0.808 | 1.08 | 0.83–1.42 | 0.567 |
| Baseline (n=1,376)b | ||||||
| No aspirin | 121 (27.9%) | 170 (31.7%) | 0.232 | 1.36 | 0.97–1.90 | 0.075 |
| Aspirin | 60 (31.5%) | 59 (25.0%) | 0.165 | 0.60 | 0.36–1.02 | 0.058 |
| Interactionc | 0.57 | 0.31–1.05 | 0.072 | |||
| Baseline and visit 2 (n=859)d | ||||||
| No aspirin | 90 (29.5%) | 98 (30.7%) | 0.745 | 1.40 | 0.93–2.09 | 0.107 |
| Aspirin | 32 (30.4%) | 33 (23.9%) | 0.295 | 0.55 | 0.26–1.18 | 0.128 |
| Interactionb | 0.43 | 0.19–0.99 | 0.047 | |||
CI, confidence interval; HRR, hazard rate ratio.
Adjusted for age, gender, study center, current smoker, diabetes, hypertension, high-density lipoprotein cholesterol, total cholesterol, and body mass index.
Aspirin utilization assessed (yes/no) at the baseline visit only. No aspirin=no. Aspirin=yes. Excluded=none.
Multiplicative G-765C by aspirin utilization interaction term.
Aspirin utilization assessed (yes/no) at the baseline and first follow-up visit. No aspirin=no at both visits. Aspirin=yes at both visits. Excluded=yes at only one visit.
PTGS1 G-1006A urinary biomarker analysis
Participants with one (G/A) or two (A/A) copies of the PTGS1 variant −1006A allele had lower urinary 11-dehydrothromboxane (Tx)B2 concentrations compared to wild-type (G/G) individuals (G/G: 0.58±0.17 ng/mg versus G/A or A/A: 0.33±0.06 ng/mg creatinine; P = 0.133) (Figure 1). These differences were most pronounced and statistically significant in Caucasians (G/G: 0.94±0.27 ng/mg versus G/A or A/A: 0.38±0.09 ng/mg creatinine; P = 0.023); however, no differences were observed in African Americans (G/G: 0.21±0.09 ng/mg versus G/A or A/A: 0.24±0.04 ng/mg creatinine; P = 0.763). Similar results were observed in the adjusted analysis (Figure 1b). No significant differences in urinary 2,3-dinor-6-keto-PGF1α concentrations were observed across G-1006A genotype in either Caucasians (P = 0.867) or African Americans (P = 0.799) (Supplementary Materials).
Figure 1.
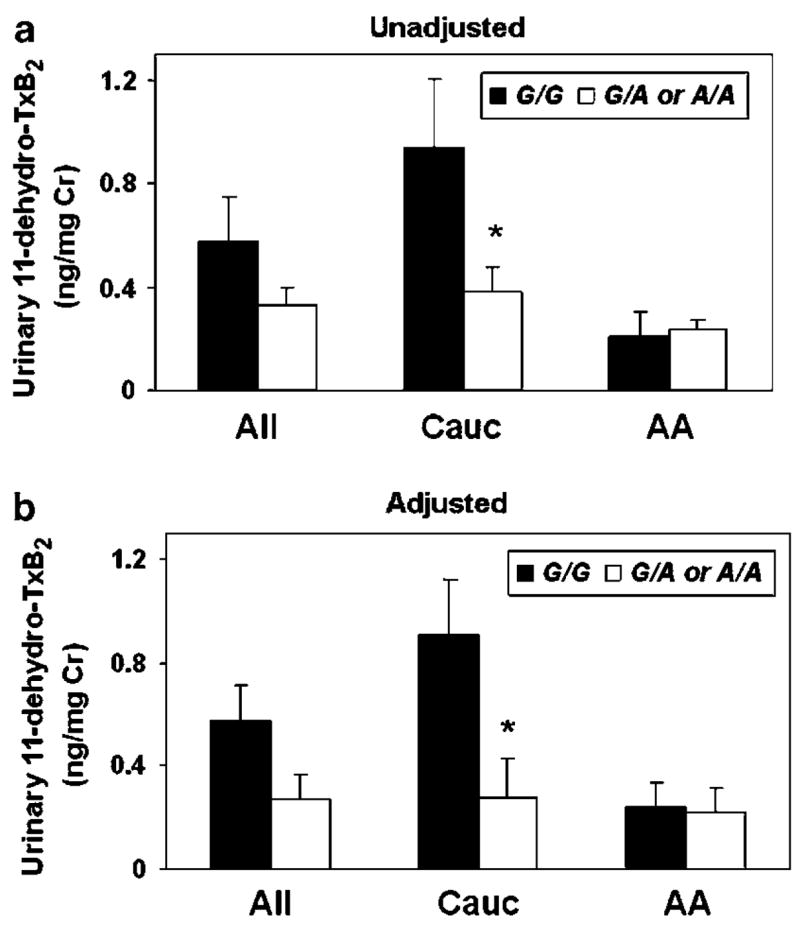
(a) Unadjusted and (b) adjusted mean±SEM urinary 11-dehydro-TxB2 concentrations, normalized to milligrams of urinary creatinine (Cr), are presented according to G-1006A genotype as wild-type (G/G) (black bars) and variant (G/A or A/A) (white bars). Data are presented separately for all participants (All, n = 33) and stratified by race (Cauc = Caucasians, n = 20; AA = African Americans, n = 13). *P<0.05 versus wild type. The values presented in b were adjusted for age, gender, race, PTGS2 G-765C genotype, CHD or stroke case status, and aspirin/non-steroidal use at the time of sample collection, using least squares regression.
