

Summary
Aberrant TGFβ signaling is common in human cancers and contributes to tumor metastasis. Here, we demonstrate that Gr-1+CD11b+ myeloid cells, are recruited into mammary carcinomas with type II TGFβ receptor gene (Tgfbr2) deletion and directly promote tumor metastasis. Gr-1+CD11b+ cells infiltrate into the invasive front of tumor tissues, and facilitate tumor cell invasion and metastasis through a process involving metalloproteinase activity. This infiltration of Gr-1+CD11b+ cells also results in increased abundance of TGFβ1 in tumors with Tgfbr2 deletion. The recruitment of Gr-1+CD11b+ cells into tumors with Tgfbr2 deletion involves two chemokine receptor axes, SDF-1/CXCR4 and CXCL5/CXCR2 axes. Together, these data indicate that Gr-1+CD11b+ cells contribute to TGFβ mediated metastasis through enhancing tumor cell invasion and metastasis.
Significance
TGFβ is a very important tumor suppressor. Inactivation of TGFβ signaling frequently occurs in human cancers and contributes to tumor metastasis. However, the contribution of host cells in this process is unclear. Here, we show that deletion of Tgfbr2 in mammary carcinoma cells results in increased chemokine signals that enhance Gr-1+CD11b+ myeloid cell infiltration into tumors, which leads to enhanced tumor invasion and metastasis. Gr-1+CD11b+ cell infiltration also results in increased TGFβ production in tumors with Tgfbr2 deletion. Thus tumor-suppressing role of TGFβ can be switched to tumor promoting through the recruitment of Gr-1+CD11b+ cells in the tumor microenvironment. Inhibition of Gr-1+CD11b+ cell production/recruitment could improve host immunosurveillance and inhibit tumor metastasis, having the effect of “killing two birds with one stone”.
Keywords: TGFβ, tumor progression, metastasis, inflammation, tumor microenvironment, bone marrow, myeloid cells, matrix metalloproteinase, chemokine
Introduction
Mounting evidence indicates that alterations in TGFβ signaling have significant effects on tumor initiation and progression (Bierie and Moses, 2006). TGFβ signals through the type I and type II TGFβ receptors (TβRI and TβRII, respectively) resulting in phosphorylation of Smad2 and Smad3 which then combine with Smad4 to enter the nucleus to modulate transcription (Bierie and Moses, 2006). In addition, TGFβ binding to its receptors activates many non-canonical signaling pathways. TGFβ can function as a tumor suppressor or a tumor promoter depending on the context and stage of tumor progression. Mutations of the genes encoding TβRI and TβRII (TGFBR1 and TGFBR2, respectively) have been reported in a number of human cancers (Akhurst and Derynck, 2001). Mutations in TGFBR2 are particularly frequent in tumors with microsatellite instability (Grady and Markowitz, 2002). Although mutations in TGFβ receptor genes are infrequent in human breast cancers, there is compelling evidence for impairment of TGFβ signaling in this disease (Bagadi et al., 2006; Chen et al., 1998; Chen et al., 2006; Seitz et al., 2003). Decreased expression of TβRII occurs frequently both in early and advanced breast cancer (de Jong et al., 1998; Gobbi et al., 1999). Further, polymorphisms in TGFBR1 that result in decreased TGFβ signaling are associated with an increased frequency and metastasis of breast cancer (Chen et al., 2006; Pasche et al., 2005). Transgenic animal studies indicate that a simple reduction of TGFβ signaling is sufficient to predispose to the development of mammary carcinomas (Tang et al., 1998).
Conditional knockout of Tgfbr2 in combination with expression of KrasG12D in pancreatic cancer (Ijichi et al., 2006) or with APC mutation in intestinal carcinomas (Munoz et al., 2006) resulted in the development of aggressive tumor phenotype. We have shown that conditional deletion of Tgfbr2 in mammary epithelial cells that also express the polyoma middle T antigen (PyVmT) under the MMTV promoter resulted in a shortened tumor latency and an increased metastases (Forrester et al., 2005; Ijichi et al., 2006; Munoz et al., 2006). However, others have shown that enhancement of TGFβ signaling by expression of a constitutively active TGFβ1 (Muraoka et al., 2003; Muraoka-Cook et al., 2004) or TβRI (Muraoka-Cook et al., 2004; Muraoka-Cook et al., 2006; Siegel et al., 2003) in mammary epithelial cells in conjunction with c-Neu of PyVmT expression increases pulmonary metastases creating a conundrum. It is unclear what mechanisms underlie these different observations.
