

Abstract
Accumulation of oxidized lipids in the arterial wall contributes to atherosclerosis. Glutathione peroxidase-4 (GPx4) is a hydroperoxide scavenger that removes oxidative modifications from lipids such as free fatty acids, cholesterols, and phospholipids. Here, we set out to assess the effect of GPx4 overexpression on atherosclerosis in apolipoprotein E-deficient (ApoE−/−) mice. The results revealed that atherosclerotic lesions in the aortic tree and aortic sinus of ApoE−/− mice overexpressing GPx4 (hGPx4Tg/ApoE−/−) were significantly smaller than those of ApoE−/− control mice. GPx4 overexpression also diminished signs of advanced lesions in the aortic sinus, as seen by a decreased occurrence of fibrous caps and acellular areas among hGPx4Tg/ApoE−/− animals. This delay of atherosclerosis in hGPx4Tg/ApoE−/− mice correlated with reduced aortic F2-isoprostane levels (R2 = 0.75, p < 0.01). In addition, overexpression of GPx4 lessened atherogenic events induced by the oxidized lipids, lysophosphatidylcholine and 7-ketocholesterol, including upregulated expression of adhesion molecules in endothelial cells, adhesion of monocytes to endothelial cells, as well as endothelial necrosis and apoptosis. These results suggest that overexpression of GPx4 inhibits the development of atherosclerosis by decreasing lipid peroxidation and inhibiting the sensitivity of vascular cells to oxidized lipids.
Keywords: Glutathione peroxidase-4, Atherosclerosis, Lipid peroxidation, Monocyte adhesion, Necrosis, Apoptosis
Accumulating evidence suggests that endogenously generated peroxides, including hydrogen peroxide (H2O2) and lipid hydroperoxides, are involved in the pathogenesis of atherosclerosis through two main mechanisms. First, peroxides have been shown to induce oxidative modifications of lipoproteins. A number of laboratories, including ours, have shown that H2O2 released from vascular cells can oxidize low-density lipoprotein (LDL) in vitro [1,2]. Oxidized LDL and its lipid components induce atherogenic events such as injury to vascular cells, increase in interactions between inflammatory and endothelial cells, and induction of vascular smooth muscle cell proliferation [reviewed in [3]. The second mechanism by which peroxides contribute to the pathogenesis of atherosclerosis is by increasing the sensitivity of vascular cells to oxidized LDL and lipid components. For example, H2O2 may function as a second messenger of oxidized lipids/lipoproteins to induce expression of a variety of proteins that are thought to be involved in the recruitment of inflammatory cells to the vessel wall and in the proliferation and death of vascular cells [4,5]. We recently showed that overexpression of catalase, an H2O2 scavenger, significantly decreased atherosclerotic lesions and levels of oxidized lipids in the arterial wall of apolipoprotein E-deficient (ApoE−/−) mice [6], providing direct evidence for the atherogenic role of H2O2. This finding suggests that increasing the cellular level of peroxide scavengers may provide a valuable strategy for the treatment of atherosclerosis.
Mammalian cells contain several peroxide scavengers, including catalase, glutathione peroxidases (GPxs), and peroxiredoxins [reviewed in [7]. Among these, GPx4 has attracted extensive attention due to its unique structural and functional characteristics. GPx4 is a 20- to 22-kDa monomer, whereas other GPx proteins are tetramers. GPx4 is widely expressed in all normal tissues, including arteries, and is distributed among multiple subcellular sites such as the cytoplasm, endoplasmic reticulum, plasma membrane, mitochondria, and nucleus [8]. In contrast, the distribution of most other peroxide scavengers is tissue- and/or organelle-specific. Catalase, for instance, is restricted to peroxisomes [9]. GPx4 can react with H2O2 [10] and, contrary to most other antioxidant enzymes, a wide range of lipid hydroperoxides including oxidized phospholipids [10] and cholesterol hydroperoxides [11]. It is well established that oxidized lipids are highly atherogenic [reviewed in [12]. In addition to detoxifying nonenzymatically oxidized lipids, GPx4 reduces lipid peroxides generated by lipoxygenases, cyclooxygenases, and acetyl-CoA:1-O-alkyl-2-lyso-sn-glycero-3-phosphocholine acetyltransferase, thereby decreasing the synthesis of leukotrienes [13], prostaglandin E [14], and platelet-activating factor [15]. These lipid mediators have been reported to play a role in inflammation and atherogenesis [reviewed in [16,17].
Overexpression of GPx4 has been shown to protect cells against oxidants and cytokines. GPx4 overexpression inhibits basal and cytokine-induced adhesion molecule expression in vascular smooth muscle cells [18] and prevents oxidative injury in various other cell types [19]. GPx4 overexpression also increases the resistance of cells to cholesterol hydroperoxides [20]. Conversely, a heterozygous mutation in GPx4 increases the sensitivity of mouse fibroblasts to oxidant-induced apoptosis [21]. Ran et al. have developed a transgenic mouse (hGPx4Tg) that overexpressed GPx4 in all tissues examined [22]. Primary cultures of cortical neurons and embryonic fibroblasts derived from hGPx4Tg mice showed increased survival and decreased apoptosis after exposure to oxidants such as tert-butyl hydroperoxide, diquat, and H2O2, compared to cells derived from wild-type mice [22,23]. Furthermore, liver damage and lipid peroxidation induced by diquat injection were significantly decreased in hGPx4Tg mice compared to wild-type mice [23].
One major difficulty in using mouse models to study atherosclerosis is that mice with normal plasma cholesterol levels are highly resistant to atherosclerosis. This has led to the development of ApoE−/− mice, which have spontaneously increased plasma cholesterol levels in response to a normal chow diet and develop atherosclerotic lesions with morphologic features and an arterial distribution resembling those in humans [24]. In the present study, we crossbred hGPx4Tg mice with ApoE−/− mice and examined the effect of GPx4 overexpression on atherosclerosis. Results of the present study showed that overexpression of GPx4 in ApoE−/− mice inhibited the development of atherosclerosis in association with a decrease in the level of oxidized lipids in the aorta. Oxidized lipids induce various atherogenic events including recruitment of inflammatory cells to the intima and induction of vascular cell death [3,25]. To further evaluate the anti-atherogenic role of GPx4, we studied the effect of GPx4 overexpression on the monocyte-endothelium interaction and on endothelial cell death induced by lysophosphatidylcholine (LPC) and 7-ketocholesterol (7-KC), which are commonly present in oxidized LDL and in atherosclerotic lesions. We observed that overexpression of GPx4 significantly inhibited LPC- and 7-KC-induced adhesion of monocytes to endothelial cells as well as necrotic and apoptotic death of endothelial cells. These observations suggest that an increase in cellular GPx4 levels inhibits the development of atherosclerosis by decreasing lipid peroxidation and increasing the resistance of vascular cells to oxidized lipids.
Materials and methods
Animals
Transgenic mice overexpressing human GPx4 (hGPx4Tg) were generated by injection of fertilized C57BL/6 mouse embryos with a fragment of human genomic DNA containing the GPx4 gene [22]. ApoE−/− mice with a 129vEv genetic background were generated [26] and backcrossed with C57BL/6 mice for more than 12 generations. Mice with heterozygous overexpression of GPx4 and a homozygous mutation of ApoE (hGPx4Tg/ApoE−/−) were obtained by crossbreeding hGPx4Tg mice with ApoE−/− mice. Only male mice were used in this study since estrogen has been shown to affect the development of atherosclerosis by decreasing LDL oxidation and other mechanisms [27]. After weaning, all mice were maintained on a chow diet containing approximately 5% fat and 19% protein by weight (Harlan Teklad, Madison, WI). The animals used in experiments were 5–6 months of age. All procedures were approved by the Institutional Animal Care and Use Committee of Meharry Medical College.