DISCUSSION
Our analysis demonstrated that genetic variation in PTGS1 and PTGS2 was associated with risk of incident ischemic cardiovascular disease events in Caucasians and African Americans enrolled in the ARIC study. Specifically, the variant −1006A allele in PTGS1 and −765C allele in PTGS2 were associated with higher risk of ischemic stroke events in Caucasians and African Americans, respectively. Moreover, the rare G230S variant allele in PTGS1 appeared to be associated with higher risk of ischemic stroke and CHD events in African Americans. Although the G-765C polymorphism in PTGS2 was not significantly associated with CHD incidence, aspirin utilization appeared to modify this relationship in Caucasians. Collectively, our findings suggest that genetic variation in PTGS1 and PTGS2 may be important modifiers of cardiovascular disease risk in humans; however, these putative relationships remain exploratory until confirmed in an independent population.
COX-derived prostaglandins are important regulators of cardio- and cerebrovascular disease risk in vivo.2–5 For instance, the balance between TxA2 and prostacyclin (PGI2) significantly modifies atherosclerotic lesion development.7 Moreover, preferentially inhibiting COX-1-derived TxA2 in platelets via low-dose aspirin administration reduces the risk of myocardial infarction, ischemic stroke, and death in high-risk patients.5 In contrast, selective COX-2 antagonists inhibit endothelial PGI2 biosynthesis without influencing platelet TxA2 production and increase risk of CHD events, particularly in high-risk patients.3,4 Selective inhibition of COX-1 significantly reduces cerebral blood flow and vasodilatory responses, and COX-1−/− mice have larger cerebral infarct volumes compared to wild-type mice after middle cerebral artery occlusion.17,18 Moreover, cerebral COX-1 gene transfer before middle cerebral artery occlusion reduces cerebral infarct volume in rats.19 Selective inhibition of COX-2 also abolishes cerebral vasodilatory responses.20 However, COX-2−/− mice have significantly smaller infarct volumes compared to wild-type mice after middle cerebral artery occlusion,21 and transgenic mice with neuronal overexpression of COX-2 have larger infarct volumes, implicating the post-ischemic inflammatory effects of increased COX-2 expression in brain injury.22 Collectively, preclinical and clinical evidence demonstrates the importance of COX-1 and COX-2 in the pathogenesis of ischemic stroke and CHD. Consequently, genetic variation in PTGS1 and PTGS2 may be important modifiers of cardiovascular disease susceptibility.