Host-derived inflammatory cells infiltrate into tumor tissues and create an environment that favors tumor progression. They promote tumor angiogenesis and tumor progression by producing angiogenic factors and matrix-degrading enzymes (Balkwill and Coussens, 2004; Coussens and Werb, 2002). The recruitment of inflammatory cells to tumor sites is regulated by the interactions of chemokines and chemokine receptors. ENA-78/CXCL5, interacting with CXCR2, is responsible for the recruitment of polymorphonuclear neutrophils into inflamed lungs (Ahuja and Murphy, 1996; Bozic et al., 1996; Jeyaseelan et al., 2005; Zineh et al., 2006). In addition, stromal derived factor 1 (SDF-1 or CXCL12) is considered as one of the key regulators of hematopoietic stem and progenitor cell trafficking between the peripheral circulation and targeted tissues. SDF-1 mediates its effects on chemotaxis through its receptor, CXCR4, which is highly expressed on putative stem and progenitor cells (Balkwill and Coussens, 2004).
Gr-1+CD11b+ myeloid cells, also called as myeloid immune suppressor cells (MISCs) or myeloid derived suppressor cells (MDSCs), are significantly over-produced in the bone marrow and spleens of tumor-bearing mice (Melani et al., 2003; Serafini et al., 2006) as well as in peripheral blood of cancer patients (Almand et al., 2001; Young and Lathers, 1999). In mice they express CD11b, a marker for myeloid cells of the macrophage lineage, and a marker for granulocytes, Gr-1, thus they are also called Gr+CD11b+ cells. In human cancer patients, Gr-1+CD11b+ myeloid cells are identified as lineage (CD3, CD14, CD19, and CD57) and HLA-DR negative (Lin−HLA-DR−), or immature dendritic cells. Gr-1+CD11b+ myeloid cells have been demonstrated to be immune suppressive since the 1980s (Almand et al., 2001; Bronte et al., 2000; Gabrilovich et al., 1996; Serafini et al., 2006; Serafini et al., 2004; Young and Lathers, 1999). They represent one mechanism of tumor escape from immune system control and compromise the efficacy of cancer immunotherapy.
Recently, Gr-1+CD11b+ cells were found to infiltrate into tumors, and promote tumor angiogenesis by producing high levels of MMP9 and by directly incorporating into tumor endothelium (Yang et al., 2004). Gr-1+CD11b+ cells have also been implicated in tumor refractoriness to anti-VEGF treatment (Shojaei et al., 2007). Despite the data defining the overproduction in tumor hosts and infiltration in tumor tissues, the molecular mechanisms for their recruitment and roles in tumor progression remain to be investigated. Here we show that Gr-1+CD11b+ cells are specifically recruited into mammary carcinomas with conditional deletion of Tgfbr2 and 4T1 tumors through two distinct chemokine mechanisms, CXCL5/CXCR2 and SDF-1/CXCR4. These immature myeloid cells directly promote tumor invasion and metastasis through increased production and function of MMPs. In addition Gr-1+CD11b+ cell infiltration also results in an increased TGFβ production in the microenvironment of tumors with Tgfbr2 deletion. These findings improve our understanding of tumor-host interaction. Therapeutic interventions on Gr-1+CD11b+ cells may not only enhance the host immune system but also inhibit tumor invasion and metastasis.
RESULTS
Gr-1+CD11b+ cells are recruited to mammary carcinomas with genetic deletion of Tgfbr2 especially in the invasive front
We have previously reported that conditional deletion of Tgfbr2 in mammary epithelial cells also expressing PyVmT results in a shortened tumor latency and an increased pulmonary metastases compared to PyVmT carcinomas with intact TGFβ signaling (Chytil et al., 2002; Forrester et al., 2005). We have also reported that Gr-1+CD11b+ myeloid cells, infiltrate into tumor tissues and contribute to tumor angiogenesis (Yang et al., 2004). To examine the role of Gr-1+CD11b+ cells in TGFβ mediated tumor progression, tumor tissues were collected one week after palpable tumors developed in a PyVmT genetic tumor model with Tgfbr2 knockout (Tgfbr2MGKO). Single cell suspension from tumor tissues of Tgfbr2MGKO and floxed PyVmT control mice (Tgfbr2flox/flox) were analyzed by flow cytometry. We observed a significant increase of Gr-1+CD11b+ cells in the PyVmT/Tgfbr2MGKO compared with their Tgfbr2flox/flox control tumors (Figure 1A & B). Gr-1+CD11b+ cells residing in tumor tissues express both Gr-1 and CD11b, and no distinct Gr-1 alone population was found (Figure 1C). This allowed us to examine the precise location of Gr-1+CD11b+ cells in tumor tissues with immunohistochemistry staining with anti Gr-1 antibody. Very interestingly, Gr-1+CD11b+ cells were found mostly in the invasive front of PyVmT/Tgfbr2MGKO mammary tumors (Figure 1D). In addition to Gr-1+CD11b+ cells, we also observed an increased infiltration of F4/80 positive and VEGFR I positive cells in the PyVmT/Tgfbr2MGKO tumors compared with their Tgfbr2flox/flox control tumors (Figure 1E and 1F), although without statistical significance. We thus concentrated our studies on Gr-1+CD11b+ myeloid cells. The significant increase of Gr-1+CD11b+ cells is consistent with increased lung metastasis in those animals (Forrester et al., 2005), suggesting that these Gr-1+CD11b+ cells may play an role in breast tumor metastasis.
