

Abstract
The RNA-binding factor HuR is a ubiquitously expressed member of the Hu protein family that binds and stabilizes mRNAs containing AU-rich elements (AREs). Hu proteins share a common domain organization of two tandemly arrayed RNA Recognition Motifs (RRMs) near the N-terminus followed by a basic hinge domain and a third RRM near the C-terminus. In this study we have engineered recombinant wild type and mutant HuR proteins lacking affinity tags to characterize their ARE-binding properties. Using combinations of electrophoretic mobility shift and fluorescence anisotropy-based binding assays, we show that HuR can bind ARE substrates as small as 13 nucleotides with low nanomolar affinity, but forms cooperative, oligomeric protein complexes on ARE substrates of at least 18 nucleotides in length. Analyses of deletion mutant proteins indicate that RRM3 does not contribute to high affinity recognition of ARE substrates, but is required for cooperative assembly of HuR oligomers on RNA. Finally, the hinge domain between RRMs 2 and 3 contributes significant binding energy to HuR:ARE complex formation in an ARE length-dependent manner. The hinge does not enhance RNA-binding activity by increased ion pair formation despite extensive positive charge within this region, nor does it thermodynamically stabilize protein folding. Together, these studies define distinct roles for the HuR hinge and RRM3 domains in formation of cooperative HuR:ARE complexes in solution.
The cytoplasmic concentration of any given mRNA is a major determinant of the rate with which its encoded protein may be produced by the translation machinery, and is governed by a balance between the rates of mRNA synthesis and degradation. Cellular decay kinetics can vary widely among the mRNA population, with different transcripts exhibiting half-lives ranging from minutes to days in mammalian cells (1, 2). Control of mRNA decay rates thus constitutes a major cellular mechanism for post-transcriptional regulation of gene expression.
Several mRNAs that are rapidly degraded, often with half-lives of less than one hour, include those encoding oncoproteins and signaling proteins such as cytokines, chemokines, and inflammatory mediators (3). A common feature of many rapidly degraded mRNAs is the presence of an AU-rich element (ARE) in their 3’ untranslated regions (3’UTR). The sequence of this cis-element is highly variable, although it frequently contains one or more AUUUA pentameric motifs within or near a U-rich region. The length of an ARE is also variable and can range from 30 to 120 nucleotides (4). AREs serve as binding sites for a variety of nuclear and cytoplasmic proteins, over twenty of which have been identified to date (3, 5). However, interactions between ARE-binding proteins and their cognate RNA targets can yield a diverse range of consequences. For example, AUF1, tristetraprolin, and KSRP can direct rapid decay of mRNAs containing high affinity binding sites (6–10). Conversely, members of the Hu family of proteins inhibit degradation of many ARE-containing mRNAs, possibly by antagonizing recruitment of competing mRNA-destabilizing factors (11–14). Finally, proteins of the TIA-1/TIAR family do not appear to influence mRNA decay kinetics directly; rather, they control the subcellular localization and translational efficiency of targeted transcripts (15, 16).
The four mammalian members of the Hu family of ARE-binding, mRNA-stabilizing proteins share significant sequence similarity to the Drosophila RNA-binding protein Elav (embryonic lethal abnormal vision) (17). HuR, alternatively called HuA, is the only ubiquitously expressed protein of the four (18). Expression of the remaining three family members (HuB - sometimes referred to as Hel-N1, HuC, and HuD) are limited to neuronal tissues (17, 19). The primary structures of the Hu proteins are well conserved (> 68% pair-wise identity among all proteins) and share an identical domain arrangement, with two tandemly arrayed RNA recognition motif (RRM) domains near the N-terminus followed by a hinge region and finally a third RRM domain near the C-terminus (18). To date, characterization of the RNA-binding activity of the Hu proteins has been largely limited to the neuronal isoforms. Binding studies with HuD deletion mutants indicated that RRM1 is essential for RNA binding, but that high affinity association with ARE substrates also requires RRM2 or RRM3 (20). Crystal structures of a HuD deletion mutant containing the two N-terminal RRMs bound to ARE fragments show that both of these RRMs specifically contact poly(U) sequences within RNA substrates, spanning a total of 8–9 nucleotides and inducing a pronounced kink in the RNA backbone between sequences contacted by RRM1 versus RRM2 (21). An NMR structure of the corresponding domain of HuC bound to an ARE fragment yielded similar data (22). However, the function of the C-terminal RRM domain is much less clear. A HuD binding study indicated ARE-binding activity for RRM3, although inclusion of this domain made relatively minor contributions (2-fold) to binding affinity (20). By contrast, an earlier report indicated that the HuR RRM3 domain facilitated association with poly(A) RNA sequences (23). The cellular role of RRM3 also remains unresolved. Overexpression studies using HuR truncation mutant proteins have alternatively demonstrated that the RRM3 domain is either required, or dispensable, for the mRNA-stabilizing activity of this protein, although these contradictory results may be cell type specific (13, 24). Finally, the basic hinge region located between RRMs 2 and 3 contains a nuclear localization sequence (NLS) which allows HuR to shuttle between the cytoplasm and the nucleus, although the protein is predominantly nuclear (25–27). To date, the hinge domain has not been implicated in the association of Hu proteins with RNA substrates.
In this study we have constructed recombinant wild type and mutant HuR proteins lacking affinity tags and characterized their RNA-binding properties using combinations of electrophoretic mobility shift assays (EMSA) and fluorescence anisotropy-based binding experiments. The use of differentially sized RNA substrates spanning the ARE from tumor necrosis factor α (TNFα) mRNA permitted resolution of the minimal RNA binding sites necessary for HuR recognition and cooperative binding. Together, these studies identify RNA and protein determinants contributing to cooperative formation of multi-subunit HuR complexes on ARE substrates, and define specific roles for both the basic hinge and C-terminal RRM domains in this process.
EXPERIMENTAL PROCEDURES
RNA Substrates
Synthesis, 2’-hydroxyl deprotection, and purification of RNA substrates used in this study were performed by Dharmacon Research or Integrated DNA Technologies. The RNA substrate ARE38 includes the core ARE sequence from TNFα mRNA (Table I). Other substrates designated ARExx are subsections of this TNFα mRNA sequence. The RNA substrate Rβ encodes a fragment of the rabbit β-globin mRNA coding sequence. Fluorescein (Fl) groups were linked to the 5’-ends of some RNA substrates during solid phase synthesis and are designated in the text using the prefix Fl- where applicable. Lyophilized RNA samples were resuspended in 10 mM TrisHCl (pH 8). Quantification of RNA yields and fluorophore labeling efficiency was performed by absorbance spectroscopy as described previously (28, 29). For preparation of 5’-32P-radiolabeled RNA substrates, 5’-hydroxyl RNA oligonucleotides were incubated with [γ-32P]ATP (PerkinElmer) and T4-polynucleotide kinase (Promega) as described (30), yielding specific activities of 3–5 × 10³ cpm/fmol.
Table I.
RNA substrates used in this study
| Name | Sequence (5’→3’)a |
|---|---|
| ARE38 | GUGAUUAUUUAUUAUUUAUUUAUUAUUUAUUUAUUUAG |
| ARE20 | UUUAUUAUUUAUUUAUUUAG |
| ARE18 | UUAUUAUUUAUUUAUUUA |
| ARE16 | AUUAUUUAUUUAUUUA |
| ARE15 | UUAUUUAUUUAUUUA |
| ARE14 | UAUUUAUUUAUUUA |
| ARE13 | AUUUAUUUAUUUA |
| ARE11 | UUAUUUAUUUA |
| Rβ | UGGCCAAUGCCCUGGCUCACAAAUACCACUG |
RNA substrate variants containing 5’-fluorescein groups are indicated by the prefix “Fl-” where applicable.
Construction of Recombinant HuR and HuR Deletion Mutant Expression Vectors
A cDNA fragment encoding the complete open reading frame of human HuR was amplified by reverse transcription-PCR using RNA purified from the monocytic leukemia cell line THP-1, and then subcloned into pGEM-7Z(+) (Promega) to generate plasmid pGW01. The fidelity of the HuR open reading frame was verified by automated DNA sequencing. HuR cDNA subfragments encoding the complete 306 amino acid open reading frame, residues 1–185 (HuRΔHinge3), or residues 1–242 (HuRΔRRM3) were amplified from the pGW01 template by PCR utilizing DNA primers incorporating unique terminal restriction sites. These cDNA fragments were then subcloned downstream (HuR, HuRΔHinge3) or upstream (HuRΔRRM3) of an intein/chitin-binding domain tag using plasmids pTYB11 and pTYB1, respectively, from the IMPACT-CN (Intein Mediated Purification with an Affinity Chitin-binding Tag) system (New England Biolabs). The sequence and orientation of all cDNA inserts were verified by automated DNA sequencing.
Preparation and Characterization of Recombinant Proteins
Recombinant HuR or HuR deletion mutant proteins (Fig. 1A) were prepared in E. coli ER2566 cells using a variation of a procedure described by Meisner et al. (31). Transformed cells were grown shaking at 37 °C in SOB medium supplemented with 10 mM MgCl2 and 50 µg/ml ampicillin until reaching OD600 ≈ 0.6–0.8. Protein production was induced by addition of isopropyl 1-thio-β-D-galactopyranoside (1 mM) and cell growth was continued at 25 °C for 5 h. Cells were recovered by centrifugation at 3,300 × g for 20 min. In pilot experiments, optimal protein buffer conditions were selected through a solubility screening test monitored by dynamic light scattering using a Zetasizer Nanoseries instrument (Malvern Instruments), essentially as described (32). This procedure indicated optimal solubility for HuR and HuR deletion mutant proteins in a high salt buffer composed of sodium phosphate (pH 7.0), 500 mM NaCl and 1 mM EDTA (data not shown), which was subsequently termed HuR column buffer.
Fig. 1. Preparation and characterization of recombinant HuR and HuR deletion mutant proteins.