Quantification of atherosclerotic lesions in mouse aortas
Mouse aortas were cut at a distance of 2 mm from the heart, and coronal sections (8 μm) were prepared from the proximal aorta attached to the heart as described previously [28]. Sections were stained with Oil Red O and viewed with a microscope (E600; Nikon Instruments Inc., Melville, NY) equipped with a color digital camera (CoolSnaps; Nikon Instruments Inc.) and a computer image acquisition system (MetaMorph Image System; Universal Imaging Corp., West Chester, PA). The average area (μm2) of the lesion as well as its morphologic features (i.e., foam cell deposition, cholesterol clefts, acellular areas, and fibrous-caps) was recorded. Sixteen sections were examined from each animal.
The distal aorta (2 mm from the heart to the iliac bifurcation) was opened longitudinally as described previously [6]. The en face preparation was fixed overnight and stained with Sudan IV stain. A photograph of the aorta was obtained with a CoolSnaps digital camera mounted on a SMZ1000 dissecting microscope (Nikon Instruments Inc.), and the area of the atherosclerotic lesion and the total area of the aorta were measured with the MetaMorph Image System. Data are expressed as the percentage of the total surface area of the aorta covered by atherosclerotic lesions.
Measurement of F2-isoprostanes in the mouse aorta
The concentration of F2-isoprostanes in the aorta was measured as an indicator of in vivo lipid peroxidation. Mouse aortas were minced in 2 ml ice-cold high-performance liquid chromatography-grade water containing 100 μM butylhydroxytoluene and 1 mM ethylenediaminetetraacetic acid (EDTA) and homogenized on ice at 500 rpm for 5 min. After addition of a known amount of [2H4] F2-isoprostane internal standard to the homogenate, the total lipids were extracted and purified by thin-layer chromatography. F2-isoprostanes were analyzed by gas chromatography/mass spectrometry [6].
Western blot analysis
Expression of GPx4, vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and β-actin protein in mouse aortas was determined by western blotting with specific antibodies obtained from Cayman Chemical Co. (Ann Arbor, MI) and Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Aortas pooled from two mice were homogenized in 20 mM Tris-Cl, and homogenates were centrifuged at 14,000 rpm for 10 min at 4°C. Supernatants containing 15 μg of protein were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. Detection of GPx4, VCAM-1, ICAM-1, and β-actin was performed according to standard western blotting protocols, using the ECL Western Blotting Detection System (Amersham Biosciences Inc., Piscataway, NJ).
GPx4 activity assay
GPx4 activity was determined as described by Maiorino et al. [29] with slight modifications. Mouse aortas were homogenized in 0.25 M sucrose and 20 mM Tris-HCl (pH 7.4), sonicated for 30 sec (Model 100 Sonic Dismembrator; Fisher Scientific International Inc., Hampton, NH), and centrifuged at 10,000 × g for 15 min at 4°C. Phosphatidylcholine hydroperoxide, the substrate of GPx4, was prepared from soybean L-α-phosphatidylcholine (P6263) (Sigma-Aldrich, St. Louis, MO) as described by Lei et al. [30]. The GPx4 activity assay was carried out in a 1-ml reaction mixture containing 100 mM Tris-HCl, 5 mM EDTA, 0.1 mM NADPH, 1 mM NaN3, 3 mM reduced glutathione, 0.2% peroxide-free Triton X-100, 1.2 × 10−6 U glutathione reductase, 10–50 μg aorta protein extract, and 78 nM phosphatidylcholine hydroperoxide. Absorbance was determined at 340 nm with a DU640 spectrophotometer (Beckman Coulter Inc., Fullerton, CA).
Plasma lipid analysis
Concentrations of plasma cholesterols and triglycerides were quantified spectrophotometrically with reagents obtained from Sigma-Aldrich. For analysis of the cholesterol distributed in various lipoproteins, 100 μl of plasma obtained from an individual mouse was injected onto a Superose-6 column and fractionated by fast-performance liquid chromatography (Äkta FPLC 900; Amersham Biosciences, Piscataway, NJ). Forty 0.5-ml fractions were collected, and fractions 11–40 were analyzed for cholesterol content of various lipoproteins as described elsewhere [31].
Quantification of hydroperoxide production in mouse aorta endothelial cells
Mouse aorta endothelial cells (MAECs) were isolated from hGPx4Tg/ApoE−/− and ApoE−/− mice by an outgrowth technique as detailed previously [32]. The production of hydroperoxides by MAECs was determined by two methods. First, intracellular hydroperoxide levels were determined with the use of 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (DCFHDA). MAECs grown to confluence in a clear, black-bottomed, 96-well plate were incubated for 1 h with 10 μg/ml DCFHDA and then with the indicated concentrations of LPC or 7-KC for 30 min at 37°C. Fluorescence was determined with a Fluoroskan Ascent AL fluorometer (Thermo Labsystems, Franklin, MA) with excitation and emission wavelengths of 485 nm and 510 nm, respectively. Second, hydroperoxides released into the culture medium were measured with an Amplex Red Hydrogen Peroxide Assay Kit (Invitrogen Corp., Carlsbad, CA). MAECs grown to confluence in a 96-well plate were pretreated with 500 U/ml catalase (Sigma-Aldrich) or culture medium alone for 5 min at 37°C. The indicated concentrations of LPC or 7-KC and 150 μl Amplex Red Reagent were then added to each well. After a 40-min incubation, fluorescence was read with the Fluoroskan Ascent AL fluorometer with excitation and emission wavelengths of 540 nm and 590 nm. The cumulative hydroperoxide concentration was extrapolated from a standard curve obtained by incubation of the Amplex Red Reagent with H2O2. The amount of hydroperoxides released was normalized to total cellular protein and were expressed as nM/mg protein/h.
Adhesion of mononuclear cells to endothelial cells
Mouse mononuclear cells (MNCs) were isolated from the blood of wild-type mice by Histopaque gradient separation and labeled with calcein acetoxymethyl ester provided by the Vybrant Cell Adhesion Assay Kit (Molecular Probes, Eugene, OR) as described previously [32]. MAECs were grown to confluence in 96-well plates and treated for 4 h with 30 μM LPC, 100 μM 7-KC, or culture medium alone as a control. The medium was then replaced with serum-free Dulbecco’s modified Eagle’s medium (DMEM), and fluorescently-labeled MNCs were added to each well (2 × 105 cells/well). After a 2-min centrifugation at 500 rpm and a 1-h incubation, nonadherent MNCs were removed by two washes with phosphate-buffered saline, and firmly adherent MNCs were lysed with 0.5 N NaOH. Fluorescence was determined with the Fluoroskan Ascent AL fluorometer with excitation and emission wavelengths of 485 nm and 510 nm. The number of adherent MNCs per well was determined according to a standard curve generated with known numbers of fluorescently-labeled MNCs [32].
Endothelial cell-surface expression of VCAM-1 and ICAM-1
Expression of VCAM-1 and ICAM-1 on the surface of MAECs was determined by enzyme-linked immunosorbent assay (ELISA) [32]. MAECs grown to confluence in 96-well plates were treated for 2 h at 37°C with 30 μM LPC, 100 μM 7-KC, or culture medium alone as a control. After fixation in 1% glutaraldehyde, cells were incubated with a primary antibody against mouse VCAM-1 or ICAM-1, then peroxidase-conjugated secondary antibody. The reaction was developed with 100 μl 0.1 mg/ml 3,3′,5,5′-tetramethylbenzidine (Sigma-Aldrich) and 0.003% H2O2 for approximately 30 min at room temperature. Absorbance at 450 nm was determined with a microplate reader (Dynex Technologies, Chantilly, VA).