The variant −1006A allele in PTGS1 was associated with higher risk of incident ischemic stroke events in Caucasians in the ARIC study. This polymorphism is one of seven variants within the PTGS1 5′ untranslated region present in substantial LD;12 however, the functional relevance of this haplotype had not been well characterized to date. Our biomarker analysis demonstrated that Caucasian variant −1006A allele carriers had significantly lower urinary 11-dehydro-TxB2 levels, indicative of lower COX-1 metabolic activity. Although lower platelet TxA2 production would be hypothesized to lower risk of ischemic stroke events, urinary levels of its stable 11-dehydro-TxB2 metabolite may represent a biomarker of COX-1 metabolic activity across various cell types, including the cerebral vasculature. As described, lower COX-1 activity impairs cerebral blood flow and enhances ischemic brain injury in preclinical models.17,18 Consequently, our analysis could suggest that variant −1006A allele carriers have lower COX-1 metabolic activity, providing a potential mechanistic explanation for the observed relationship between the −1006A allele and higher risk of incident ischemic stroke events in Caucasians. Owing to the known LD structure within the PTGS1 5′ untranslated region, the G-1006A polymorphism may not be the functionally relevant locus driving the observed associations. The G-1006A polymorphism was in near-perfect LD with the P17L polymorphism in Caucasians, consistent with our previous findings,12 and the reconstructed haplotype containing both the variant −1006A and P17L alleles demonstrated an association with higher ischemic stroke risk similar to that observed with the variant −1006A allele alone. Although the P17L variant has normal basal COX-1 metabolic activity in vitro,12 the lack of an association between the G-1006A polymorphism and both urinary 11-dehydro-TxB2 levels and ischemic stroke risk in African Americans could be due to the lack of LD with the P17L polymorphism and/or other genetic and environmental factors not accounted for in our analysis. Confirmation of these findings in an independent population and additional mechanistic studies characterizing the functional effects of this variant haplotype are necessary.
The rare G230S variant allele in PTGS1, which is monomorphic in Caucasians, appeared to be associated with higher risk of ischemic cardiovascular events in African Americans. The G230S variant has significantly lower COX-1 metabolic activity in vitro compared to wild-type enzyme, and molecular modeling suggests that this variant may disrupt the active conformation of COX-1.12 Association between the G230S variant allele and higher ischemic stroke incidence is also consistent with preclinical data, implicating the protective role of COX-1 in cerebrovascular function.17–19 Owing to the rare frequency of the G230S variant allele and subsequent fewer incident events than covariates included in the regression analysis, the observed association carried wide CIs and should be interpreted with caution.
A significant association between the variant −765C allele in PTGS2 and higher risk of ischemic stroke incidence was observed in African Americans. This variant disrupts an Sp1 binding site in the PTGS2 proximal promoter, reduces COX-2 transcription and expression, and consequently carries significant anti-inflammatory effects.9,13–15,23 The −765C allele was previously associated with lower risk of prevalent myocardial infarction or ischemic stroke in a high-risk Italian population,15 contrasting epidemiological and clinical trial evidence demonstrating associations between selective COX-2 inhibitor use and increased risk of cardiovascular events.3,4 Although preclinical evidence suggests that selective COX-2 inhibition abolishes cerebral vasodilatory responses,20 lower COX-2 expression also reduces the extent of ischemic brain injury after a cerebral infarct.21 Future studies evaluating the influence of the G-765C polymorphism on prognosis after stroke events appear necessary. A subsequent analysis from the Physicians’ Health Study demonstrated no significant relationship between the −765C allele and risk of either myocardial infarction or ischemic stroke.24 No significant association between the −765C allele and risk of incident CHD events was observed in the ARIC study. However, aspirin utilization may have modified this relationship in Caucasians, such that the −765C allele appeared to be associated with higher CHD risk in aspirin non-users and lower risk in those reporting aspirin use. This observation would be consistent with the hypothesis that concomitant aspirin utilization mitigates the cardiovascular hazard associated with selective COX-2 inhibition via restoration of PGI2-TxA2 balance.3 Presence of a gene–aspirin interaction could help explain, at least in part, the association between the −765C allele and lower cardiovascular risk observed by Cipollone et al.15 This analysis targeted a high-risk population and 67% were receiving aspirin therapy.15 Perhaps aspirin use in −765C allele carriers could help restore PGI2-TxA2 balance, allow the plaque stabilizing effects related to suppression of COX-2-mediated PGE2 synthesis to predominate,15 and result in lower risk of CHD events. However, due to the power limitations in our interaction analysis, these hypothesis-generating findings must be interpreted with caution and confirmed in larger populations. Future studies evaluating the mechanisms underlying this potential pharmacogenetic interaction will also be necessary, particularly as recent evidence suggests that the G-765C polymorphism does not influence celecoxib-mediated suppression of PGE2 biosynthesis.25