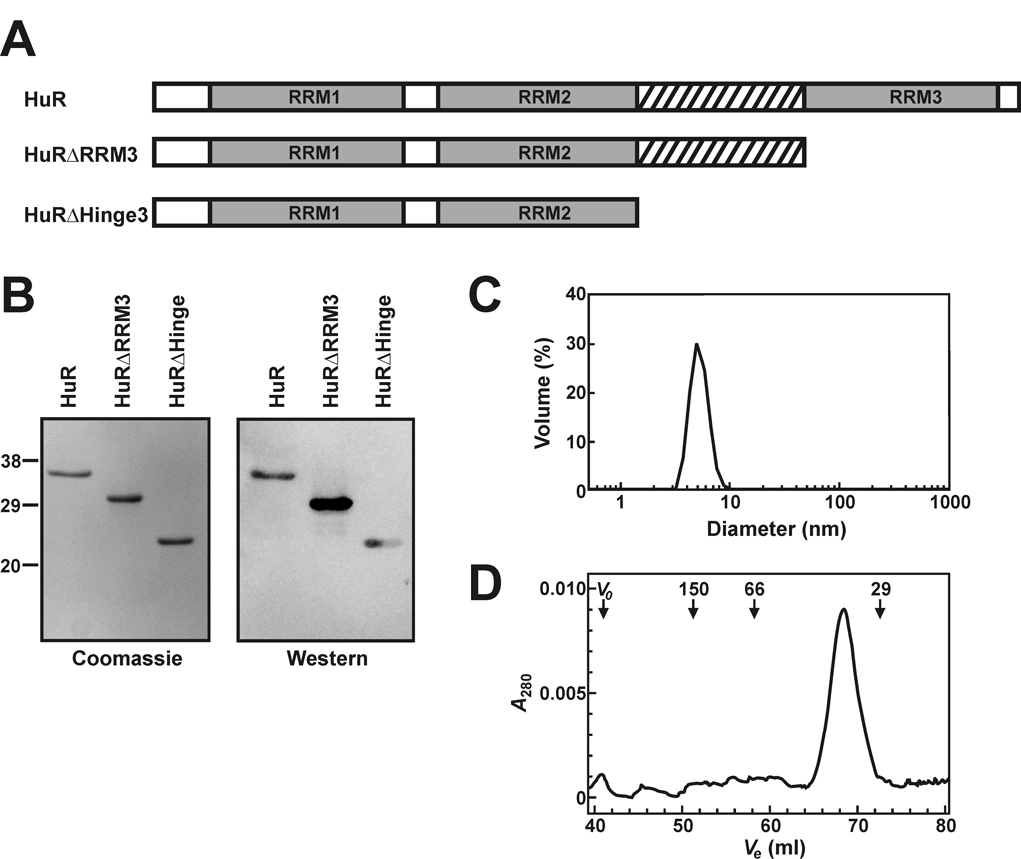
A, domain organization of HuR and HuR deletion mutant proteins. RRM domains are shown as grey boxes and the hinge region is designated by crosshatch. B, a Commassie Blue stained 15% SDS-PAGE gel of recombinant HuR and HuR deletion mutant proteins (left panel). The positions of molecular weight markers (masses in kDa) are listed to the left of the gel. All proteins were purified by the IMPACT-CN system as described under “Experimental Procedures” and 1 µg of each protein was loaded onto the gel. An identical gel was blotted onto nitrocellulose and probed with anti-HuR antibodies (right panel). C, a dynamic light scattering volume trace of purified HuRΔHinge3 protein indicating that the protein sample exists in solution as a monodisperse species with an average diameter of 5.2 nm. D, recombinant full length HuR was fractionated through a Sephacryl S-200 High Resolution column as described under “Experimental Procedures”. Elution volumes (Ve) were monitored by absorbance at 280 nm. The column void volume (Vo) and the elution volumes of proteins standards are indicated, with molecular masses given in kDa.
Cells were resuspended in 15 ml chilled HuR column buffer containing 20 µM phenylmethanesulfonyl fluoride and disrupted on ice by sonication. All subsequent purification steps were performed at 4 °C. Cell lysates were clarified by centrifugation at 20,000 × g for 20 min. The supernatant was loaded onto a chitin affinity column (New England Biolabs) equilibrated in HuR column buffer. The column was washed with 10 bed volumes of column buffer to remove unbound or nonspecifically bound proteins, followed by addition of 3 bed volumes of cleavage buffer (HuR column buffer containing 50 mM DTT). The column was then capped and DTT-induced self-cleavage of the intein tag was allowed to proceed for 40 hours. Cleaved protein was eluted in HuR column buffer and concentrated using an Amicon Ultra-4 concentrator (10 kDa MWCO). DTT remaining in the sample was removed by repeated concentration in HuR column buffer before a final centrifugation step (20,000 × g for 15 min) to remove any particulate matter. Recombinant proteins were quantified using Coomassie Blue-stained SDS-PAGE gels against a titration of bovine serum albumin.
The purity of all recombinant proteins was judged to be > 95% by Coomassie Blue-stained SDS-PAGE (Fig. 1B, left). Western blot analyses using anti-HuR antibodies (Santa Cruz Biotechnology) further verified that each recombinant protein was immunologically related to HuR (Fig. 1B, right). UV absorbance spectra indicated no significant nucleic acid contamination of HuRΔRRM3 or HuRΔHinge3 (Supplemental Fig. S1A). By contrast, absorbance of the full length HuR protein was enhanced at wavelengths below 270 nm, characteristic of co-purifying nucleic acids. Comparisons to HuR spectra measured following addition of varying amounts of an RNA substrate containing a single HuR binding site (ARE16) indicated that the total number of nucleic acid binding sites co-purifying with recombinant HuR constitutes less than 0.1 mol equivalents (Supplemental Fig. S1B).
Dynamic light scattering verified that recombinant protein preparations were free of high molecular weight complexes or aggregates. Typically, protein preparations returned peaks representing particle diameters of 5.1 nm to 7.5 nm, constituting > 98% of the total sample volume (Fig. 1C). Finally, the oligomerization status of purified recombinant proteins was monitored by gel filtration chromatography (Fig. 1D). A HiPrep 16/60 column (GE Biosciences) packed with Sephacryl S-200 High Resolution resin was equilibrated with HuR column buffer before loading protein samples (50 – 100 µg). Chromatography was performed at 30 ml/h with protein elution monitored by absorbance at 280 nm. Column void volume was determined using Blue Dextran. The column was calibrated by monitoring elution of the following protein standards (Sigma) by A280: alcohol dehydrogenase (150 kDa), bovine serum albumin (66 kDa), carbonic anhydrase (29 kDa), and cytochrome c (12.4 kDa). Apparent molecular weights calculated for each of the HuR, HuRΔRRM3 and HuRΔHinge3 proteins resolved to within 10% of their predicted molecular weights calculated from amino acid sequence (Supplemental Table S1), indicating that each recombinant HuR protein is a monomer in solution.
Analyses of protein folded stability by chemical denaturation
For all recombinant proteins, the thermodynamic stability of protein folding was estimated by equilibrium denaturation in guanidine HCl (GnHCl). Protein samples (2 µM) were incubated at room temperature in RNA binding buffer (10 mM TrisHCl (pH 8) containing 50 mM KCl, 2 mM DTT, and 0.5 mM EDTA) in the absence or presence of varying concentrations of GnHCl (60 µl total volume). After 60 minutes, the extent of protein unfolding was assessed by measurement of protein fluorescence (λex = 270 nm; λem = 290 – 400 nm; 10 nm bandwidth) at 21°C using a Cary Eclipse spectrofluorometer equipped with a micro-cell. Because the HuRΔRRM3 and HuRΔHinge3 proteins do not contain tryptophan, the excitation wavelength was blue-shifted to 270 nm to preferentially excite tyrosine residues (33). Protein denaturation was considered as a two-state, GnHCl-dependent transition between native and unfolded conformations yielding distinct fluorescence emission intensities Fnative and Funfolded, respectively. Thermodynamic parameters describing the denaturant-induced protein unfolding transition were estimated from the change in protein fluorescence measured at 305 nm (F305) as a function of GnHCl concentration using Equation 1 and Equation 2 adapted from the linear extrapolation method of Santoro and Bolen (34), as modified by Manyusa and Whitford (35).
| (Eq. 1) |
| (Eq. 2) |
Here, ΔGu represents the free energy of protein denaturation at each concentration of GnHCl. ΔGuw is the extrapolated free energy of protein unfolding in the absence of denaturant, while meq relates the sensitivity of ΔGu to GnHCl concentration. All parameters were resolved from F305 versus [GnHCl] plots by nonlinear regression using PRISM version 3.03 software (GraphPad).
RNA-Protein Binding Assays
Electrophoretic mobility shift assays (EMSA) were used to qualitatively assess the formation of complexes between recombinant HuR proteins and 32P-labeled RNA substrates. EMSAs were performed essentially as described (30) but with EDTA (0.5 mM) instead of MgCl2 in the binding buffer. Quantitative measurement of protein:RNA binding equilibria was performed using fluorescence anisotropy essentially as described (28, 36). Reactions containing limiting concentrations of 5’ Fl-labeled RNA substrates (0.2 nM) and varying concentrations of protein were assembled in 10 mM TrisHCl (pH 8) containing 50 mM KCl, 2 mM DTT, 0.5 mM EDTA, and 0.1 µg/µl acetylated bovine serum albumin (100 µl final volume). Alternative monovalent salt or RNA substrate concentrations are indicated where applicable. Heparin (1 µg/µl) was also included in all binding reactions to suppress non-specific interactions between proteins and RNA substrates (30). In the absence of this polyanionic competitor, HuR displayed modest binding to irrelevant RNA substrates (data not shown). Following incubation at 25 °C for 60 minutes, fluorescence anisotropy of the Fl-RNA substrates was measured using a Beacon 2000 Fluorescence Polarization System (Panvera) equipped with fluorescein excitation (490 nm) and emission (535 nm) filters. Total fluorescence emission from each reaction was concomitantly measured to verify that the fluorescence quantum yields of RNA substrates were not significantly affected by protein binding events. Equilibrium binding constants describing formation of protein:RNA complexes were resolved from plots of total measured anisotropy versus protein concentration by nonlinear regression using PRISM, employing analytical functions describing one or more of the binding models defined below (equations derived in Refs. 36, 37). The appropriateness of each binding model was evaluated by the coefficient of determination (R²) from individual binding experiments and analysis of residual plot non-randomness to detect any bias for data subsets (PRISM). When multiple models were considered for a common data set, pair-wise comparisons of sum-of-squares deviations were performed using the F test (PRISM), with differences exhibiting P < 0.05 considered significant.
Reactions best described by cooperative binding were resolved by a variant of the Hill model. Under conditions of constant fluorescence quantum yield and limiting RNA substrate, measured anisotropy (At) varies with total protein concentration ([P]) as a function of the protein concentration yielding half-maximal binding ([P]1/2) and the Hill coefficient (h) by Equation 3. Here, AR represents the intrinsic anisotropy of the free Fl-RNA substrate measured directly from sample reactions assembled without protein (n ≥ 4), while APxR represents the intrinsic anisotropy of the saturated HuR:Fl-RNA complex.
| (Eq. 3) |
In some cases, assembly of protein:RNA complexes involving two distinct protein binding events without cooperativity was indicated. Here, a sequential binding model given by Equation 4 resolves explicit equilibrium association binding constants for each stage of complex assembly, provided: [i] the affinity of the first binding step (K1) is at least 5-fold greater than the second (K2), [ii] the concentration of the Fl-RNA substrate is significantly lower than 1/K1, and [iii] the fluorescence quantum yield of the Fl-RNA substrate is not significantly influenced by protein binding (36, 37). Additional resolved parameters are the intrinsic anisotropy values of the Fl-RNA substrate bound to one (APR) or two (AP2R) protein molecules.
| (Eq. 4) |
If a single or multiple identical binding events are indicated, Equation 4 may be simplified by substitution of K2 = 0 to yield Equation 5.
| (Eq. 5) |
In cases where apparent ARE-binding affinity was very high (1/K < 1 nM), data sets were also resolved by Equation 6. This function describes a quadratic binding algorithm incorporating protein depletion, and includes terms for the total concentrations of RNA ([R]tot) and protein ([P]tot).
| (Eq. 6) |
Where indicated, the free energy of protein binding was extracted from equilibrium association constants as ΔG = −RTln(K), where R = 1.987 × 10−3 kcal·mol−1·K−1.