Cell viability
The viability of MAECs was determined with a Bioluminescence Detection Kit for ATP (Promega Corp., Madison, WI) as described elsewhere [33]. MAECs grown to confluence in a clear, white-bottomed, 96-well plate were incubated at 37°C in serum-free DMEM (100 μl/well) containing 75 μM LPC or 200 μM 7-KC. At the indicated times, 100 μl of kit reagent was added to each well, and luminescence was quantified with a BL10000 Lumicount luminometer (PerkinElmer Life and Analytical Sciences, Downers Grove, IL). The intensity of the luminescence signal generated in this assay is proportional to cell viability. The viability of cells treated with oxidized lipids was expressed a percentage of control cell viability.
Caspase activity assay
Caspase activity was determined with an EnzoLyte Homogeneous Rh110 Caspase-3/7 Assay Kit (AnaSpec Inc., San Jose, CA), in which cleavage of (Asp-Glu-Val-Asp)2-Rhodamine (Rh) 110 ([DEVD]2-Rh110 by caspases-3 and -7 liberates Rh110 to generate a fluorescence signal. Fluorescence intensity is proportional to caspase-3/7 activity. MAECs that had been grown to confluence in a clear, black-bottomed, 96-well plates were incubated with 75 μM LPC, 200 μM 7-KC, or serum-free culture medium alone as a control. After a 4-h incubation, 50 μl substrate solution was added to each well, and fluorescence was determined with the Fluoroskan Ascent AL fluorometer with excitation and emission wavelengths of 485 nm and 510 nm, respectively. Caspase activity was normalized to the total amount of cellular protein per well.
Statistical analysis
All data are reported as mean ± standard error of the mean (SEM). Data distribution was examined by the Shapiro-Wilk normality test. Comparisons between hGPx4Tg/ApoE−/− and ApoE−/− mice were performed with Student’s t-test, and differences were considered significant at a P value less than 0.05. All statistical analyses were performed with Stastix software (Analytical Software, Tallahassee, FL). For experiments in 96-well plates, results represent the mean of four duplicate wells in the same plate.
Results
hGPx4Tg/ApoE−/− mice overexpress GPx4 and show elevated GPx4 activity in the aorta
Ran et al. [22] reported that expression of GPx4 protein in hGPx4Tg mice was increased approximately 2–4 fold in all tissues studied. We found that the GPx4 protein levels were approximately 2.5- higher in the aorta of hGPx4Tg/ApoE−/− mice than in ApoE−/− littermates, and enzyme activity was 2.3-fold higher (Fig. 1). Similarly, the protein level and enzyme activity of GPx4 were both approximately 2.5-fold higher in MAECs from hGPx4Tg/ApoE−/− mice than in those from ApoE−/− mice (data not shown).
Fig. 1.
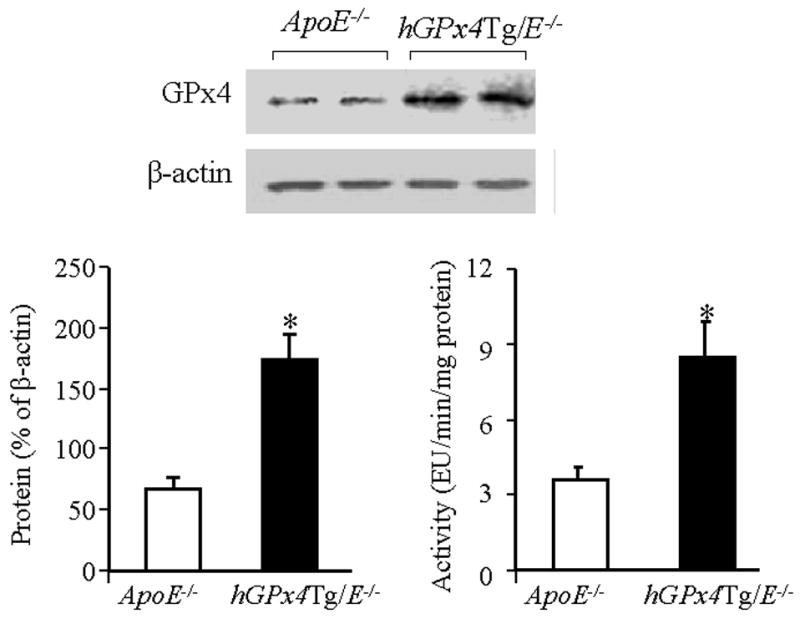
GPx4 is overexpressed in the aorta of hGPx4Tg/ApoE−/− mice. Aortic extracts from hGPx4Tg/ApoE−/− and ApoE−/− mice were subjected to western blot analysis for GPx4. GPx4 levels were normalized to β-actin expression. Alternatively, extracts were assayed for GPx4 activity. One enzyme unit (EU) is defined as the amount of enzyme that oxidizes 1 nM of reduced glutathione per minute. Data are derived from three to four independent experiments in which aorta extracts were pooled from two mice. *P < 0.01 vs. ApoE−/− mice.
Overexpression of GPx4 decreases atherosclerosis but does not affect plasma lipid levels
The effect of GPx4 overexpression on atherosclerosis in ApoE−/− mice is shown in Fig. 2 and the Table 1. The surface area of the entire aortic tree affected by atherosclerotic lesions and the size of atherosclerotic lesions in the aortic sinus were significantly smaller in hGPx4Tg/ApoE−/− mice than in ApoE−/− control mice. Aortic sinus sections from both transgenic lines showed early stage atherosclerotic lesions as seen by the presence of foam cells and free lipids (data not shown). All ApoE−/− mice showed advanced lesions (e.g., fibrous caps and acellular areas). In contrast, only 5 of 14 hGPx4Tg/ApoE−/− mice showed acellular areas in atherosclerotic lesions located in the aortic sinus (Table 1). These observations suggest that overexpression of GPx4 inhibits the development of atherosclerosis in ApoE−/− mice.
Fig. 2.
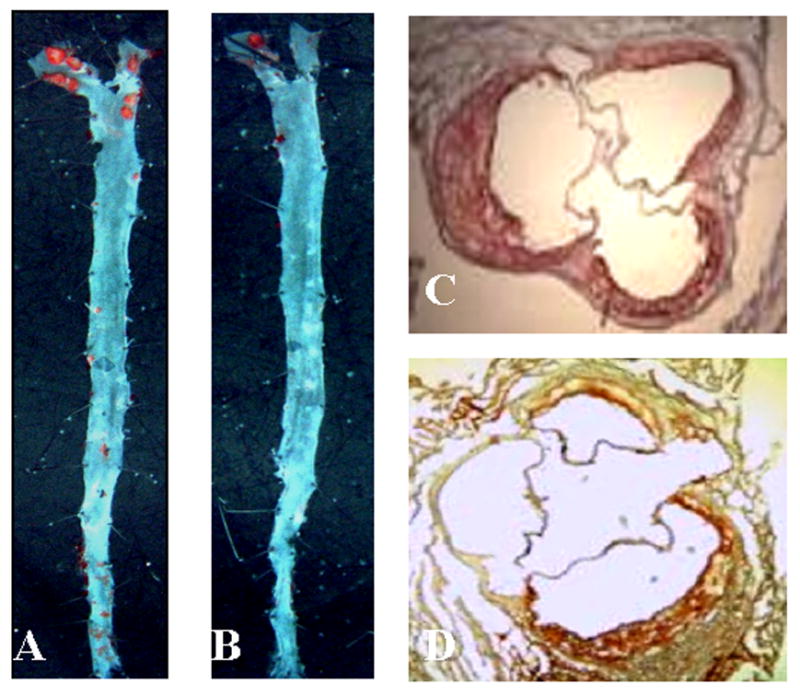
GPx4 overexpression reduces atherosclerosis. Representative images of (A, B) en face preparations and (C, D) coronal sections of mouse aorta. The en face preparations and coronal sections were stained with Sudan IV and Oil Red O, respectively. Red-stained areas indicate atherosclerotic lesions. A and C: ApoE−/− mice; B and D: hGPx4Tg/ApoE−/− mice.