Although our study evaluated rigorously ascertained incident events, we are unable to elucidate mechanisms underlying the observed associations. We also recognize the limitations related to the evaluation of aspirin utilization at baseline in an observational study. Patients were not randomized at baseline, and aspirin therapy was likely initiated in those at highest risk of cardiovascular events. We attempted to adjust for potential confounders in our regression analysis; however, additional factors such as misclassification bias, changes in aspirin utilization throughout follow-up, duration of therapy, and dose could have influenced our results. In order to enhance our confidence that participants reporting aspirin use at baseline were on a stable regimen, most likely for primary prevention, we repeated the interaction analysis after restriction to only those reporting aspirin use or no aspirin use at both their baseline and first follow-up visit. Similar results were obtained, enhancing our confidence in the observed putative interaction. However, these findings should be considered exploratory and hypothesis generating until confirmed in a larger population. Moreover, even though the ARIC study is one of the largest biethnic cohorts assembled for the evaluation of incident cardiovascular disease risk, our gene association analysis also carried limitations in statistical power. For the PTGS1 G-1006A and PTGS2 G-765C polymorphisms, we estimated that greater than 80 and 90% power was present to detect an HRR of 2.0 for the stroke and CHD end points, respectively, in both Caucasians and African Americans (Supplementary Materials). However, substantially less power was present to detect an HRR of 1.5, which may represent a more plausible magnitude for an association between a genetic polymorphism and risk of a complex, polygenic disease.
We also acknowledge that it may be difficult to gauge the statistical significance of our findings considering the number of comparisons completed. Consequently, we assessed the false discovery rate across all completed tests in our association analysis. Although no gold-standard q-value threshold has been established to identify “true” associations, incorporation of this statistical approach into candidate gene association studies has become an increasingly recognized method to account for multiple comparisons and to enhance confidence in observed associations.26 As q-values related to the association between the G-1006A (Caucasians) and G-765C (African Americans) polymorphisms and ischemic stroke risk were estimated to be <0.15, we have a higher level of confidence in our reported findings. However, validation in a well-powered, independent population will be ultimately necessary to confidently conclude that genetic variation in PTGS1 and PTGS2 is significantly associated with cardiovascular disease susceptibility. Moreover, molecular, biochemical, and physiological studies will also be necessary to elucidate the mechanisms underlying the observed associations.
METHODS
Study population
Participants were selected from the ARIC study, a longitudinal, population-based cohort study of 15,792 men and women aged 45–64 years from four US communities (Forsyth County, NC; Jackson, MS; Minneapolis, MN; and Washington County, MD) enrolled between 1987 and 1989.27 Since enrollment, participants have been followed prospectively via annual phone interviews, clinic examinations approximately every 3 years through 1998, and ongoing abstraction of hospital and death certificate records. The study protocol was approved by the Institutional Review Board of each center, and consent was obtained from each participant.
Ascertainment of incident CHD and stroke cases
All incident cases that occurred between baseline and December 31, 1998 were evaluated (median follow-up 9.1 years), excluding participants with a history of a physician-diagnosed CHD or stroke event at baseline. Incident CHD (n = 1,085) was defined as (1) definite or probable myocardial infarction (n = 520), (2) electrocardiographic evidence of silent myocardial infarction (n = 112), (3) definite CHD death (n = 110), or (4) coronary revascularization procedure (n = 343). Incident stroke was defined as a definite or probable ischemic stroke (n = 300). Of note, n = 68 individuals experienced an incident CHD and ischemic stroke event on separate occasions.
The ascertainment of cases and criteria for classification have been described previously.28,29 All potential events were systematically reviewed and adjudicated by the ARIC Morbidity and Mortality Classification Committee.27–29 Briefly, hospitalized myocardial infarction was classified as definite or probable based on chest pain symptoms, cardiac enzyme levels, and electrocardiographic changes. Definite CHD death was classified based on chest pain symptoms, underlying cause of death, hospitalization records, and medical history. Coronary revascularization procedures included coronary artery bypass grafting and percutaneous coronary intervention. Using the National Survey of Stroke criteria,30 a stroke event was classified as definite or probable if there was (1) rapid onset of neurological symptoms that lasted >24 h or led to death within 24 h; (2) no evidence of pathology that could have mimicked stroke; and (3) one major (e.g., aphasia or hemiparesis) or two minor (e.g., diplopia or dysarthria) neurological deficits. Qualifying cases were further classified as ischemic (thrombotic brain infarction, cardioembolic stroke) or hemorrhagic (subarachnoid hemorrhage, intracerebral hemorrhage) on the basis of neuroimaging studies and autopsy, when available.29
Baseline measurements
Detailed demographic, clinical, and biochemical data were obtained from each participant at baseline, as described.31 Race was self-reported. Aspirin utilization (yes/no), including aspirin-containing products, was assessed through a detailed medication history at baseline and each follow-up visit.32 Dose and duration of use were not determined.