HuR Binding Density Analysis
The size of the HuR binding site on the Fl-ARE38 substrate was calculated using the model-independent ligand binding density analysis approach of Bujalowski et al. (38, 39). Briefly, quantitative estimates of the average binding density (Σν, defined as the average number of HuR proteins bound per fluorescein-labeled RNA) and free HuR protein concentration ([P]f) were extracted from the measured fluorescence anisotropy of the Fl-ARE38 substrate across a titration of HuR. The analysis holds that each possible HuR complex with the Fl-ARE38 RNA affects the experimentally observable net change in anisotropy (ΔA = Ax − AR, where AR is the intrinsic anisotropy of the Fl-ARE38 substrate in the absence of protein and Ax is the anisotropy in the presence of x nM HuR). As such, each HuR:Fl-ARE38 complex displays an intrinsic anisotropy (ΔAi) which contributes to the population weighted ensemble average (ΔAobs). A common value of ΔAobs in experiments containing two different RNA concentrations ([R]T1 and [R]T2) indicates that each system possesses similar distributions of complexes, yielding the same values of Σν and [P]f. The average binding density and free HuR protein concentration is related to the total protein concentrations under these conditions ([P]T1 and [P]T2), and can be extracted from the slope and intercept given by equation 7 and equation 8, respectively, where i = 1 or 2 in the intercept determination.
| (Eq. 7) |
| (Eq. 8) |
In this study, HuR titrations were performed across three concentrations of fluorescein-labeled RNA ([R]Ti) to permit estimation of errors in the resolved values of Σν and [P]f. For a series of ΔAobs values, slopes and intercepts were determined using linear least squares analysis of [R]Ti versus interpolated [P]Ti values across the set of titration curves for each given ΔAobs. An estimate of the average HuR binding density on the RNA substrate at saturation (Σνsat) was provided by linear extrapolation of ΔAobs versus Σν to the net anisotropy change at binding saturation (ΔAmax), determined from the HuR titration isotherms. The maximal binding site size (n) was then calculated from the ratio of RNA nucleotide length (N) to saturation binding density as n = N/Σνsat.
Bioinformatic Analyses
Amino acid sequences for the human HuR/A, HuB/HelN-1, HuC and HuD proteins (GenBank accession numbers NP_001410, Q12926, NP_001411, and NP_068771, respectively) were retrieved from the National Center for Biotechnology Information website (www.ncbi.nlm.nih.gov) using the Entrez Protein utility. Sequence alignments were performed using the ClustalW program at the San Diego Supercomputer Center Biology Workbench (http://workbench.sdsc.edu). The molecular weights and the theoretical isoelectric points (pI) of the HuR and HuR deletion mutant proteins were calculated using the ExPASy Proteomics Server, Compute pI/Mw tool (www.expasy.org/tools/pi_tool.html).
RESULTS
Wild Type HuR Forms a Cooperative, Oligomeric Complex on the TNFα ARE38 RNA Substrate
Previously, we and others have employed GST-tagged versions of HuR to characterize its RNA-binding properties (18, 40, 41). However, several features of this protein raised concerns that its RNA-binding activity might not accurately reflect functions of cellular HuR. GST-HuR binding to an ARE substrate exhibited significant cooperativity in fluorescence anisotropy-based binding assays (40), which had not been reported previously for HuR or any other Hu family member. Given that GST domains are capable of self-association (42), it was also possible that free energy from GST:GST contacts could be responsible for the apparent cooperativity of GST-HuR binding to an ARE substrate by enhancing the second protein binding step. Conversely, the GST-tag could sterically occlude adjacent binding sites on some RNA substrates, since this moiety contributes 26 kDa to the chimeric protein.
To address these issues, we adapted a procedure by Meisner et al. (31) to produce untagged versions of full-length HuR, or deletion variants lacking the C-terminal RRM but either retaining (HuRΔRRM3) or lacking (HuRΔHinge3) the hinge domain (Fig. 1A). By EMSA, the untagged wild type HuR protein binds specifically to the ARE38 substrate, since no HuR:RNA comples were formed with the Rβ substrate, which lacks an ARE sequence (Fig. 2A). Interestingly, association of HuR with the ARE38 substrate yielded three distinct complexes at protein concentrations between 2 and 5 nM. Since HuR exists as a monomer in solution, the three binding events between HuR and ARE38 likely indicate three separate monomers binding to the RNA substrate. Weakly resolved bands migrating slightly ahead of the largest HuR:ARE38 complex at 20 nM protein may indicate additional protein-binding events, but more likely result from complex dissociation during electrophoresis. Alternatively, this band may represent protein binding to a small proportion of degraded RNA (43). Although only two complexes were detected in EMSAs using GST-HuR (40), we cannot rule out the possibility that the size of the GST-tag may occlude a third binding site on the RNA, or that additional GST-HuR:ARE complexes are too dynamic to be retained during gel loading and/or electrophoresis.
Fig. 2. Evaluation of HuR binding to the ARE38 RNA substrate in vitro.
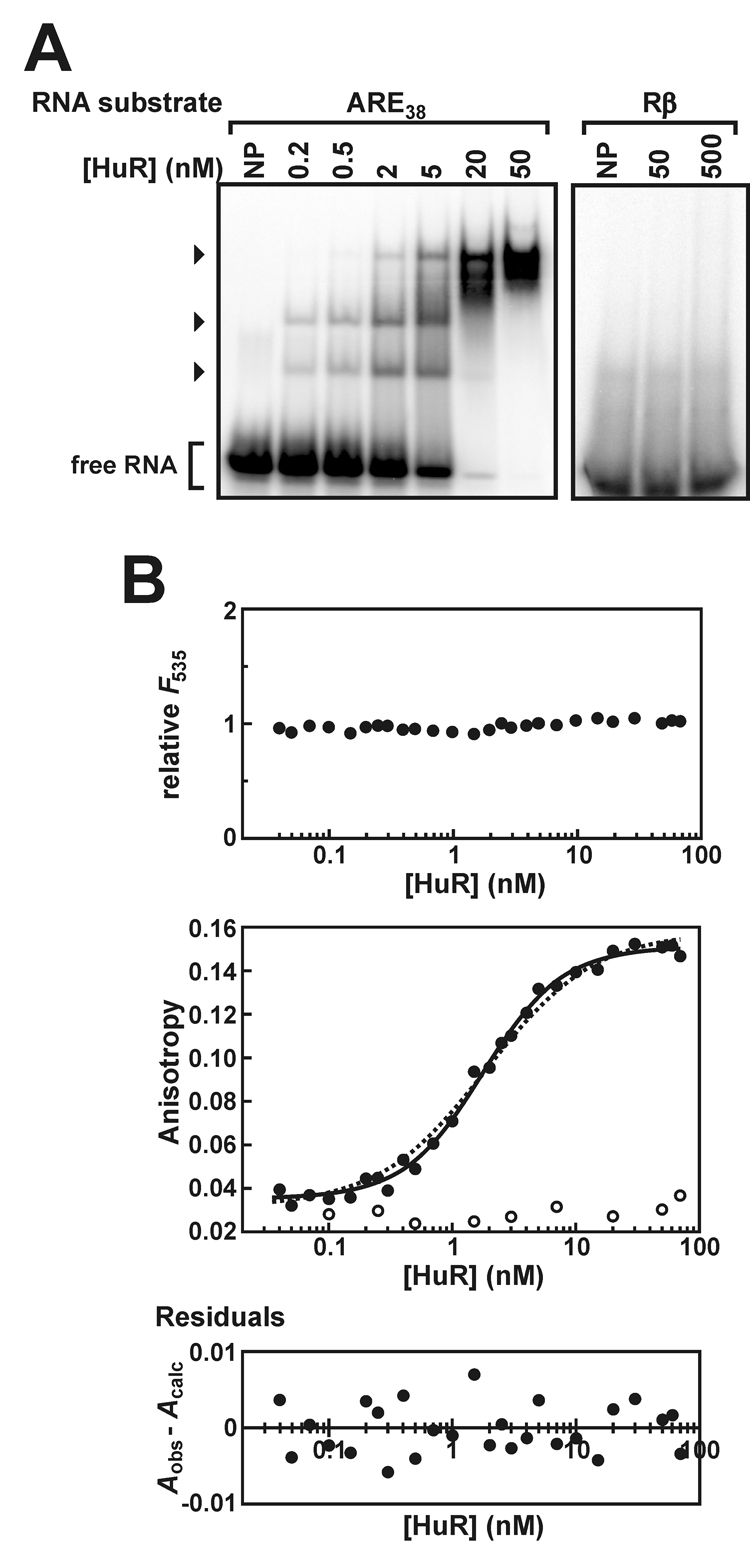
A, EMSAs were performed using 5'-32P-labeled ARE38 substrate (0.2 nM) and a titration of recombinant HuR protein (left panel). Additional experiments used 32P-Rβ RNA substrate and a reduced number of HuR protein concentrations (right panel). Lanes indicated NP contained no recombinant protein. The arrowheads indicate bands corresponding to complexes containing the 32P-ARE38 substrate and HuR. Unbound probe is designated as free RNA. B, association of HuR with the Fl-ARE38 (solid circles) or Fl-Rβ RNA substrates (open circles) was analyzed by fluorescence anisotropy as described under “Experimental Procedures” and plotted as a function of protein concentration (middle panel). Total fluorescence emission from the Fl-ARE38 substrate (λem = 535 nm) was also measured across a titration of HuR and is reported relative to Fl-ARE38 samples lacking protein (top panel). HuR:Fl-ARE38 anisotropy plots were resolved by a cooperative binding model using Equation 3 (solid line). For comparison, binding data were also fit to a single site binding model using Equation 5 (dotted line). A residuals plot for the HuR:Fl-ARE38 binding data resolved using Equation 3 (bottom panel) showed no bias for data subsets.