Table 1.
Overexpression of glutathione peroxidase-4 inhibits the formation of atherosclerotic lesions
| Transgenic (n = 14) | Lesion areaa (aortic tree, %) | Lesion areab (aortic sinus, 103 μm2) | Acellular aread | Fibrous capd |
|---|---|---|---|---|
| ApoE−/− | 12.0 ± 4.1 | 37 ± 8 | 14/14 | 14/14 |
| hGPx4Tg/ApoE−/− | 4.7 ± 2.3c | 22 ± 3c | 5/14 | 12/14 |
Lesion area expressed as percentage of the total aortic surface area.
Lesion area was measured in sections of aortic sinus.
P < 0.05 vs. ApoE−/− mice.
Data expressed as the number of mice showing these atherosclerotic features/the total number of mice examined. Although acellular areas and fibrous caps were present in a subset of animals, foam cells and free lipids were present in all animals (not shown).
Increased plasma levels of cholesterol and triglycerides are risk factors for atherosclerosis [34]. Therefore, to determine whether the reduction in atherosclerotic lesions in hGPx4Tg/ApoE−/− mice was due to a change in plasma cholesterol or triglyceride levels, we measured the concentration of plasma cholesterol and triglycerides in ApoE−/− mice in the presence or absence of GPx4 overexpression. The hGPx4Tg/ApoE−/− and ApoE−/− mice showed comparable plasma concentrations of total cholesterol (513 ± 43 mg/dl vs. 519 ± 32 mg/dl, respectively) and triglycerides (91 ± 15 mg/dl vs. 82 ± 8 mg/dl, respectively). The cholesterol and triglyceride contents in the very low-density lipoprotein (VLDL), LDL and high-density lipoprotein were also comparable in hGPx4Tg/ApoE−/− and ApoE−/− mice (data not shown). These results indicate that the decrease in atherosclerotic lesions in hGPx4Tg/ApoE−/− mice is not caused by an alteration in plasma lipid levels.
Overexpression of GPx4 decreases lipid peroxidation in the aorta
Accumulation of oxidized lipids in the arterial wall may give rise to atherosclerosis [3,25]. To determine the effect of GPx4 overexpression on lipid peroxidation, we compared the concentration of F2-isoprostanes in the aorta of hGPx4Tg/ApoE−/− and ApoE−/− mice. The concentration of F2-isoprostanes was significantly lower in the aorta of hGPx4Tg/ApoE−/− mice than in that of the ApoE−/− control mice (Fig. 3, left panel). In addition, F2-isoprostane concentration and atherosclerotic lesion area were significantly correlated (Fig. 3, right panel).
Fig. 3.
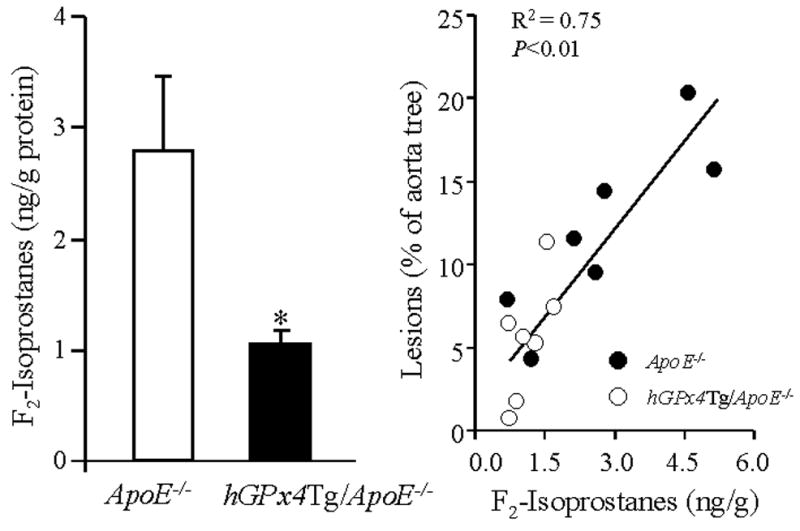
GPx4 overexpression of is associated with a decreased concentration of F2-isoprostanes in the aorta. The concentration of F2-isoprostanes in the aorta of hGPx4Tg/ApoE−/− mice and ApoE−/− littermates (n = 5 per group) was measured by gas chromatography/mass spectrometry (left panel). *P < 0.01 vs. ApoE−/− mice. Aortic F2-isoprostane concentration and atherosclerotic lesion size in the aortic sinus were strongly correlated (right panel).
Overexpression of GPx4 decreases LPC- and 7-KC-induced hydroperoxide production in mouse aorta endothelial cells
Increasing evidence suggests that the atherogenic action of oxidized lipids is associated, at least in part, with the ability of the lipids to induce the production of reactive oxygen species in vascular cells [6,35,36]. Therefore, we tested the effect of GPx4 overexpression on hydroperoxide production in endothelial cells. Under control conditions, MAECs obtained from hGPx4Tg/ApoE−/− mice and ApoE−/− littermates showed comparable levels of intracellular hydroperoxides and hydroperoxide release (Fig. 4). However, the level of peroxide production elicited by LPC or 7-KC differed significantly between each cell type. For example, treatment of MAECs from ApoE−/− mice with 100 or 200 μM 7-KC increased the intracellular hydroperoxide level 5- and 8-fold, respectively. On the other hand, treatment of MAECs from hGPx4Tg/ApoE−/− mice with the same concentrations of 7-KC increased the intracellular hydroperoxide levels only 2-and 3-fold, respectively (Fig. 4). Treatment of MAECs from ApoE−/− mice with 30 or 75 μM LPC increased the intracellular hydroperoxide level 6- and 24-fold, respectively, suggesting that MAECs are more sensitive to LPC than to 7-KC. The production of hydroperoxides in response to LPC was significantly lower in hGPx4Tg/ApoE−/−-derived MAECs than in ApoE−/−-derived MAECs (Fig. 4). Thus, overexpression of GPx4 inhibits 7-KC- and LPC-induced hydroperoxide production in MAECs.
Fig. 4.
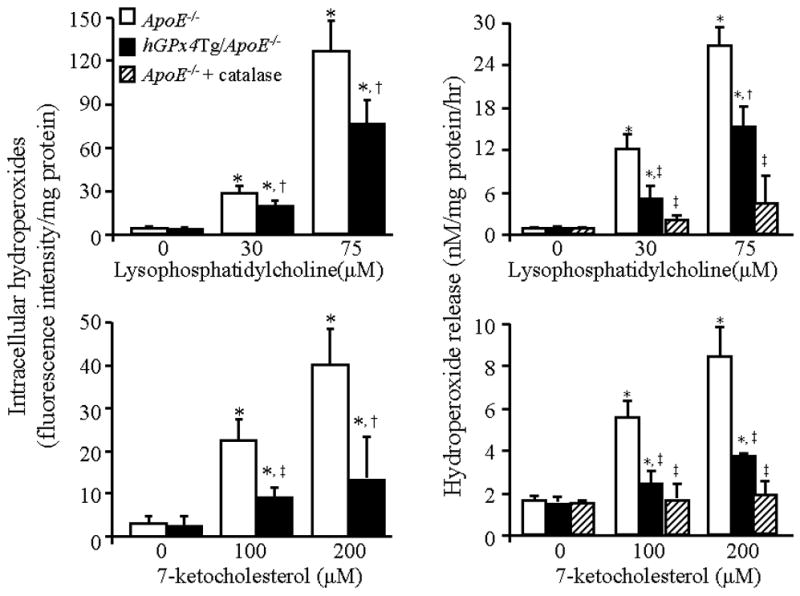
GPx4 overexpression of decreases hydroperoxide production in mouse aorta endothelial cells (MAECs) in response to lysophosphatidylcholine (LPC) and 7-ketocholesterol (7-KC). Intracellular hydroperoxides and hydroperoxide release were determined in MAECs from hGPx4Tg/ApoE−/− and ApoE−/− mice after incubation with the indicated concentrations of LPC and 7-KC in the presence or absence of 500 U/ml catalase. Data are derived from five separate experiments in which MAECs were pooled from four mice. *P < 0.01 vs. cells without LPC or 7-KC treatment; †P < 0.05 vs. ApoE−/− cells treated with the same concentration of LPC or 7-KC; ‡P < 0.01 vs. ApoE−/− cells treated with the same concentration of LPC or 7-KC.