Cohort random sample
A random sample of all ARIC participants without history of CHD or stroke at baseline was assembled to serve as the reference group for the case–cohort comparisons (n = 1,065). Of note, n = 85 and n = 29 of these individuals became incident CHD and stroke cases, respectively, during follow-up. Sampling of the cohort was stratified on age (⩾55 or <55 years), gender, and race (Caucasian or African American). Sampling proportions varied across each stratum.
Genotyping
Available genomic DNA from all incident CHD (n = 1,023) and ischemic stroke cases (n = 270) and non-cases in the cohort random sample (n = 919) was genotyped for the G-1006A, R8W, P17L, R53H, G230S, and L237M polymorphisms in PTGS1 and the V511A polymorphism in PTGS2 using multiplex matrix-assisted laser desorption/ionization time-of-flight mass spectrometry methods (Sequenom, San Diego, CA), as described (Supplementary Materials).33 As matrix-assisted laser desorption/ionization time-of-flight methods were unsuccessful, the G-765C polymorphism in PTGS2 was genotyped using the BeadArray system (Illumina, San Diego, CA), as described.34 Blind replicates were included for quality control for both methods, and fewer than 5% of individuals had missing genotype data.
These polymorphisms were identified from our resequencing effort as part of the NIEHS Environmental Genome Single Nucleotide Polymorphism program (https://dir-apps.niehs.nih.gov/egsnp/home.htm) and/or the published literature (Table 1) to include the most frequent promoter/5′ untranslated region or non-synonymous polymorphisms identified to date in each gene.8–13,15 The G-1006A polymorphism is one of seven frequent polymorphisms in the 5′ untranslated region of PTGS1 present in significant LD and on the same haplotype in both European/Caucasian (D′ = 1.0, r2 = 0.73–1.0) and African (D′ = 1.0, r2 = 1.0) individuals.12 The G-1006A polymorphism was selected to capture variation at each of these loci. The other selected polymorphisms in PTGS1 include infrequent variants with known functional relevance (R53H, G230S, L237M) and the most frequent non-synonymous variants identified to date (R8W, P17L).10–12 In PTGS2, the G-765C polymorphism was selected, as this polymorphism is frequent and functionally relevant.9,13,15 The other selected polymorphism in PTGS2 (V511A) is the most frequent non-synonymous variant identified to date.8
Urinary biomarker analysis
Urine from the most recent clinic visit was obtained from 33 participants selected based on their PTGS1 G-1006A genotype (G/G (n = 12), G/A (n = 10), A/A (n = 11)), and matched based on age, gender, race, and aspirin/non-steroidal use. In order to evaluate the functional relevance of this polymorphism, systemic biosynthesis of TxA2 and PGI2 was assessed by measurement of their stable urinary metabolites, 11-dehydro-TxB2 and 2,3-dinor-6-keto-PGF1α, respectively, using gas chromatography-mass spectrometry methods, as described.35,36
Data analysis
Inverse sampling fractions from each stratum were used as weights in estimation of adjusted covariate means and proportions by linear and logistic regression, respectively. Cohort random sample allele frequencies were evaluated for deviation from Hardy–Weinberg equilibrium, and pairwise LD statistics were calculated (Haploview 3.3).37 HRRs and 95% CIs for the development of incident CHD or ischemic stroke in relation to genotype were calculated by weighted proportional hazards regression, using Barlow’s method to account for the stratified random sampling and case–cohort design.38 Model 1 included baseline age, gender, and study center as covariates. Model 2 also included significant clinical covariates, which were current smoking status, diabetes, hypertension, high-density lipoprotein cholesterol, total cholesterol, and body mass index at baseline for the CHD end point, and current smoking status, diabetes, and hypertension for the stroke end point. Assuming an autosomal-dominant mode of inheritance, individuals with one or two variant alleles were combined for comparison to wild-type individuals. This assumption was based on previous studies demonstrating significant functional effects of the PTGS2 G-765C polymorphism in individuals carrying one or two variant alleles,9,13,15 and our current urinary 11-dehydro-TxB2 biomarker analysis with the PTGS1 G-1006A polymorphism (Figure 1). All analyses were completed separately in Caucasians and African Americans.