Fluorescence anisotropy-based binding assays were used to quantitatively characterize the association between HuR and the fluorescein-tagged Fl-ARE38 RNA substrate. Total fluorescence emission from the Fl-ARE38 substrate did not significantly vary as a function of HuR concentration (Fig. 2B, top), indicating that HuR binding does not influence the quantum yield of the fluorophore and validating assumptions required to derive analytical solutions for binding models (37). The increase in fluorescence anisotropy of the Fl-ARE38 substrate as a function of HuR concentration was best resolved by the cooperative binding model described by Equation 3 (Fig. 2B, middle), based on random distribution of residuals (Fig. 2B, bottom) and high coefficients of determination (R² > 0.99 across each of 6 independent experiments). By comparison, these data were not resolved by Equation 5, which could describe a single-site binding model or multiple identical but non-interacting HuR binding sites on the Fl-ARE38 substrate (Fig. 2B, middle, dashed line). Inappropriateness of non-cooperative models was indicated by significant residual nonrandomness (P = 0.0027) and by significant increases in the sum-of-squares deviation when compared to the cooperative binding model using the F test (P < 0.0001). Cooperative HuR binding to the ARE38 substrate is also qualitatively supported by EMSA data (Fig. 2A), since the abundance of the two fastest migrating HuR:RNA complexes accumulate concomitantly as a function of protein concentration, rather than sequentially. Assembly of HuR oligomers on the Fl-ARE38 substrate is a positively cooperative process, since resolution of anisotropy data to Equation 3 yielded a Hill coefficient significantly greater than unity (Table II). The concentration of HuR conferring 50% binding saturation under these conditions was 2.0 ± 0.3 nM, indicating that HuR targets the TNFα ARE with an affinity similar to that of the ARE-binding protein p37AUF1 and the RNA-binding domain of tristetraprolin (40, 44). Finally, and consistent with the EMSA data, the fluorescence anisotropy assays also support specific binding of HuR binding to AU-rich RNA substrates, since the presence of HuR did not significantly influence the anisotropy of a fluorescent RNA substrate lacking AU-rich sequences (Fl-Rβ; Fig. 2B, middle, open circles)
Table II.
ARE substrate length requirements for association and oligomerization of wild-type HuR
| RNA substrate | Apparent shifted speciesa | [HuR]1/2 or Kd-appb (nM) | hb | n |
|---|---|---|---|---|
| ARE38 | 3 | 2.0 ± 0.3 | 1.41 ± 0.06 | 6 |
| ARE20 | 2 | 0.44 ± 0.08 | 1.38 ± 0.04 | 3 |
| ARE18 | 2 | 0.83 ± 0.09 | 1.47 ± 0.09 | 3 |
| ARE16 | 1 | 2.5 ± 0.6 | 1 | 5 |
| ARE15 | n/d | 2.5 ± 0.4 | 1 | 3 |
| ARE14 | n/d | 4.8 ± 0.2 | 1 | 3 |
| ARE13 | 1 | 8.9 ± 1.4 | 1 | 3 |
| ARE11 | n/d | 82 ± 9 | 1 | 2 |
the number of protein:RNA complexes formed between HuR and each RNA substrate assessed by EMSA experiments (Fig. 2, Fig. 4, and data not shown); n/d, not determined.
the concentration of HuR required for half-maximal binding ([HuR]1/2) to Fl-labeled RNA substrates and associated Hill coefficients (h) were calculated from At versus [HuR] data sets (eg: Fig. 2 and Fig. 3) using Equation 3. For RNA substrates ≤ 16 bases in length, the Hill coefficient did not significantly differ from unity. In these cases, solution of Equation 3 with h = 1 yielded apparent bimolecular dissociation constants (Kd-app). All resolved constants are expressed as the mean ± σn-1 for n ≥ 3 or mean ± spread for n = 2 independent experiments.
ARE Substrate Length Requirements for Cooperative Assembly of HuR Oligomers
Model-independent ligand binding density analysis (38, 39) was used to determine the occluded site size of HuR binding to RNA substrates. By this method, the site size is calculated from an estimate of the maximal number of HuR proteins associated with a given RNA substrate at binding saturation. Association of HuR with the Fl-ARE38 substrate was monitored by the fluorescence anisotropy-based binding assay across a range of RNA concentrations. As the RNA substrate concentration ([R]T) was increased, greater concentrations of total HuR ([P]T) were required to obtain equivalent net changes in measured anisotropy (ΔAobs) (Fig. 3A). The population weighted average binding density (Σν) was calculated from the linear dependence of interpolated ([P]T, [R]T) pairs obtained at constant ΔAobs as described under Experimental Procedures. Σν was resolved across 80 distinct values of ΔAobs spanning the lower 75% of the anisotropy isotherms to minimize the inherent inaccuracies in the derivation of this function near saturating protein concentrations reported by Bujalowski and Jezewska (45). Linear extrapolation of Σν to the maximal ΔAobs (ΔAmax) of 0.119 ± 0.003 (Fig. 3B) yielded a saturation binding density (Σνsat) of 4.2 ± 0.3 HuR molecules bound per Fl-ARE38 substrate. The error associated with this determination was propagated from the linear least squares analyses of [P]T versus [R]T data sets. Given that the Fl-ARE38 substrate contains N = 38 nucleotides, the HuR site size thus resolves to 9.1 ± 0.3 nucleotides at binding saturation. Consistent results were observed with a truncated ARE substrate spanning 20 nucleotides near the 3’-end of the TNFα ARE sequence (Fl-ARE20), where binding density analysis revealed a HuR site size of 8.2 ± 1.3 nucleotides (Supplemental Fig. S2).
Fig. 3. Ligand binding density analysis of HuR oligomers on the Fl-ARE38 substrate.
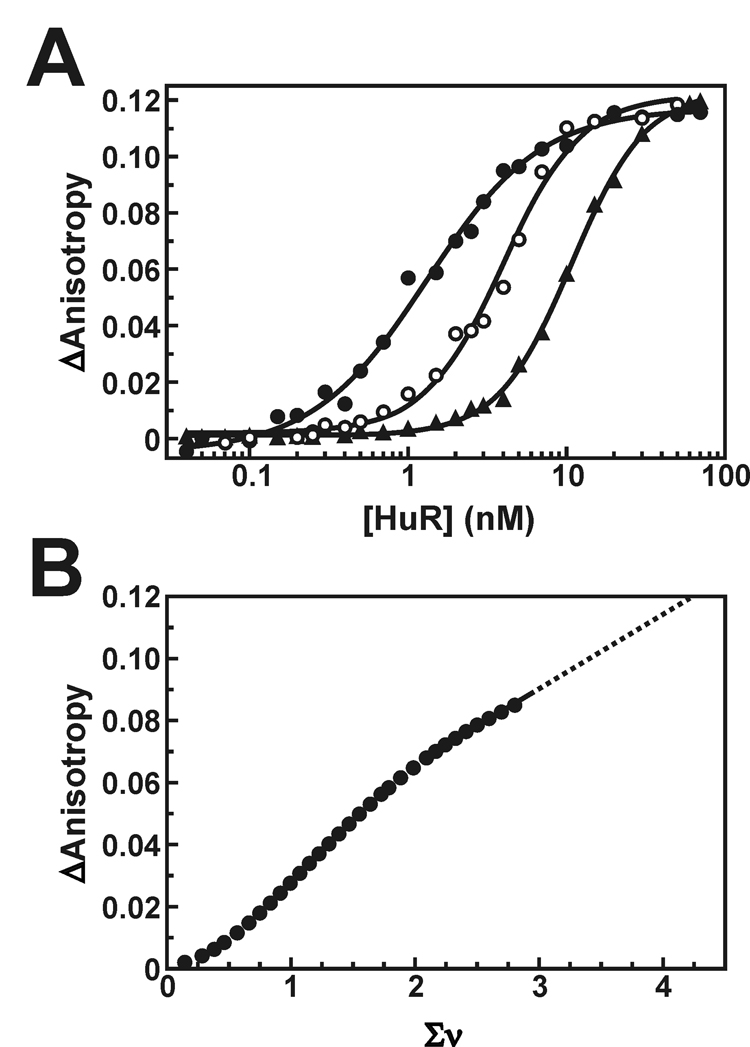
A, anisotropy plots showing HuR binding across vaying concentrations of the Fl-ARE38 substrate (0.2 nM RNA, solid circles; 1 nM RNA, open circles; 5 nM RNA, triangles). The solid lines are nonlinear least squares fits to cooperative Hill isotherms using Equation 3. The average increase in fluorescence anisotropy at binding saturation (ΔAmax) is 0.119 ± 0.003. B, the HuR-dependent change in measured anisotropy (ΔA) was calculated as a function of average number of bound HuR molecules per RNA strand (Σν) as described under “Experimental Procedures”. For clarity, only every third point is shown. The solid line is a linear least squares interpolation and has no theoretical basis. The dotted line is an extrapolation to the maximal anisotropy shift observed in A, estimating the Σνsat at 4.2 ± 0.2. This value was used to calculate the site size for saturated HuR binding to the Fl-ARE38 RNA substrate (see text).
As an orthogonal approach to define minimal RNA substrate requirements for cooperative HuR binding, an extensive series of ARE38 truncation mutants were employed for both qualitative and quantitative in vitro binding studies (Table I). By EMSA, multiple HuR binding events were not observed on RNA substrates less than 18 nucleotides in length. While two HuR:RNA complexes were readily assembled on the ARE20 and ARE18 substrates, consistent with the HuR site size of 8–9 nucleotides resolved by binding density analysis, only a single protein:RNA complex was detectable with either the ARE16 or ARE13 substrates (Fig. 4A and data not shown). Fluorescence anisotropy experiments were also consistent with loss of HuR oligomer formation on ARE substrates shorter than 18 nucleotides. HuR binding to the Fl-ARE20 and Fl-ARE18 substrates was well resolved by the cooperative binding model of Equation 3 (Fig. 4B, left and center, solid lines), exhibiting Hill coefficients significantly greater than unity (Table II). Comparative regression solutions using single site or multiple identical binding site models incorporating protein depletion (Equation 6) were less favorable (Fig. 4B, left and center, dotted lines). For the Fl-ARE20 substrate, the non-cooperative quadratic solution yielded a random distribution of residuals (P = 0.2487), however, the sum-of-squares deviation was significantly improved using the cooperative binding model (P < 0.0011 by F test). For HuR binding to the Fl-ARE18 substrate, the non-cooperative quadratic model was even less favorable, since regression solutions using Equation 6 exhibited both nonrandom residuals (P = 0.0201) and significantly poorer sum-of-squares deviation relative to the cooperative binding model (P = 0.0002 by F test). By contrast, no cooperativity was detected in HuR binding to any Fl-ARE substrate of 16 nucleotides or less, since resolved Hill coefficients did not significantly differ from 1 (Table II) and regression to cooperative binding models did not significantly improve sum-of-squares deviations. Finally, interactions with HuR did not influence the fluorescence quantum yield of any Fl-labeled RNA substrate tested (data not shown), thus validating use of the binding algorithms described by Equation 3–Equation 6.
Fig. 4. In vitro evaluation of HuR binding to truncated ARE substrates.