Measurement of hydroperoxide concentrations in culture media revealed that the level of externalized hydroperoxides was increased in LPC- or 7-KC-treated MAECs, and that overexpression of GPx4 inhibited this increase (Fig. 4). The addition of the H2O2 scavenger catalase to culture media decreased the level of externalized hydroperoxides in LPD- or 7-KC-treated cells in a concentration-dependent manner (data not shown). At a concentration of 500 U/ml, catalase nearly eliminated the released hydroperoxides in culture media. Similarly, a 2.5-fold overexpression of catalase in MAECs [6] abolished the increase in intracellular hydroperoxides elicited by 100 μM LPC or 30 μM 7-KC, as measured by DCFHDA (data not shown). These results suggest that the hydroperoxide species produced in response to LPC and 7-KC consisted primarily of H2O2.
Overexpression of GPx4 decreases LPC- and 7-KC-induced monocyte adhesion to endothelial cells
An initial step of atherogenesis involves the adhesion of leukocytes to the endothelium in response to deposition of plasma lipoproteins in the intima. We investigated the effect of GPx4 overexpression on LPC- and 7-KC-induced MNC adhesion to MAECs. Under control conditions, the number of MNCs adherent to MAECs from hGPx4Tg/ApoE−/− and ApoE−/− mice was comparable, with approximately 25 MNCs/mm2 firmly adhered to MAECs (Fig. 5). Pretreatment of MAECs with 30 μM LPC or 100 μM 7-KC significantly increased the number of MNCs adhered to MAECs from ApoE−/− mice but did not significantly alter the number of MNCs adhered to MAECs from hGPx4Tg/ApoE−/− mice. These results suggest that overexpression of GPx4 inhibits LPC- and 7-KC-induced adhesion of MNCs to MAECs.
Fig. 5.
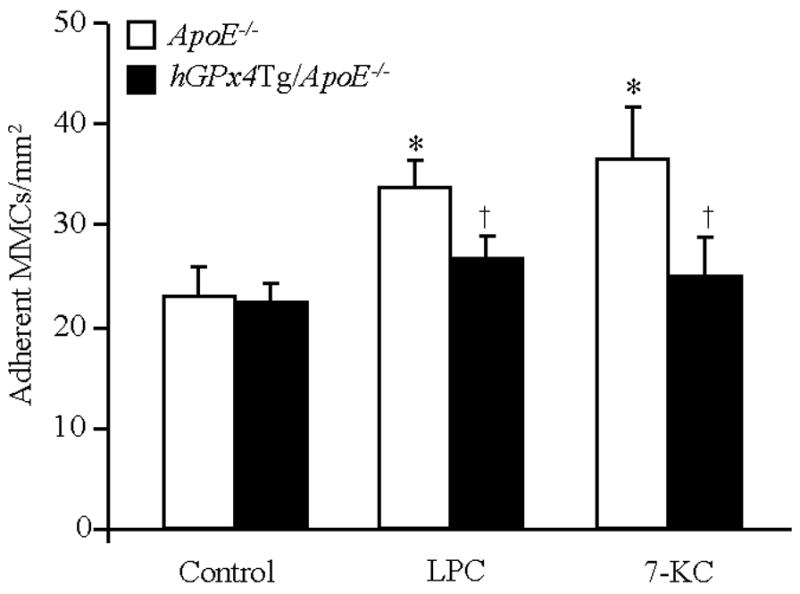
GPx4 overexpression of inhibits monocyte (MNC) adhesion to mouse aorta endothelial cells (MAECs). Confluent monolayers of MAECs from hGPx4Tg/ApoE−/− and ApoE−/− mice were treated with 30 μM lysophosphatidylcholine (LPC), 100 μM 7-ketocholesterol (7-KC), or culture medium alone (control) and then incubated with MNCs. Adherence of MNCs to MAECs was determined by fluorochrome adhesion assay. Data are derived from five separate experiments in which MAECs were pooled from four mice. *P < 0.05 vs. cells without LPC or 7-KC treatment; †P < 0.05 vs. ApoE−/− cells treated with LPC or 7-KC.
Overexpression of GPx4 decreases expression of cell adhesion molecules
Increased expression of cell adhesion molecules is known to enhance leukocyteendothelium interactions [37]. We determined the effect of GPx4 overexpression on VCAM-1 and ICAM-1 expression in cultured MAECs (Fig. 6A) and mouse aortas (Fig. 6B and C). Under control conditions, expression of VCAM-1 and ICAM-1 proteins on the surface of MAECs was relatively low, and no significant difference was observed between hGPx4Tg/ApoE−/− and ApoE−/− mice (Fig. 6A). Addition of 30 μM LPC or 100 μM 7-KC to culture media increased the expression of VCAM-1 and ICAM-1 on the surface of MAECs from ApoE−/− mice approximately 3-fold (Fig. 6A). LPC- and 7-KC-induced surface expression of VCAM-1 and ICAM-1 was significantly lower in MAECs from hGPx4Tg/ApoE−/− mice than in those from ApoE−/− mice (Fig. 6A). Consistent with these results, VCAM-1 and ICAM-1 levels were significantly lower in the aortas of hGPx4Tg/ApoE−/− mice than in those of ApoE−/− mice. These results provide evidence that overexpression of GPx4 inhibits in vitro and in vivo expression of cell adhesion molecules.
Fig. 6.
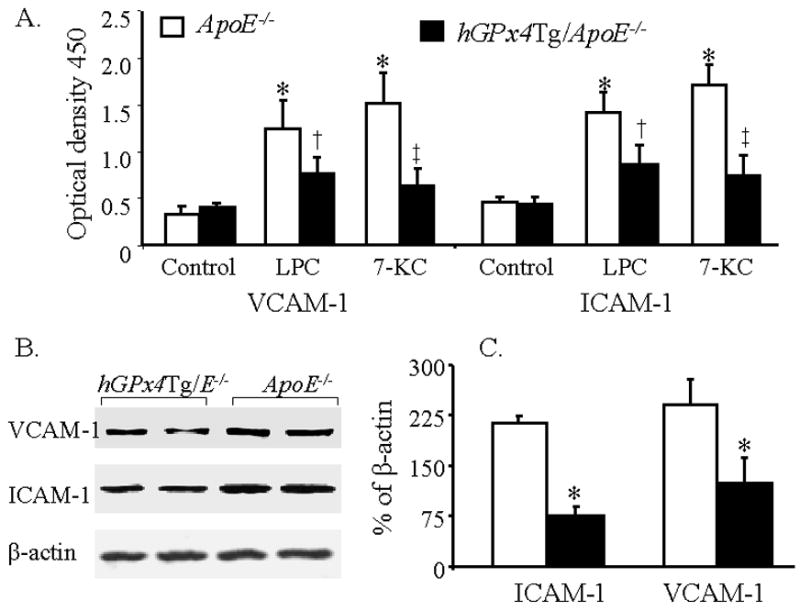
GPx4 overexpression inhibits expression of vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1). (A) Mouse aorta endothelial cells (MAECs) from hGPx4Tg/ApoE−/− and ApoE−/− mice were incubated with 30 μM lysophosphatidylcholine (LPC), 100 μM 7-ketocholesterol (7-KC), or culture medium alone (control), and the surface expression of VCAM-1 and ICAM-1 was measured by enzyme-linked immunosorbent assay. Values are derived from five separate experiments in which MAECs were pooled from four mice. *P < 0.01 vs. cells without LPC or 7-KC treatment; †P < 0.05 vs. ApoE−/− cells treated with LPC; ‡P < 0.01 vs. ApoE−/− cells treated with 7-KC. (B and C) VCAM-1 and ICAM-1 protein levels in aorta extracts from hGPx4Tg/ApoE−/− and ApoE−/− mice were measured by Western blot analysis. VCAM-1 and ICAM-1 protein levels are expressed as a percentage of β-actin expression levels. Data are derived from four separate experiments in which aorta extracts were pooled from two mice. *P < 0.01 vs. ApoE−/− mice.