Interaction testing was completed on a multiplicative scale between baseline aspirin utilization (yes/no) and both G-1006A (PTGS1) and G-765C (PTGS2) genotype for both end points in Caucasians, using a Wald χ2 test for significance of the estimated β-coefficient for the interaction term.39 Stratified weighted proportional hazards regression was also completed to further explore potential interactions. Gene–aspirin interaction testing was not completed in African Americans because of low aspirin utilization and sample size limitations.
To minimize the impact of the multiple statistical tests conducted and to enhance our confidence in the conclusions drawn from the observed associations, we estimated the false discovery rate q-value of our findings, separately for each end point, which is defined as the expected proportion of statistical tests deemed significant that are actually false-positives (QVALUE).40 We considered statistical tests from the unadjusted, model 1 and model 2 association analyses for each PTGS1 and PTGS2 polymorphism. Additional q-value estimates were also calculated for the gene–aspirin interaction analysis. Only q-values for statistically significant findings are presented.
Urinary 11-dehydro-TxB2 and 2,3-dinor-6-keto-PGF1α concentrations were compared across G-1006A genotype by analysis of variance. All data are normalized to milligrams of urinary creatinine, and presented as mean±SEM. These comparisons were repeated after adjusting for age, gender, race, PTGS2 G-765C genotype, CHD/stroke case status, and aspirin/non-steroidal use at the time of sample collection, using least squares regression.
Supplementary Material
SUPPLEMENTARY INFORMATION is linked to the online version of the paper at www.cptjournal.org.
Acknowledgments
We gratefully acknowledge Dr Jason Morrow, Dr Ginger Milne, and Mr Tyler Koestner for their contributions to the biomarker analysis, and the staff and participants of the ARIC study for their important contributions. This publication was made possible by Grant ES012856 to Dr Lee, HL073366 to Dr Bray, and funds from the Intramural Research Program of the NIH, NIEHS to Dr Zeldin. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS, NIH. The ARIC study is carried out as a collaborative study supported by NHLBI contracts N01-HC-55015, N01-HC-55016, N01-HC-55018, N01-HC-55019, N01-HC-55020, N01-HC-55021, and N01-HC-55022.
Footnotes
CONFLICT OF INTEREST
The authors declared no conflict of interest.
References
- 1.Rosamond W, et al. Heart Disease and Stroke Statistics – 2007 Update. A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115:e69–e171. doi: 10.1161/CIRCULATIONAHA.106.179918. [DOI] [PubMed] [Google Scholar]
- 2.Davidge ST. Prostaglandin H synthase vascular function. Circ Res. 2001;89:650–660. doi: 10.1161/hh2001.098351. [DOI] [PubMed] [Google Scholar]
- 3.Grosser T, Fries S, Fitzgerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116:4–15. doi: 10.1172/JCI27291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antman EM, DeMets D, Loscalzo J. Cyclooxygenase inhibition and cardiovascular risk. Circulation. 2005;112:759–770. doi: 10.1161/CIRCULATIONAHA.105.568451. [DOI] [PubMed] [Google Scholar]
- 5.Garcia Rodriguez LA, Landolfi R, Baigent C, Garcia Rodriguez LA, Landolfi R, Baigent C. Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med. 2005;353:2373–2383. doi: 10.1056/NEJMra052717. [DOI] [PubMed] [Google Scholar]
- 6.Camitta MG, et al. Cyclooxygenase-1 and -2 knockout mice demonstrate increased cardiac ischemia/reperfusion injury but are protected by acute preconditioning. Circulation. 2001;104:2453–2458. doi: 10.1161/hc4401.098429. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi T, et al. Roles of thromboxane A(2) and prostacyclin in the development of atherosclerosis in apoE-deficient mice. J Clin Invest. 2004;114:784–794. doi: 10.1172/JCI21446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fritsche E, Baek SJ, King LM, Zeldin DC, Eling TE, Bell DA. Functional characterization of cyclooxygenase-2 polymorphisms. J Pharmacol Exp Ther. 2001;299:468–476. [PubMed] [Google Scholar]