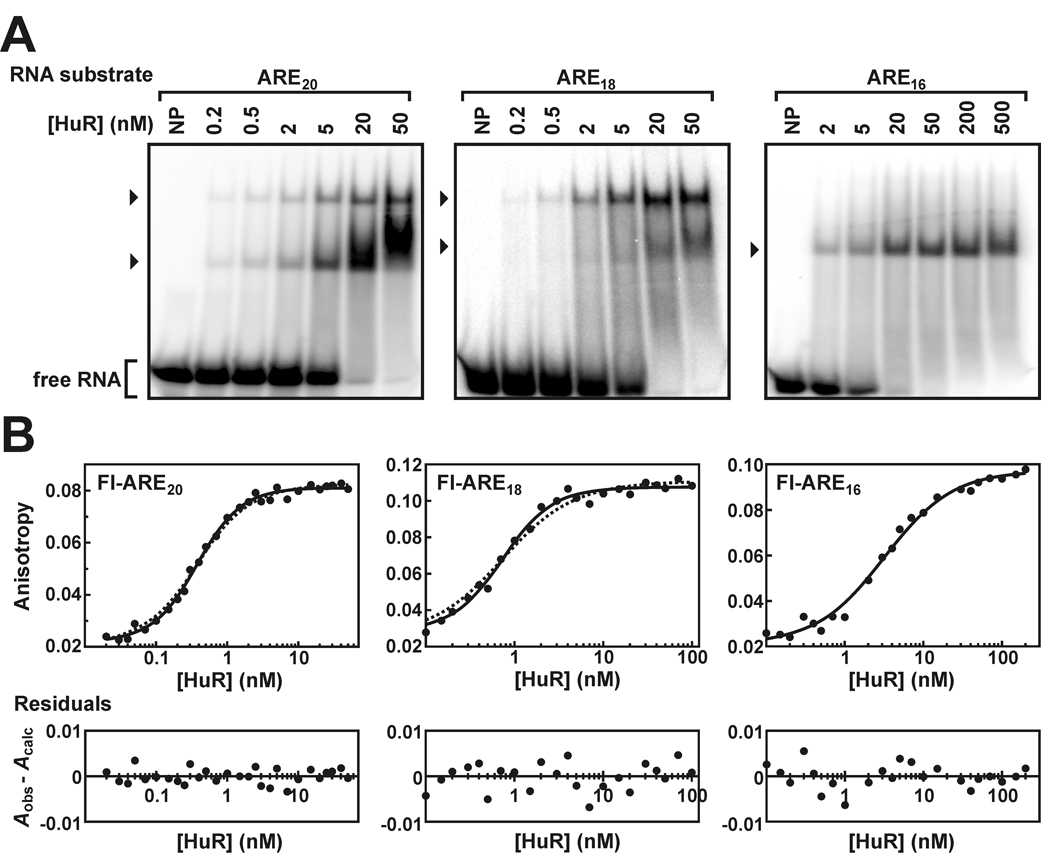
A, EMSAs performed using either 32P-labeled ARE20, ARE18 or ARE16 RNA substrates (0.2 nM) and a titration of recombinant HuR protein. Lanes indicated NP contained no recombinant protein. The arrowheads to the left of each gel indicate bands corresponding to complexes containing 32P-RNA substrates and one or more HuR molecules. Unbound probe is designated as free RNA. B, association of HuR with the Fl-ARE20 (left panel), Fl-ARE18 (middle panel), or Fl-ARE16 RNA substrates (right panel) analyzed by fluorescence anisotropy. All plots were fit to a cooperative binding model using Equation 3 (solid lines). For comparison, the regression solutions for the Fl-ARE20 and Fl-ARE18 substrates are also shown for single site or multiple independent site binding models incorporating protein depletion using Equation 6 (dotted lines). Single site and cooperative binding models yielded essentially identical fits for HuR binding to the Fl-ARE16 substrate. For all data sets, residuals plots are presented as described in Figure 2.
The concurrence of a single HuR:RNA complex containing either the ARE16 or ARE13 substrates by EMSA (Fig. 4A, right and data not shown), together with a confident single site binding solution by anisotropy-based assays (Fig. 4B, right and Table II), indicates that HuR forms 1:1 complexes with ARE substrates ≤ 16 nucleotides in length. Truncation of a single nucleotide (Fl-ARE15) did not alter HuR binding activity, while removal of one or two additional bases (Fl-ARE14 and Fl-ARE13) only modestly diminished the affinity of HuR binding (Table II). By contrast, shortening the ARE substrate to 11 nucleotides (Fl-ARE11) significantly decreased HuR binding activity. Together, these data demonstrate that wild type HuR optimally recognizes ARE substrates of approximately 15 nucleotides in length, but that slightly larger sequences are required to promote cooperative formation of HuR oligomers. A potential mechanism reconciling differences in the RNA substrate requirements for initial versus subsequent cooperative HuR binding events is considered under Discussion.
The RRM3 and Hinge Domains Contribute to the Affinity and Mechanism of ARE Substrate Binding by HuR
Published structures of peptide:RNA complexes containing RRMs 1 and 2 of HuD or HuC indicated that these domains are sufficient for specific recognition of ARE substrates (21, 22). However, these structures indicated peptide contact with only 8–9 contiguous RNA bases, while data reported in this work show that association of a single HuR monomer is significantly enhanced for longer RNA substrates (Table II). These observations suggest that peptide sequences outside of RRMs 1 and 2 may make substantive contributions to the stability of HuR:ARE complexes. To test whether additional C-terminal domains of the HuR protein might contribute to its ARE-binding activity, we generated HuR truncation mutant proteins that removed the RRM3 domain alone (HuRΔRRM3) or the hinge and RRM3 domains together (HuRΔHinge3) (Fig. 1), and then tested their ability to bind ARE substrates. EMSA experiments revealed that the recombinant HuRΔRRM3 protein retains the ability to form multi-subunit complexes with the ARE38 substrate, consistently yielding at least two distinct protein:ARE complexes (Fig. 5A, left panel). By contrast, binding of the HuRΔHinge3 protein to the ARE38 substrate only generated a single RNA:protein complex by EMSA (Fig. 5A, right panel). RNA binding by either HuR deletion mutant remained dependent on the AU-rich sequence, since neither protein formed complexes on the Rβ substrate (data not shown).
Fig. 5. HuRΔRRM3 and HuRΔHinge3 binding to the ARE38 RNA substrate in vitro.
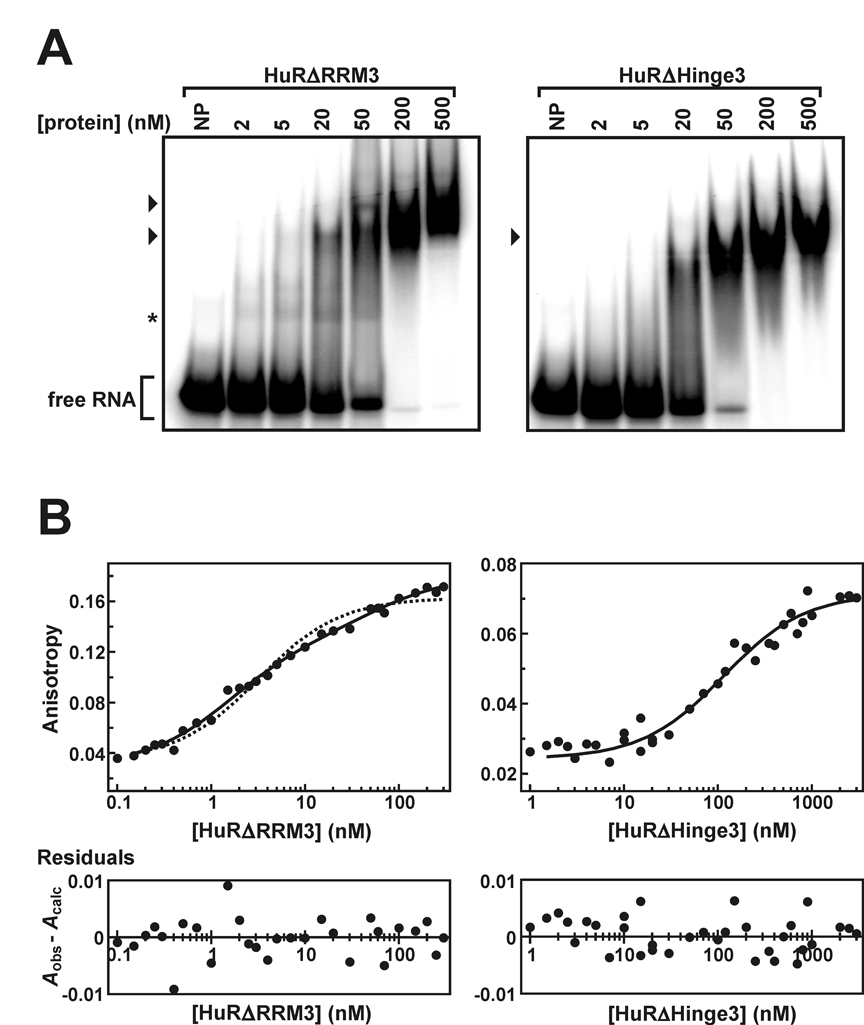
A, EMSAs performed using the 32P-ARE38 substrate (0.2 nM) and a titration of recombinant HuRΔRRM3 (left panel) or HuRΔHinge3 (right panel) proteins. Lanes indicated NP contained no recombinant protein. The arrowheads to the left of each gel indicate bands corresponding to complexes containing 32P-ARE38 substrates and recombinant proteins. The complex labeled by the asterisk was faintly but reproducibly observed in binding experiments containing the 32P-ARE38 substrate with HuRΔRRM3 but not HuRΔHinge3. Unbound probe is designated as free RNA. B, association of HuRΔRRM3 (left panel) or HuRΔHinge3 (right panel) with the Fl-ARE38 RNA substrate analyzed by fluorescence anisotropy as described in Figure 2. HuRΔRRM3:Fl-ARE38 binding data were resolved with a two-step binding model using Equation 4 (left panel, solid line). By comparison, this data set was also fit to a single site binding model using Equation 5 (left panel, dotted line). HuRΔHinge3:Fl-ARE38 binding data were fit to a single site binding model using Equation 5 (right panel, solid line). For each data set, residuals plots (bottom) are presented as described in Figure 2.