Overexpression of GPx4 protects MAECs from LPC- and 7-KC-induced cytotoxicity
We have previously shown that LPC causes cell membrane lysis and induces necrotic death in MAECs at concentrations greater than the critical micellar concentration (50 μM), while 7-KC induces MAEC apoptosis at concentrations greater than 200 μM [33]. To determine whether GPx4 overexpression protects MAECs against LPC- and 7-KC-induced cytotoxicity, we monitored changes in intracellular ATP levels following exposure to each agent. Incubation of MAECs with 75 μM LPC decreased cell viability within 10 min, whereas incubation with 200 μM 7-KC did not significantly affect cell viability until 12 h (Fig. 7). MAECs from ApoE−/− mice were significantly more sensitive to both LPC and 7-KC than MAECs from hGPx4Tg/ApoE−/− mice (Fig. 7). We also examined LPC- and 7-KC-induced cytotoxicity by staining MAEC nuclei with propidium iodide (PI) and Hoechst 33258 (HT) as described previously [33]. Normal and apoptotic nuclei were easily distinguished by HT staining, as chromatin in the former was loosely organized, while that in latter was condensed and/or fragmented. Necrotic cells were easily identified by their large, PI-positive nuclei. Treatment with either 7-KC or LPC induced apoptosis and necrosis in MAECs from ApoE−/− mice and hGPx4Tg/ApoE−/− mice; however, this increase was less pronounced in hGPx4Tg/ApoE−/− MAECs (data not shown).
Fig. 7.

Inhibitory effect of GPx4 overexpression on lysophosphatidylcholine (LPC)- or 7-ketocholesterol (7-KC)-induced death of mouse aorta endothelial cells (MAECs). (A and B) MAECs from hGPx4Tg/ApoE−/− and ApoE−/− mice were incubated with 75 μM LPC or 200 μM 7-KC for the indicated time periods. The viability of MAECs treated with LPC or 7-KC was expressed as a percentage of the viability of untreated control cells. †P < 0.05 and *P < 0.01 vs. ApoE−/− cells treated with LPC or 7-KC for same time period. (C) MAECs were incubated with 70 μM LPC or 200 μM 7-KC for 4 h, and caspase-3/7 activity was measured. Data are derived from five separate experiments in which MAECs were pooled from four mice. *P < 0.01 vs. cells without LPC or 7-KC treatment; †P < 0.05 vs. ApoE−/− cells treated with 7-KC.
The anti-apoptotic effect of GPx4 overexpression was confirmed by measuring the activation of caspase-3/7, an event characteristic of apoptotic death. Treatment of MAECs with 200 μM 7-KC increased caspase-3/7 activity in MAECs from both ApoE−/− mice and hGPx4Tg/ApoE−/− mice (Fig. 7). This increase was significantly greater in ApoE−/− MAECs (Fig. 7). In contrast, treatment of MAECs with LPC did not alter caspase activity. These results suggest that 7-KC may induce MAEC death via a mechanism involving activation of caspase-3/7, whereas LPC may induce a caspase-independent form of death in these cells.
Discussion
Results of this study provide the first evidence that overexpression of GPx4 inhibits the development of atherosclerosis in ApoE−/− mice. Development of atherosclerotic lesions in the muscular arteries is believed to be an inflammatory process activated in response to the accumulation of oxidized lipids in the intima [reviewed in [3,25]. These oxidized lipids may be derived from the oxidative modification of cellular lipids and plasma lipoproteins. Very few lipoproteins in the circulation are oxidized; therefore, the pool of oxidized lipoproteins in the intima are thought to be derived, at least in part, from LDLs that enter the intima from the plasma and that are oxidized locally by cells in the arterial wall [25]. In this regard, all cell types within the arterial wall (i.e., endothelial cells [1,38], smooth muscle cells [1], and macrophages [39]) are able to mediate LDL oxidation. Oxidized LDLs are then internalized by macrophages, inducing foam cell formation, which is an early stage of atherogenesis. Results of the present study show that the delayed development of atherosclerosis in hGPx4Tg/ApoE−/− mice is correlated with a decreased level of aorta F2-isoprostanes, an in vivo marker of lipid peroxidation. These observations suggest that GPx4 delays the development of atherosclerosis via inhibition of lipid peroxidation in the arterial wall.
Oxidized lipids induce the adhesion of leukocytes to the endothelium by upregulating the expression of cell adhesion molecules in vascular cells, particularly endothelial cells. This promotes migration of leukocytes to the intima, which in turn initiates the atherogenic process [3,25]. In the present study, we observed that expression of VCAM-1 and ICAM-1 proteins was significantly lower in the aorta of hGPx4Tg/ApoE−/− mice than in ApoE−/− mice. It has been suggested that accumulation of oxidized LDL in the arterial wall induces expression of cell adhesion molecules and triggers adhesion of inflammatory cells to the endothelium [40]. Tests of the components in oxidized LDL have revealed that the lipid-soluble fraction of oxidized LDL induces monocyte-endothelial interactions, whereas the water-soluble fraction (containing apolipoprotein B) is inactive [40]. Here, we investigated the effect of LPC and 7-KC, two major lipid components of oxidized LDL, on cell adhesion molecule expression and MNC adhesion to MAECs. Our results showed that overexpression of GPx4 significantly inhibited LPC- and 7-KC-induced surface expression of VCAM-1 and ICAM-1 in endothelial cells and significantly decreased LPC- and 7-KC-induced MNC adhesion to MAECs. These observations suggest that inhibition of oxidized lipid-induced cell adhesion molecule expression in vascular cells is responsible, at least in part, for the anti-atherogenic role of GPx4.
Increasing evidence suggests that the ability of oxidized lipids to regulate the expression of cell adhesion molecules is associated with their ability to induce the production of ROS such as superoxide and H2O2 [32,41]. We have previously shown that 7-KC and LPC induce ROS generation in MAECs via different mechanisms. The former activates NADPH oxidase, which generates O2− and provides a source of oxygen-centred radicals, such as H2O2. LPC, on the other hand, increases O2− and peroxide generation via mechanisms other than activation of NADPH oxidase, such as the induction of mitochondrial dysfunction [33]. It is well established that ROS, in particular H2O2, induce expression of cell adhesion molecules by activating redox-sensitive transcription factors such as NFκB [18,41]. Overexpression of GPx4 in vascular smooth muscle cells has been shown to inhibit VCAM-1 expression via inhibition of NFκB [18]. Our results show that overexpression of GPx4 efficiently decreases LPC- and 7-KC-induced hydroperoxide generation in MAECs. Thus, the inhibitory effect of GPx4 on LPC- and 7-KC-induced cell adhesion molecule expression may be explained by its ability to decrease H2O2 production in endothelial cells. In addition, GPx4 has been shown to decrease the levels of enzymatically generated lipid peroxides. A number of these lipid peroxides, such as leukotrienes [13], prostaglandin E [14], and platelet-activating factor [15], have been reported to function as inflammatory or atherogenic stimuli and to upregulate cell adhesion molecule expression.