- 9.Papafili A, et al. Common promoter variant in cyclooxygenase-2 represses gene expression: evidence of role in acute-phase inflammatory response. Arterioscler Thromb Vasc Biol. 2002;22:1631–1636. doi: 10.1161/01.atv.0000030340.80207.c5. [DOI] [PubMed] [Google Scholar]
- 10.Ulrich CM, et al. Cyclooxygenase 1 (COX1) polymorphisms in African-American and Caucasian populations. Hum Mutat. 2002;20:409–410. doi: 10.1002/humu.9080. [DOI] [PubMed] [Google Scholar]
- 11.Halushka MK, Walker LP, Halushka PV. Genetic variation in cyclooxygenase 1: effects on response to aspirin. Clin Pharmacol Ther. 2003;73:122–130. doi: 10.1067/mcp.2003.1. [DOI] [PubMed] [Google Scholar]
- 12.Lee CR, et al. Identification and functional characterization of polymorphisms in human cyclooxygenase-1 (PTGS1) Pharmacogenet Genomics. 2007;17:145–160. doi: 10.1097/01.fpc.0000236340.87540.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hill MR, et al. Functional prostaglandin-endoperoxide synthase 2 polymorphism predicts poor outcome in sarcoidosis. Am J Respir Crit Care Med. 2006;174:915–922. doi: 10.1164/rccm.200512-1839OC. [DOI] [PubMed] [Google Scholar]
- 14.Lee YS, Kim H, Wu TX, Wang XM, Dionne RA. Genetically mediated interindividual variation in analgesic responses to cyclooxygenase inhibitory drugs. Clin Pharmacol Ther. 2006;79:407–418. doi: 10.1016/j.clpt.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 15.Cipollone F, et al. A polymorphism in the cyclooxygenase 2 gene as an inherited protective factor against myocardial infarction and stroke. JAMA. 2004;291:2221–2228. doi: 10.1001/jama.291.18.2221. [DOI] [PubMed] [Google Scholar]
- 16.Lee CR, et al. NOS3 polymorphisms, cigarette smoking, and cardiovascular disease risk: The Atherosclerosis Risk in Communities study. Pharmacogenet Genomics. 2006;16:891–899. doi: 10.1097/01.fpc.0000236324.96056.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niwa K, Haensel C, Ross ME, Iadecola C. Cyclooxygenase-1 participates in selected vasodilator responses of the cerebral circulation. Circ Res. 2001;88:600–608. doi: 10.1161/01.res.88.6.600. [DOI] [PubMed] [Google Scholar]
- 18.Iadecola C, Sugimoto K, Niwa K, Kazama K, Ross ME. Increased susceptibility to ischemic brain injury in cyclooxygenase-1-deficient mice. J Cereb Blood Flow Metab. 2001;21:1436–1441. doi: 10.1097/00004647-200112000-00008. [DOI] [PubMed] [Google Scholar]
- 19.Lin H, et al. Cyclooxygenase-1 and bicistronic cyclooxygenase-1/prostacyclin synthase gene transfer protect against ischemic cerebral infarction. Circulation. 2002;105:1962–1969. doi: 10.1161/01.cir.0000015365.49180.05. [DOI] [PubMed] [Google Scholar]
- 20.Brian JE, Jr, Faraci FM, Moore SA. COX-2-dependent delayed dilatation of cerebral arterioles in response to bradykinin. Am J Physiol Heart Circ Physiol. 2001;280:H2023–2029. doi: 10.1152/ajpheart.2001.280.5.H2023. [DOI] [PubMed] [Google Scholar]
- 21.Iadecola C, et al. Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proc Natl Acad Sci USA. 2001;98:1294–1299. doi: 10.1073/pnas.98.3.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dore S, et al. Neuronal overexpression of cyclooxygenase-2 increases cerebral infarction. Ann Neurol. 2003;54:155–162. doi: 10.1002/ana.10612. [DOI] [PubMed] [Google Scholar]