Anisotropy-based analyses of HuRΔRRM3 binding to the Fl-ARE38 substrate were well resolved by the sequential two-step binding model described by Equation 4 (Fig. 5B, left panel, solid line), consistent with the formation of two or more protein:RNA complexes indicated by the EMSA experiment. The utility of this model was further supported by random distribution of residuals and a high coefficient of determination (R² > 0.99). By contrast, residual nonrandomness (P = 0.00025) and pair-wise model comparisons using the F test (P < 0.0001) indicated that a single site binding model (Equation 5) was clearly inappropriate for these data (Fig. 5B, left panel, dashed line). Using the two-step binding model, the apparent affinity of the initial interaction between HuRΔRRM3 and the Fl-ARE38 substrate (Kd-1 = 1.7 nM; Table III) was similar to that between the full length HuR protein and optimal single-site substrates Fl-ARE15 and Fl-ARE16 (Kd = 2.5 nM; Table II). Analyses of HuRΔRRM3 binding to truncated Fl-ARE substrates indicated that the affinity of this initial protein binding event was similar to the affinity of wild type HuR for comparably sized ARE substrates, with apparent Kd values ≤ 10 nM for all substrates of 13 nucleotides or longer (Table III). While the EMSA experiment suggested that higher protein concentrations were required to drive the first HuRΔRRM3:ARE38 binding step, it is possible that this initial protein:RNA complex is dynamic in nature, and does not remain associated during gel loading and/or prolonged electrophoresis. A similar discrepancy between binding affinities resolved by EMSA versus anisotropy was observed for truncation mutants of p37AUF1, which form highly dynamic complexes with RNA substrates (36). Removal of RRM3 from the HuD protein also accelerated the kinetics of RNA binding and dissociation in surface plasmon resonance experiments (20). However, although anisotropy-based experiments indicated that initial contact between HuRΔRRM3 and the Fl-ARE38 substrate was not impaired by the absence of RRM3, the apparent affinity of the second protein binding event was inhibited by a factor of nearly 30 (Table III; cf. Kd-1 versus Kd-2). This second HuRΔRRM3 binding event was also sensitive to RNA substrate size, since it was not detectable by either EMSA (data not shown) or anisotropy (Table III) with ARE substrates ≤ 20 nucleotides in length. Together, these data show that HuRΔRRM3 remains capable of high affinity ARE recognition. However, subsequent formation of multi-subunit protein complexes on ARE substrates is significantly impaired by loss of RRM3, indicating that this domain is essential for the cooperative assembly of protein oligomers observed with full length HuR.
Table III.
Association of HuRΔRRM3 or HuRΔHinge3 with TNFα ARE-based RNA substrates.
| Protein | RNA substrate | Apparent shifted speciesa | Kd-app-1b (nM) | Kd-app-2b (nM) | n |
|---|---|---|---|---|---|
| HuRΔRRM3 | ARE38 | 2 | 1.7 ± 0.2 | 47 ± 6 | 3 |
| ARE20 | 1 | 1.6 ± 0.2 | - | 3 | |
| ARE18 | n/d | 2.2 ± 0.3 | - | 3 | |
| ARE16 | 1 | 3.5 ± 0.9 | - | 3 | |
| ARE15 | n/d | 4.4 ± 0.6 | - | 3 | |
| ARE14 | n/d | 5.3 ± 0.6 | - | 3 | |
| ARE13 | 1 | 10.1 ± 1.4 | - | 6 | |
| ARE11 | n/d | 26 ± 2 | - | 2 | |
| HuRΔHinge3 | ARE38 | 1 | 98 ± 14 | - | 4 |
| ARE20 | 1 | 65 ± 9 | - | 3 | |
| ARE16 | 1 | 99 ± 12 | - | 2 | |
| ARE13 | 1 | 64 ± 14 | - | 3 | |
the number of protein:RNA complexes formed between HuR deletion mutant proteins and specific RNA substrates assessed by EMSA experiments (Fig. 4 and data not shown); n/d, not determined.
association binding constants were solved from At versus [protein] data sets (eg: Fig. 4) using Equation 4 (for sequential two-step binding) or Equation 5 (for single site binding), then converted to dissociation constants using Kd-app = 1/K. All resolved constants are expressed as the mean ± σn-1 for n ≥ 3 or mean ± spread for n = 2 independent experiments.
The formation of a single complex between the HuRΔHinge3 protein and the ARE38 substrate suggested by EMSA experiments was also reflected in quantitative anisotropy-based binding assays, since these data were well resolved by the single site binding model described by Equation 5 (Fig. 5B, right panel). However, the apparent affinity of HuRΔHinge3 for the Fl-ARE38 substrate was reduced approximately 60-fold relative to the HuRΔRRM3 protein (Table III). Also, no significant differences in the ARE-binding affinity of the HuRΔHinge3 protein were observed for RNA substrates between 13 and 38 nucleotides in length. Comparing the apparent ARE-binding affinities of the HuRΔHinge3 and HuRΔRRM3 proteins indicates that the hinge domain contributes significant binding energy (ΔΔG = −2.4 kcal/mol) to the protein:RNA complex.
The Thermodynamic Stability of Protein Folding is not Impaired by Removal of the RRM3 and Hinge Domains.
A trivial explanation for the defects in ARE-binding affinity and cooperative oligomer assembly exhibited by the HuRΔHinge3 and HuRΔRRM3 proteins, respectively, is that HuR proteins do not fold properly in the absence of the hinge and/or RRM3 domains. To test this possibility, the folded stability of each recombinant protein was measured through chemical denaturation experiments. Here, denaturant-induced release of protein structure was monitored by changes in the fluorescence of tyrosine residues as each protein transitioned from folded to unfolded conformations across a titration of guanidine hydrochloride (GnHCl) (Fig. 6A). The fluorescence of free tyrosine is not sensitive to GnHCl (Supplemental Fig. S3). Thermodynamic parameters describing the folded stability of each protein were then calculated from the GnHCl concentration-dependence of tyrosine fluorescence using Equation 1 and Equation 2 (Fig. 6B–D). Resolved constants describing the extrapolated free energy of protein unfolding in the absence of denaturant (ΔGuw) and the sensitivity of each protein to GnHCl (meq) are listed in Table IV. No statistically significant differences were observed among values of ΔGuw or meq for any of the three recombinant proteins, indicating that loss of either the RRM3 and/or hinge domains does not compromise the overall stability of HuR protein folding.
Fig. 6. Chemical denaturation of HuR and HuR deletion mutant proteins.
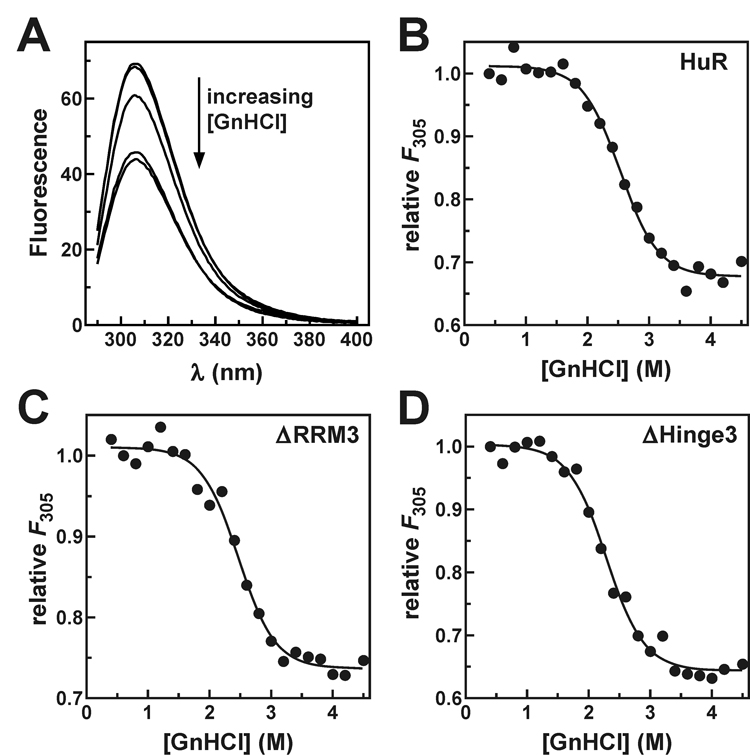
A, blank-corrected fluorescence emission spectra (λex = 270 nm) of HuRΔHinge3 (2 µM) measured in the presence of 0.4 M (top trace), 1 M, 2 M, 3 M, or 4 M GnHCl. Fluorescence emission at 305 nm from (B) HuR, (C) HuRΔRRM3, and (D) HuRΔHinge3 plotted as a function of GnHCl concentration. Thermodynamic parameters describing the folded stability of each protein were resolved by nonlinear regression using Equation 1 and Equation 2 as indicated under “Experimental Procedures” and are quoted in Table IV.
Table IV.
Thermodynamic parameters describing GnHCl-induced unfolding of HuR and HuR deletion mutant proteins
| protein | ΔGuwa (kcal·mol−1) | meqa (kcal·mol−1·M−1) |
|---|---|---|
| HuR | 4.8 ± 0.6 | 1.9 ± 0.2 |
| HuRΔRRM3 | 4.6 ± 0.3 | 1.9 ± 0.1 |
| HuRΔHinge3 | 4.1 ± 0.2 | 1.8 ± 0.1 |
the free energy of protein unfolding in the absence of denaturant (ΔGuw) and the sensitivity of protein folding free energy to GnHCl (meq) were solved from F305 versus [GnHCl] data sets (Fig. 5) using Equation 1 and Equation 2 and expressed as the mean ± spread of duplicate independent experiments.
The Basic Hinge Region does not Stabilize Protein:ARE Complexes via Enhanced Electrostatic Contacts
Reversible transport of HuR between the nucleus and cytoplasm is mediated by an NLS element within the hinge region (25). However, loss of the hinge region also results in a significant decrease in ARE-binding affinity that is not coupled to global protein folding defects (detailed above), indicating that this domain might have additional roles in HuR function beyond housing the NLS. One mechanism by which the hinge domain might contribute to ARE-binding affinity was indicated by a significant decrease in the calculated isoelectric point of HuRΔHinge3 relative to the HuRΔRRM3 protein (Supplemental Table SI). The hinge domain of HuR includes five basic amino acid residues that are largely conserved among the Hu protein family (Fig. 7A), which could potentially enhance the stability of Hu protein:ARE complexes by formation of intra- or intermolecular ion pairs. Such ionic interactions frequently contribute to the stability of complexes between RNA-binding proteins and their polyanionic substrates (46, 47). To test this hypothesis, we measured the apparent binding affinities of the HuRΔRRM3 and HuRΔHinge3 proteins for the Fl-ARE20 RNA substrate across varying concentrations of monovalent cation (K+) using the fluorescence anisotropy assay. The Fl-ARE20 substrate was selected for these experiments to avoid cation-dependent effects on the folding of larger ARE substrates (48) and potential influences of ARE folding on protein binding affinity (40). Apparent binding constants describing HuRΔRRM3:Fl-ARE20 and HuRΔHinge3:Fl-ARE20 complex assembly at each cation concentration were plotted as log(Kapp) versus −log[K+] and resolved by linear regression (Fig. 7B). The net number of ion pairs formed during assembly of a protein:RNA complex is reflected in the slope of this line (49). If the basic hinge domain contributed to the thermodynamic stability of protein:ARE complexes by formation of unique ionic interactions, removal of this region should result in a decrease in the net number of ion pairs formed. However, no statistically significant difference was observed in the affinity of HuRΔRRM3 versus HuRΔHinge3 binding to the Fl-ARE20 substrate as a function of monovalent cation concentration (Fig. 7B; cf. slopes of 2.2 ± 0.3 for HuRΔRRM3 versus 2.0 ± 0.3 for HuRΔHinge3). As such, the enhanced ARE-binding activity conferred by inclusion of the HuR hinge region is unlikely to result from nonspecific ionic interactions involving this basic protein domain.