Atherogenesis is believed to be an inflammatory process activated in the arterial wall in response to endothelial injury [42]. One important finding of the present study was that 7-KC and LPC significantly decreased the viability of aorta endothelial cells from ApoE−/− mice but were less cytotoxic toward cells from hGPx4Tg/ApoE−/− mice. These observations suggest that the decrease in atherosclerotic lesions in hGPx4Tg/ApoE−/− mice may result from inhibition of oxidized lipid-induced endothelial cell injury by GPx4. Our laboratory has shown that oxidized LDL and its lipid components, such as 7-KC and LPC, cause injury of mouse aorta smooth mouse cells and endothelial cells via separate mechanisms, with oxidized LDL and 7-KC inducing apoptosis, as evidenced by caspase-3/7 activation, and LPC inducing necrotic death, as characterized by cell membrane rupture and nuclear expansion [2,33]. We have also shown that overexpression of catalase or addition of chemical peroxide scavenger to culture media attenuates oxidized LDL- and 7-KC-induced apoptosis [2,33] and LPC-induced necrotic cell death [33], suggesting that endogenously generated H2O2 is a common mediator of cell death pathways activated by oxidized lipids [2,33]. Results of the present study showed that overexpression of GPx4 inhibited 7-KC- and LPC-induced cell death and hydroperoxide generation in MAECs. Thus, the protective role of GPx4 against LPC- and 7-KC-induced injury in MAECs can be explained, at least in part, by its ability to reduce H2O2. Other lipid peroxides have been proposed to play roles in cell death. For example, cardiolipin hydroperoxides generated in the mitochondria can trigger cytochrome c release from the inner membrane of the mitochondria and induce apoptosis. It has been suggested that GPx4 may inhibit apoptosis by decreasing cardiolipin peroxidation [8].
In summary, this study provides evidence that GPx4 inhibits the development of atherosclerosis by decreasing lipid peroxidation and decreasing the sensitivity of vascular cells to oxidized lipids. Moreover, it suggests that decreasing intracellular peroxide levels may be a viable therapeutic strategy for atherosclerosis-related vascular diseases.
Acknowledgments
The authors thank Dr. Lee Limbird for critical reading of the manuscript and for useful suggestions. This study was supported by NIH grants HL076623 (Hong Yang) and ES014471 and GM08037 (ZhongMao Guo).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Morel DW, Dicorleto PE, Chisolm GM. Endothelia and smooth muscle cells alter low density lipoprotein in vitro by free radical oxidation. Arteriosclerosis. 1984;4:357–364. doi: 10.1161/01.atv.4.4.357. [DOI] [PubMed] [Google Scholar]
- 2.Guo ZM, Van Remmen H, Yang H, Chen XL, Mele J, Vijg J, Epstein CJ, Ho Y-S, Richardson A. Changes in expression of antioxidant enzymes affect cell-mediated LDL oxidation and oxLDL-induced apoptosis in mouse aorta cells. Arterioscler Thromb Vasc Biol. 2001;21:1131–1138. doi: 10.1161/hq0701.092092. [DOI] [PubMed] [Google Scholar]
- 3.Witztum JL, Steinberg D. The oxidative modification hypothesis of atherosclerosis: does it hold for humans? Trends Cardiovasc Med. 2001;11:93–102. doi: 10.1016/s1050-1738(01)00111-6. [DOI] [PubMed] [Google Scholar]
- 4.Yokote K, Morisaki N, Zenibayashi M, Ueda S, Kanzaki T, Saito Y, Yoshida S. The phospholipase-A2 reaction leads to increased monocyte adhesion of endothelial cells vial the expression of adhesion molecules. Eur J Bioc. 1993;217:723–729. doi: 10.1111/j.1432-1033.1993.tb18298.x. [DOI] [PubMed] [Google Scholar]
- 5.Stiko-Rahm A, Hultgardh-Nilsson A, Regnstrom J, Hamsten A, Nilson J. Native and oxidized LDL enhances production of PDGF AA and the surface expression of PDGF receptors in cultured human smooth muscles cells. Arterioscler Thromb. 1992;12:1099–1109. doi: 10.1161/01.atv.12.9.1099. [DOI] [PubMed] [Google Scholar]
- 6.Yang H, Roberts LJ, Shi MJ, Zhou LC, Ballard BR, Richardson A, Guo ZM. Retardation of atherosclerosis by overexpression of catalase or both Cu/Znsuperoxide dismutase and catalase in mice lacking apolipoprotein E. Circ Res. 2004;95:1075–1081. doi: 10.1161/01.RES.0000149564.49410.0d. [DOI] [PubMed] [Google Scholar]
- 7.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 8.Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med. 2003;34:145–169. doi: 10.1016/s0891-5849(02)01197-8. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Z, Kang YJ. Cellular and subcellular localization of catalase in the heart of transgenic mice. J Histochem Cytochem. 2000;48:585–594. doi: 10.1177/002215540004800502. [DOI] [PubMed] [Google Scholar]
- 10.Takebe G, Yarimizu J, Saito Y, Hayashi T, Nakamura H, Yodoi J, Nagasawa S, Takahashi K. A comparative study on the hydroperoxide and thiol specificity of the glutathione peroxidase family and selenoprotein P. J Biol Chem. 2002;277:41254–41258. doi: 10.1074/jbc.M202773200. [DOI] [PubMed] [Google Scholar]
- 11.Thomas JP, Maiorino M, Ursini F, Girotti AW. Protective action of phospholipid hydroperoxide glutathione peroxidase against membrane-damaging lipid peroxidation. J Biol Chem. 1990;265:454–461. [PubMed] [Google Scholar]
- 12.Ridgway ND, Byers DM, Cook HW, Storey MK. Integration of phospholipid and sterol metabolism in mammalian cells. Prog Lipid Res. 1999;38:337–360. doi: 10.1016/s0163-7827(99)00007-7. [DOI] [PubMed] [Google Scholar]
- 13.Imai H, Narashima K, Arai M, Sakamoto H, Chiba N, Nakagawa Y. Suppression of leukotriene formation in RBL-2H3 cells that overexpressed phospholipid hydroperoxide glutathione peroxidase. J Biol Chem. 1998;273:1990–1997. doi: 10.1074/jbc.273.4.1990. [DOI] [PubMed] [Google Scholar]
- 14.Chen CJ, Huang HS, Chang WC. Inhibition of arachidonate metabolism in human epidermoid carcinoma a431 cells overexpressing phospholipid hydroperoxide glutathione peroxidase. J Biomed Sci. 2002;9:453–459. doi: 10.1007/BF02256540. [DOI] [PubMed] [Google Scholar]
- 15.Sakamoto H, Tosaki T, Nakagawa Y. Overexpression of phospholipid hydroperoxide glutathione peroxidase modulates acetyl-CoA, 1-O-alkyl-2-lyso-sn-glycero-3-phosphocholine acetyltransferase activity. J Biol Chem. 2002;277:50431–50438. doi: 10.1074/jbc.M204190200. [DOI] [PubMed] [Google Scholar]
- 16.Leventhal AR, Leslie CC, Tabas I. Suppression of macrophage eicosanoid synthesis by atherogenic lipoproteins is profoundly affected by cholesterol-fatty acyl esterification and the Niemann-Pick C pathway of lipid trafficking. J Biol Chem. 2004;279:8084–8092. doi: 10.1074/jbc.M310672200. [DOI] [PubMed] [Google Scholar]