- 23.Orbe J, Beloqui O, Rodriguez JA, Belzunce MS, Roncal C, Paramo JA. Protective effect of the G-765C COX-2 polymorphism on subclinical atherosclerosis and inflammatory markers in asymptomatic subjects with cardiovascular risk factors. Clin Chim Acta. 2006;368:138–143. doi: 10.1016/j.cca.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 24.Hegener HH, Diehl KA, Kurth T, Gaziano JM, Ridker PM, Zee RY. Polymorphisms of prostaglandin-endoperoxide synthase 2 gene, and prostaglandin-E receptor 2 gene, C-reactive protein concentrations and risk of atherothrombosis: a nested case–control approach. J Thromb Haemost. 2006;4:1718–1722. doi: 10.1111/j.1538-7836.2006.02054.x. [DOI] [PubMed] [Google Scholar]
- 25.Skarke C, et al. The cyclooxygenase 2 genetic variant −765G>C does not modulate the effects of celecoxib on prostaglandin E2 production. Clin Pharmacol Ther. 2006;80:621–632. doi: 10.1016/j.clpt.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 26.Smith NL, et al. Association of genetic variations with nonfatal venous thrombosis in postmenopausal women. JAMA. 2007;297:489–498. doi: 10.1001/jama.297.5.489. [DOI] [PubMed] [Google Scholar]
- 27.The ARIC Investigators. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. Am J Epidemiol. 1989;129:687–702. [PubMed] [Google Scholar]
- 28.White AD, et al. Community surveillance of coronary heart disease in the Atherosclerosis Risk in Communities (ARIC) Study: methods and initial two years’ experience. J Clin Epidemiol. 1996;49:223–233. doi: 10.1016/0895-4356(95)00041-0. [DOI] [PubMed] [Google Scholar]
- 29.Rosamond WD, et al. Stroke incidence and survival among middle-aged adults: 9-year follow-up of the Atherosclerosis Risk in Communities (ARIC) cohort. Stroke. 1999;30:736–743. doi: 10.1161/01.str.30.4.736. [DOI] [PubMed] [Google Scholar]
- 30.National Institute of Neurological and Communicative Disorders and Stroke. The National Survey of Stroke. Stroke. 1981;12:I1–91. [PubMed] [Google Scholar]
- 31.Lee CR, et al. Genetic variation in soluble epoxide hydrolase (EPHX2) and risk of coronary heart disease: The Atherosclerosis Risk in Communities (ARIC) study. Hum Mol Genet. 2006;15:1640–1649. doi: 10.1093/hmg/ddl085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shahar E, et al. Patterns of aspirin use in middle-aged adults: the Atherosclerosis Risk in Communities (ARIC) Study. Am Heart J. 1996;131:915–922. doi: 10.1016/s0002-8703(96)90173-8. [DOI] [PubMed] [Google Scholar]
- 33.Bray MS, Boerwinkle E, Doris PA. High-throughput multiplex SNP genotyping with MALDI-TOF mass spectrometry: practice, problems and promise. Hum Mutat. 2001;17:296–304. doi: 10.1002/humu.27. [DOI] [PubMed] [Google Scholar]
- 34.Shen R, et al. High-throughput SNP genotyping on universal bead arrays. Mutat Res. 2005;573:70–82. doi: 10.1016/j.mrfmmm.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 35.Morrow JD, Minton TA. Improved assay for the quantification of 11-dehydrothromboxane B2 by gas chromatography-mass spectrometry. J Chromatogr. 1993;612:179–185. doi: 10.1016/0378-4347(93)80161-v. [DOI] [PubMed] [Google Scholar]
- 36.Daniel VC, Minton TA, Brown NJ, Nadeau JH, Morrow JD. Simplified assay for the quantification of 2, 3-dinor-6-keto-prostaglandin F1 alpha by gas chromatography-mass spectrometry. J Chromatogr B Biomed Appl. 1994;653:117–122. doi: 10.1016/0378-4347(93)e0432-p. [DOI] [PubMed] [Google Scholar]
- 37.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 38.Barlow WE, Ichikawa L, Rosner D, Izumi S. Analysis of case–cohort designs. J Clin Epidemiol. 1999;52:1165–1172. doi: 10.1016/s0895-4356(99)00102-x. [DOI] [PubMed] [Google Scholar]
- 39.Li R, Folsom AR, Sharrett AR, Couper D, Bray M, Tyroler HA. Interaction of the glutathione S-transferase genes and cigarette smoking on risk of lower extremity arterial disease: the Atherosclerosis Risk in Communities (ARIC) study. Atherosclerosis. 2001;154:729–738. doi: 10.1016/s0021-9150(00)00582-7. [DOI] [PubMed] [Google Scholar]
- 40.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY INFORMATION is linked to the online version of the paper at www.cptjournal.org.