Fig. 7. Contributions of ionic interactions to the ARE-binding activity of HuR truncation mutant proteins.
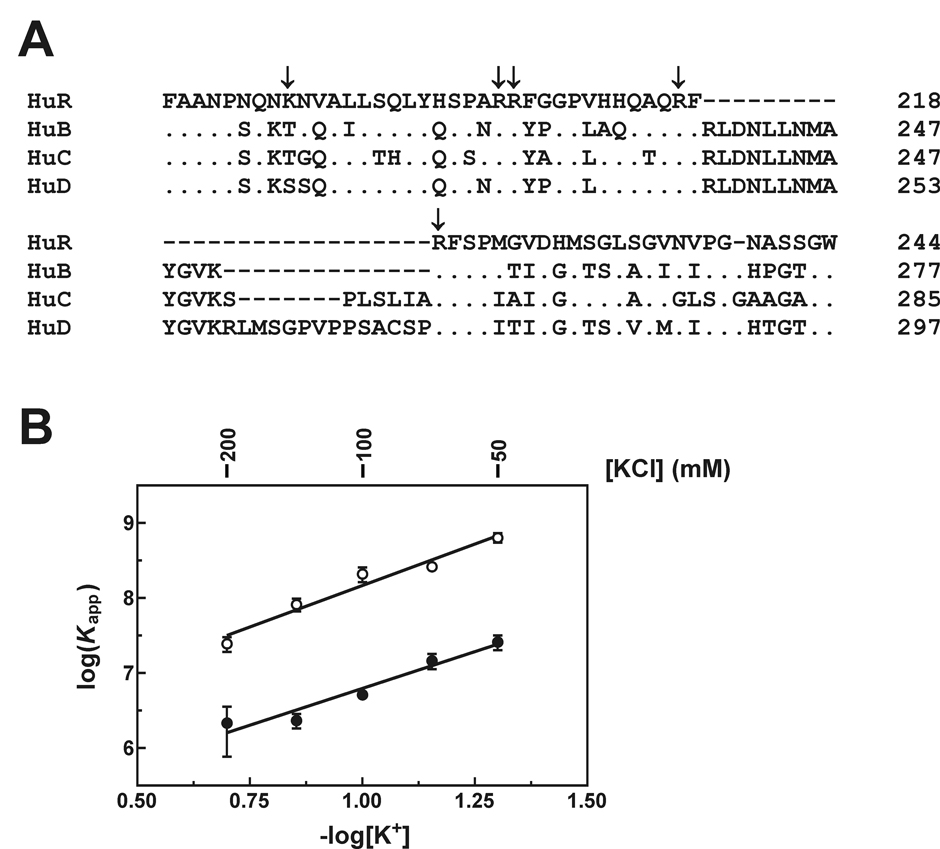
A, ClustalW homology alignment of the hinge domains from human Hu protein family members. Residues are numbered relative to the initiator methionine for each protein. Conserved amino acids are given by a dot, dashes indicate deleted residues, and amino acid substitutions are given where applicable. Positively charged residues within the HuR hinge domain are indicated by arrows. B, anisotropy binding experiments were performed as described under “Experimental Procedures” under varying concentrations of KCl. Apparent equilibrium binding constants (Kapp) describing single site complex formation between the Fl-ARE20 substrate and HuRΔRRM3 (open circles) or HuRΔHinge3 (closed circles) were calculated using Equation 5, and then plotted as log(Kapp ± spread) versus −log[K+] based on at least two independent experiments for each concentration of KCl. An index of ionic sensitivity for each protein:RNA complex was calculated as −∂log(Kapp)/∂log[K+] (solid lines).
DISCUSSION
The HuR basic hinge domain contains the NLS element required for nucleocytoplasmic shuttling by this protein (25). However, data presented in this report also indicate that this domain contributes to the stability of HuR complexes with ARE substrates (Table III). This enhancement in ARE-binding activity was significantly greater than that observed in comparable experiments with HuD truncation mutants, where inclusion of the hinge region only yielded a two-fold improvement in binding to a TNFα ARE-derived RNA substrate (20). However, two factors may account for the apparent differences in contributions of hinge domains to the ARE-binding activities of HuR versus HuD. First, the HuD RRM1+RRM2 protein included the first 13 residues of the hinge domain, which may have contributed to its strong ARE-binding activity (Kd = 5.4 nM by surface plasmon resonance analysis) (20). Second, the hinge domain is the least conserved region in the Hu protein family with the possible exception of the extreme N-terminal end. The HuR hinge shares only 47% amino acid sequence identity with other family members (Fig. 7A), and also lacks approximately 10–30 residues retained within the other three proteins. It is conceivable that sequence characteristics unique to the HuR hinge domain are required for enhancement of ARE-binding activity.
The HuR hinge domain does not include sequence motifs characteristic of any known RNA-binding domain. However, based on several findings reported in this study, we predict that the hinge domain stabilizes HuR:ARE ribonucleoprotein complexes through formation of specific protein:RNA contacts. First, equilibrium binding experiments demonstrated that removal of the hinge and RRM3 domains (HuRΔHinge3) significantly decreased binding affinity for ARE substrates relative to the HuRΔRRM3 protein (Table III). Second, enhanced ARE binding in the presence of the hinge domain requires additional RNA sequences. Binding of the HuRΔRRM3 protein was significantly enhanced for ARE substrates ≥ 14 nucleotides in length, while the ARE-binding activity of HuRΔHinge3 was not improved by increasing the length of the RNA substrate from 13 to 38 bases (Table III). Third, GnHCl denaturation studies demonstrated that the hinge domain does not enhance the thermodynamic stability of protein folding, indicating that the conformations of the RRM1 and RRM2 domains are likely similar whether alone or in the context of the full length protein (Table IV). Finally, inclusion of the HuR hinge domain does not stabilize protein:ARE complexes by increasing the net number of ion pairs formed during complex assembly (Fig. 7B). Ionic interactions can make significant contributions to the free energy of protein:nucleic acid interactions in an RNA/DNA sequence-independent manner (49, 50). However, enhanced ARE binding by HuRΔRRM3 relative to the HuRΔHinge3 protein without formation of new ion pairs suggests that the hinge domain can make specific but hitherto undefined non-ionic contacts with RNA substrates. Coincident RNA-binding and nuclear localization functions within the basic hinge domain also raise the intriguing possibility that these events may be mutually exclusive. Future studies will address whether association with RNA precludes nuclear import, and by extension, whether HuR relocalization to the nucleus is a default consequence of dissociation from cytoplasmic RNA substrates.
Several observations indicate that full length HuR cooperatively forms multimers on ARE substrates ≥ 18 nucleotides in length: [i] multiple HuR:ARE complexes were detected by EMSA (Fig. 2A and Fig. 4A), [ii] anisotropy-based binding assays resolved Hill coefficients significantly greater than unity (Table II), and [iii] apparent binding affinity was significantly improved relative to ARE substrates supporting only a single HuR binding event (Table II, cf. Fl-ARE20 and Fl-ARE18 versus Fl-ARE16 and Fl-ARE15). Cooperative assembly of HuR oligomers on ARE-based RNA substrates also required both the third RRM of the protein and some RNA sequence flanking the first high-affinity HuR binding site. It is currently unclear whether HuR binding cooperativity is mediated by protein:protein interactions, likely involving RRM3, and/or by protein-induced remodeling of local RNA structure. However, while optimal binding of a single HuR monomer required approximately 15 nucleotides (Table II), both binding density (Fig. 3) and EMSA experiments (Fig. 4A) support a HuR binding site size of 8–9 nucleotides in the cooperative oligomeric complex. This site size is similar to the binding interface of the tandem RRM1+RRM2 domains of HuC and HuD (21, 22), suggesting that RNA contacts involving the hinge and/or RRM3 domains may be released before or during association of the second HuR monomer. Data from this study also indicate that cooperative HuR binding may be limited to pairs of HuR monomers. Binding density analysis indicated that as many as four HuR binding events were possible on ARE38 substrate, three of which were resolved by EMSA (Fig. 2A). However, the decreased [HuR]1/2 observed with the Fl-ARE38 substrate versus Fl-ARE18 and Fl-ARE20 suggests that additional binding steps on the longest RNA substrate may occur with lower affinity, since the resolved [HuR]1/2 value is an aggregate parameter describing all protein binding events contributing to formation of saturated HuR:ARE complexes. The EMSA showing the concentration-dependence of HuR binding to the ARE38 substrate (Fig. 2A) was also consistent with this idea, since formation of the third detectable HuR:ARE complex was only observed at substantially higher protein concentrations.
It is intriguing that the mechanism of ARE substrate binding employed by the mRNA-stabilizing trans-factor HuR is significantly different from that utilized by the cytoplasmic isoforms of AUF1, which may contribute to the destabilization of selected mRNA targets. In this work, we have shown that HuR forms oligomers on the TNFα ARE38 substrate in a cooperative fashion. By contrast, while the p37AUF1 and p40AUF1 proteins bind the same ARE sequence via a sequential two-step binding mechanism, the second binding event typically displays 20- to 50- fold lower affinity than the first (28, 51). Additional studies have indicated that HuR and AUF1 are co-expressed in many cell types and that they associate with many of the same cellular mRNAs (27, 52, 53), suggesting that the equilibrium between HuR versus AUF1 binding may dictate the catabolic fate of specific ARE-containing transcripts. Supporting this model are observations that many cellular stresses increase cytoplasmic HuR concentrations by subcellular relocalization from the nucleus, concomitant with stabilization of HuR-targeted mRNAs (26, 27, 54, 55). The cooperative ARE-binding activity of HuR would facilitate this process, since modest changes in cytoplasmic HuR concentrations could thus dramatically influence the fractional binding density of this protein on ARE-containing mRNAs. Finally, loss of binding cooperativity in HuR proteins lacking RRM3 may explain discrepancies in the mRNA-stabilizing activity of this mutant (13, 24), since significantly higher concentrations of ΔRRM3 proteins would be required to effectively displace competing mRNA-destabilizing factors from mRNA substrates. Overexpression experiments that fail to achieve this threshold cytoplasmic HuRΔRRM3 concentration would thus be less likely to stabilize these targeted transcripts.
ACKNOWLEDGEMENTS
We would like to thank Noelle Vargas for expert technical assistance with the Western blot analyses.
The abbreviations used are
- ARE
AU-rich element
- EMSA
electrophoretic mobility shift assay
- Fl
fluorescein
- GnHCl
guanidine hydrochloride
- GST
glutathione S-transferase
- NLS
nuclear localization sequence
- RRM
RNA recognition motif
- TNFα
tumor necrosis factor α
- UTR
untranslated region
Footnotes
This work was supported by NCI, National Institutes of Health grant R01 CA102428 (to G.M.W.).