- 17.Ninio E. Phospholipid mediators in the vessel wall: involvement in atherosclerosis. Curr Opin Clin Nutr Metab Care. 2005;8:123–131. doi: 10.1097/00075197-200503000-00004. [DOI] [PubMed] [Google Scholar]
- 18.Banning A, Schnurr K, Bol GF, Kupper D, Muller-Schmehl K, Viita H, Yla-Herttuala S, Brigelius-Flohe R. Inhibition of basal and interleukin-1-induced VCAM-1 expression by phospholipid hydroperoxide glutathione peroxidase and 15-lipoxygenase in rabbit aortic smooth muscle cells. Free Radic Biol Med. 2004;36:135–144. doi: 10.1016/j.freeradbiomed.2003.10.027. [DOI] [PubMed] [Google Scholar]
- 19.Yagi K, Komura S, Kojima H, Sun Q, Nagata N, Ohishi N, Nishikimi M. Expression of human phospholipid hydroperoxide glutathione peroxidase gene for protection of host cells from lipid hydroperoxide-mediated injury. Biochem Biophys Res Commun. 1996;219:486–491. doi: 10.1006/bbrc.1996.0260. [DOI] [PubMed] [Google Scholar]
- 20.Hurst R, Korytowski W, Kriska T, Esworthy RS, Chu FF, Girotti AW. Hyperresistance to cholesterol hydroperoxide-induced peroxidative injury and apoptotic death in a tumor cell line that overexpresses glutathione peroxidase isotype-4. Free Radic Biol Med. 2001;31:1051–1065. doi: 10.1016/s0891-5849(01)00685-2. [DOI] [PubMed] [Google Scholar]
- 21.Ran Q, Van RH, Gu M, Qi W, Roberts LJ, Prolla T, Richardson A. Embryonic fibroblasts from Gpx4+/− mice: a novel model for studying the role of membrane peroxidation in biological processes. Free Radic Biol Med. 2003;35:1101–1109. doi: 10.1016/s0891-5849(03)00466-0. [DOI] [PubMed] [Google Scholar]
- 22.Ran Q, Liang H, Gu M, Qi W, Walter CA, Roberts LJ, Herman B, Richardson A, Van RH. Transgenic mice overexpressing glutathione peroxidase 4 are protected against oxidative stress-induced apoptosis. J Biol Chem. 2004;279:55137–55146. doi: 10.1074/jbc.M410387200. [DOI] [PubMed] [Google Scholar]
- 23.Ran Q, Gu M, Van RH, Strong R, Roberts JL, Richardson A. Glutathione peroxidase 4 protects cortical neurons from oxidative injury and amyloid toxicity. J Neurosci Res. 2006;84:202–208. doi: 10.1002/jnr.20868. [DOI] [PubMed] [Google Scholar]
- 24.Reddick RL, Zhang S, Maeda N. Atherosclerosis in Mice Lacking Apo E: Evaluation of lesional Development and Progression. Arterioscler Thromb. 1994;14:141–147. doi: 10.1161/01.atv.14.1.141. [DOI] [PubMed] [Google Scholar]
- 25.Chisolm GM, III, Penn MS. Oxidized Lipoproteins and Atherosclerosis. In: Fuster V, Ross R, Topol EJ, editors. Atherosclerosis and Coronary Artery Disease. Philadelphia: Lippincott-Raven; 1996. pp. 129–149. [Google Scholar]
- 26.Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutatnt apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci USA. 1992;89:4471–4475. doi: 10.1073/pnas.89.10.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nathan L, Chaudhuri G. Estrogens and atherosclerosis. Annual Reviews of Pharmacology and Toxicology. 1997;37:477–515. doi: 10.1146/annurev.pharmtox.37.1.477. [DOI] [PubMed] [Google Scholar]
- 28.Guo ZM, Mitchell-Raymundo F, Yang H, Ikeno Y, Nelson J, Diaz V, Richardson A, Reddick R. Dietary restriction reduces atherosclerosis and oxidative stress in the aorta of apolipoprotein E-deficient mice. Mech Ageing Dev. 2002;123:1121–1131. doi: 10.1016/s0047-6374(02)00008-8. [DOI] [PubMed] [Google Scholar]
- 29.Maiorino M, Gregolin C, Ursini F. Phospholipid hydroperoxide glutathione peroxidase. Methods Enzymol. 1990;186:448–457. doi: 10.1016/0076-6879(90)86139-m. [DOI] [PubMed] [Google Scholar]
- 30.Lei XG, Evenson JK, Thompson KM, Sunde RA. Glutathione peroxidase and phospholipid hydroperoxide glutathione peroxidase are differentially regulated in rats by dietary selenium. J Nutr. 1995;125:1438–1446. doi: 10.1093/jn/125.6.1438. [DOI] [PubMed] [Google Scholar]
- 31.Wu D, Yang H, Xiang W, Zhou L, Shi M, Julies G, Laplante JM, Ballard BR, Guo Z. Heterozygous mutation of ataxia-telangiectasia mutated gene aggravates hypercholesterolemia in apoE-deficient mice. J Lipid Res. 2005;46:1380–1387. doi: 10.1194/jlr.M400430-JLR200. [DOI] [PubMed] [Google Scholar]
- 32.Yang H, Shi MJ, Richardson A, Vijg J, Guo ZM. Attenuation of leukocyteendothelium interaction by antioxidant enzymes. Free Radic Biol Med. 2003;35:266–276. doi: 10.1016/s0891-5849(03)00277-6. [DOI] [PubMed] [Google Scholar]
- 33.Zhou LC, Shi MJ, Guo ZM, Hoover R, Yang H. Different cytotoxic injuries induced by lysophosphatidylcholine and 7-ketocholesterol in mouse endothelial cells. Endothelium. 2006;13:213–226. doi: 10.1080/10623320600780926. [DOI] [PubMed] [Google Scholar]
- 34.McGill HC. Overview. In: Fuster V, Ross R, Topol EJ, editors. Atherosclerosis and coronary artery disease. Philadelphia: Lippincott-Raven Publishers; 1999. pp. 25–41. [Google Scholar]
- 35.Alexander RW. Theodore Cooper Memorial Lecture. Hypertension and the pathogenesis of atherosclerosis. Oxidative stress and the mediation of arterial inflammatory response: a new perspective. Hypertension. 1995;25:155–161. doi: 10.1161/01.hyp.25.2.155. [DOI] [PubMed] [Google Scholar]
- 36.Kunsch C, Medford RM. Oxidative stress as a regulator of gene expression in the vasculature. Circ Res. 1999;85:753–766. doi: 10.1161/01.res.85.8.753. [DOI] [PubMed] [Google Scholar]
- 37.Rosenfeld ME. Inflammation, Lipids, and Free Radicals: Lessons Learned from the atherogenic process. Seminars in Reproductive Endocrinology. 1998;16:249–261. doi: 10.1055/s-2007-1016285. [DOI] [PubMed] [Google Scholar]
- 38.Steinbrecher UP. Role of superoxide in endothelial-cell modification of low-density lipoproteins. Biochem Biophys Acta. 1988;959:20–30. doi: 10.1016/0005-2760(88)90145-2. [DOI] [PubMed] [Google Scholar]
- 39.Parthasarathy S, Printz DJ, Boyd D, Joy L, Steinberg D. Macrophage oxidation of low density lipoprotein generate a form recognized by the scavenger receptor. Arteriosclerosis. 1986;6:505–510. doi: 10.1161/01.atv.6.5.505. [DOI] [PubMed] [Google Scholar]
- 40.Berliner JA, Territo MC, Sevanian A, Ramin S, Kim JA, Bamshad B, Esterson M, Fogelman AM. Minimally modified low density lipoprotein stimulates monocyte endothelial interactions. J Clinical Invest. 1990;85:1260–1266. doi: 10.1172/JCI114562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cominacini L, Garbin U, Pasini AF, Davoli A, Campagnola M, Pastorino AM, Gaviraghi G, Lo CV. Oxidized low-density lipoprotein increases the production of intracellular reactive oxygen species in endothelial cells: inhibitory effect of lacidipine. J Hypertens. 1998;16:1913–1919. doi: 10.1097/00004872-199816121-00010. [DOI] [PubMed] [Google Scholar]
- 42.Ross R. The pathogenesis of atherosclerosis: a perspective for 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]