REFERENCES
- 1.Ross J. Microbiol. Rev. 1995;59:423–450. doi: 10.1128/mr.59.3.423-450.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guhaniyogi J, Brewer G. Gene. 2001;265:11–23. doi: 10.1016/s0378-1119(01)00350-x. [DOI] [PubMed] [Google Scholar]
- 3.Barreau C, Paillard L, Osborne HB. Nucleic Acids Res. 2005;33:7138–7150. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen C-YA, Shyu A-B. Trends Biochem. Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- 5.Wilson GM, Brewer G. Prog. Nucleic Acids Res. Mol. Biol. 1999;62:257–291. doi: 10.1016/s0079-6603(08)60510-3. [DOI] [PubMed] [Google Scholar]
- 6.Carballo E, Lai WS, Blackshear PJ. Science. 1998;281:1001–1005. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 7.Chen C-Y, Gherzi R, Ong S-E, Chan EL, Raijmakers R, Pruijn GJM, Stoecklin G, Moroni C, Mann M, Karin M. Cell. 2001;107:451–464. doi: 10.1016/s0092-8674(01)00578-5. [DOI] [PubMed] [Google Scholar]
- 8.Lu J-Y, Sadri N, Schneider RJ. Genes Dev. 2006;20:3174–3184. doi: 10.1101/gad.1467606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raineri I, Wegmueller D, Gross B, Certa U, Moroni C. Nucleic Acids Res. 2004;32:1279–1288. doi: 10.1093/nar/gkh282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gherzi R, Lee K-Y, Briata P, Wegmüller D, Moroni C, Karin M, Chen C-Y. Mol. Cell. 2004;14:571–583. doi: 10.1016/j.molcel.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 11.Peng SSY, Chen C-YA, Xu N, Shyu A-B. EMBO J. 1998;17:3461–3470. doi: 10.1093/emboj/17.12.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dean JLE, Wait R, Mahtani KR, Sully G, Clark AR, Saklatvala J. Mol. Cell. Biol. 2001;21:721–730. doi: 10.1128/MCB.21.3.721-730.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen C-YA, Xu N, Shyu A-B. Mol. Cell. Biol. 2002;22:7268–7278. doi: 10.1128/MCB.22.20.7268-7278.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mobarak CD, Anderson KD, Morin M, Beckel-Mitchener A, Rogers SL, Furneaux H, King P, Perrone-Bizzozero NI. Mol. Biol. Cell. 2000;11:3191–3203. doi: 10.1091/mbc.11.9.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez dSI, Galban S, Martindale JL, Yang X, Mazan-Mamczarz K, Indig FE, Falco G, Zhan M, Gorospe M. Mol. Cell Biol. 2005;25:9520–9531. doi: 10.1128/MCB.25.21.9520-9531.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piecyk M, Wax S, Beck A, Kedersha N, Gupta M, Maritim B, Chen S, Gueydan C, Kruys V, Streuli M, Anderson P. EMBO J. 2000;19:4154–4163. doi: 10.1093/emboj/19.15.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Antic D, Keene JD. Am. J. Hum. Genet. 1997;61:273–278. doi: 10.1086/514866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma W-J, Cheng S, Campbell C, Wright A, Furneaux H. J. Biol. Chem. 1996;271:8144–8151. doi: 10.1074/jbc.271.14.8144. [DOI] [PubMed] [Google Scholar]
- 19.Antic D, Lu N, Keene JD. Genes Dev. 1999;13:449–461. doi: 10.1101/gad.13.4.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park S, Myszka DG, Yu M, Littler S, Laird-Offringa IA. Mol. Cell. Biol. 2000;20:4765–4772. doi: 10.1128/mcb.20.13.4765-4772.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Tanaka Hall TM. Nat. Struct. Biol. 2001;8:141–145. doi: 10.1038/84131. [DOI] [PubMed] [Google Scholar]
- 22.Inoue M, Muto Y, Sakamoto H, Yokoyama S. Nucl. Acids Res. 2000;28:1743–1750. doi: 10.1093/nar/28.8.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma W-J, Chung S, Furneaux H. Nucleic Acids Res. 1997;25:3564–3569. doi: 10.1093/nar/25.18.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan XC, Steitz JA. EMBO J. 1998;17:3448–3460. doi: 10.1093/emboj/17.12.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan XC, Steitz JA. Proc. Natl. Acad. Sci. USA. 1998;95:15293–15298. doi: 10.1073/pnas.95.26.15293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallouzi I-E, Steitz JA. Science. 2001;294:1895–1901. doi: 10.1126/science.1064693. [DOI] [PubMed] [Google Scholar]
- 27.Lal A, Mazan-Mamczarz K, Kawai T, Yang X, Martindale JL, Gorospe M. EMBO J. 2004;23:3092–3102. doi: 10.1038/sj.emboj.7600305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilson GM, Sutphen K, Chuang K, Brewer G. J. Biol. Chem. 2001;276:8695–8704. doi: 10.1074/jbc.M009848200. [DOI] [PubMed] [Google Scholar]
- 29.Heyduk T, Ma Y, Tang H, Ebright RH. Methods Enzymol. 1999;274:492–503. doi: 10.1016/s0076-6879(96)74039-9. [DOI] [PubMed] [Google Scholar]
- 30.Wilson GM, Brewer G. Methods. 1999;17:74–83. doi: 10.1006/meth.1998.0709. [DOI] [PubMed] [Google Scholar]
- 31.Meisner NC, Hackermuller J, Uhl V, Aszodi A, Jaritz M, Auer M. Chembiochem. 2004;5:1432–1447. doi: 10.1002/cbic.200400219. [DOI] [PubMed] [Google Scholar]
- 32.Jancarik J, Pufan R, Hong C, Kim SH, Kim R. Acta Crystallogr. D Biol. Crystallogr. 2004;60:1670–1673. doi: 10.1107/S0907444904010972. [DOI] [PubMed] [Google Scholar]
- 33.Lakowicz JR. Principles of Fluorescence Spectroscopy. New York, NY: Kluwer Academic/Plenum; 1999. [Google Scholar]
- 34.Santoro MM, Bolen DW. Biochemistry. 1988;27:8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
- 35.Manyusa S, Whitford D. Biochemistry. 1999;38:9533–9540. doi: 10.1021/bi990550d. [DOI] [PubMed] [Google Scholar]
- 36.Wilson GM, Sun Y, Lu H, Brewer G. J. Biol. Chem. 1999;274:33374–33381. doi: 10.1074/jbc.274.47.33374. [DOI] [PubMed] [Google Scholar]
- 37.Wilson GM. RNA folding and RNA-protein binding analyzed by fluorescence anisotropy and resonance energy transfer. In: Geddes CD, Lakowicz JR, editors. Reviews in Fluorescence Vol. 2. New York: Springer Science+Business Media, Inc.; 2005. [Google Scholar]
- 38.Lohman TM, Bujalowski W. Methods Enzymol. 1991;208:258–290. doi: 10.1016/0076-6879(91)08017-c. [DOI] [PubMed] [Google Scholar]
- 39.Bujalowski W, Jezewska MJ. Biochemistry. 1995;34:8513–8519. doi: 10.1021/bi00027a001. [DOI] [PubMed] [Google Scholar]
- 40.Fialcowitz EJ, Brewer BY, Keenan BP, Wilson GM. J. Biol. Chem. 2005;280:22406–22417. doi: 10.1074/jbc.M500618200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nabors LB, Gillespie GY, Harkins L, King PH. Cancer Res. 2001;61:2154–2161. [PubMed] [Google Scholar]
- 42.Tudyka T, Skerra A. Protein Science. 1997;6:2180–2187. doi: 10.1002/pro.5560061012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keller RW, Kuhn U, Aragon M, Bornikova L, Wahle E, Bear DG. J. Mol. Biol. 2000;297:569–583. doi: 10.1006/jmbi.2000.3572. [DOI] [PubMed] [Google Scholar]
- 44.Brewer BY, Malicka J, Blackshear PJ, Wilson GM. J. Biol. Chem. 2004;279:27870–27877. doi: 10.1074/jbc.M402551200. [DOI] [PubMed] [Google Scholar]
- 45.Bujalowski W, Jezewska MJ. Quantitative determination of equilibrium binding isotherms for multiple ligand-macromolecule interactions using spectroscopic methods. In: Gore MG, editor. Spectrophotometry and Spectrofluorimetry. Oxford, UK: Oxford University Press; 2000. [Google Scholar]
- 46.Frankel AD, Mattaj IW, Rio DC. Cell. 1991;67:1041–1046. doi: 10.1016/0092-8674(91)90282-4. [DOI] [PubMed] [Google Scholar]
- 47.Fisher BM, Ha J-H, Raines RT. Biochemistry. 1998;37:12121–12132. doi: 10.1021/bi980743l. [DOI] [PubMed] [Google Scholar]
- 48.Wilson GM, Sutphen K, Moutafis M, Sinha S, Brewer G. J. Biol. Chem. 2001;276:38400–38409. doi: 10.1074/jbc.M106509200. [DOI] [PubMed] [Google Scholar]
- 49.Draper DE. Annu. Rev. Biochem. 1995;64:593–620. doi: 10.1146/annurev.bi.64.070195.003113. [DOI] [PubMed] [Google Scholar]
- 50.Misra VK, Honig B. Proc. Natl. Acad. Sci. USA. 1995;92:4691–4695. doi: 10.1073/pnas.92.10.4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilson GM, Lu J, Sutphen K, Suarez Y, Sinha S, Brewer B, Villanueva-Feliciano EC, Ylsa RM, Charles S, Brewer G. J. Biol. Chem. 2003;278:33039–33048. doi: 10.1074/jbc.M305775200. [DOI] [PubMed] [Google Scholar]
- 52.Gouble A, Morello D. Oncogene. 2000;19:5377–5384. doi: 10.1038/sj.onc.1203910. [DOI] [PubMed] [Google Scholar]
- 53.Lu J-Y, Schneider RJ. J. Biol. Chem. 2004;279:12974–12979. doi: 10.1074/jbc.M310433200. [DOI] [PubMed] [Google Scholar]
- 54.Zou T, Mazan-Mamczarz K, Rao JN, Liu L, Marasa BS, Zhang AH, Xiao L, Pullmann R, Gorospe M, Wang JY. J. Biol. Chem. 2006;281:19387–19394. doi: 10.1074/jbc.M602344200. [DOI] [PubMed] [Google Scholar]
- 55.Wang W, Yang X, Kawai T, Lopez de Silanes I, Mazan-Mamczarz K, Chen P, Chook YM, Quensel C, Köhler M, Gorospe M. J. Biol. Chem. 2004;279:48376–48388. doi: 10.1074/jbc.M409014200. [DOI] [PubMed] [Google Scholar]