Figure 1. Gr-1+CD11b+ myeloid cells are recruited to mammary carcinomas with genetic deletion of Tgfbr2 (PyVmT/Tgfbr2MGKO).

A: Flow cytometry analysis of infiltrating Gr-1+CD11b+ cells in PyVmT/Tgfbr2MGKO tumors and PyVmT/Tgfbr2flox/flox control tumors. Shown are representative flow cytometry plots. B: Quantitative data for the presence of Gr-1+CD11b+ cells in tumors as shown in 1A. Five to seven week old mice were analyzed. C: Flow cytometry analysis of tumor-residing Gr-1+CD11b+ cells in PyVmT/Tgfbr2MGKO tumors. D: IHC of Gr-1+CD11b+ cells in the invasive front of PyVmT/Tgfbr2MGKO mammary carcinomas compared with PyVmT/Tgfbr2flox/flox tumors. Scale bars are indicated in the figures. E and F: Flow cytometry analysis of infiltrating F4/80 and VEGFR1 positive cells in PyVmT/Tgfbr2MGKO tumors and PyVmT/Tgfbr2flox/flox control tumors. Representative flow cytometry plots are shown on left and quantitative data on the right. m.g: adjacent mammary gland tissue; tu: tumor tissues. All quantitative data are presented with the mean ± standard error.
Gr-1+CD11b+ cells promote mammary carcinoma metastasis
To confirm our observation in PyVmT/Tgfbr2MGKO tumors, we utilized a 4T1 orthotopic tumor model. This tumor model shares many characteristics with human breast cancer, particularly its ability to spontaneously metastasize to lungs. In addition, 4T1 tumors showed significantly decreased growth inhibition by TGFβ relative to normal mammary epithelial cells or PyVmT/Tgfbr2flox.flox carcinoma cells (data not shown). Importantly, Gr-1+CD11b+ cells were significantly overproduced in bone marrow and spleens of mice bearing large 4T1 tumors (Figure 2A). The number of Gr-1+CD11b+ cells increased with the growth of the tumor, with over 50% in the spleen, and over 80% in the bone marrow 28–35 days after tumor inoculation. This allows us to be able to sort large number of Gr-1+CD11b+ cells to investigate their roles in breast tumor metastasis using both in vivo and in vitro assays. In 4T1 breast tumors, Gr-1+CD11b+ cells comprised an average of 7.2% of total cells (Figure 2B). Gr-1+CD11b+ cells were mostly present in the invasive front (Figure 2C), which is similar to the MMTV PyVmT model.
Figure 2. Gr-1+CD11b+ cells promote tumor metastasis.

A: Increased Gr-1+CD11b+ cells in spleens and bone marrow of 4T1 tumor-bearing mice after tumor inoculation. Left panel: Flow cytometry analysis of Gr-1+CD11b+ cells in the spleen of tumor bearing mice 28 days following engraftment (4 or more mice per time point). Middle panel: spleen; Right panel: bone marrow (b.m.). B: Gr-1+CD11b+ cells in 4T1 tumors with flow cytometry analysis. C: IHC showing Gr-1+CD11b+ cells in the invasive front of 4T1 tumors 15 day after tumor inoculation. Scale bar, 100 μM. D: Single cell sorting of Gr-1+CD11b+ cells from tumors and spleens of 4T1 tumor-bearing mice 35 days after tumor inoculation. Flow cytometry analysis of Gr-1+CD11b+ cells after sorting is shown. E: Metastasis was significantly increased when 4T1 cells were co-injected with tumor derived Gr-1+CD11b+ cells including Gr-1+CD11b+ cells derived from tumor tissues (tu inf Gr-1+CD11b+ cells) spleens of tumor-bearing mice (tu sp Gr-1+CD11b+ cells). 4T1 cells alone and Gr-1+CD11b+ cells from normal mice (nor Gr-1+CD11b+ cells) were used as controls. Number of animals used is indicated in the bar. m.g: adjacent mammary gland tissue; tu: tumor tissues. Results are presented as the mean ± SE.
To examine the role of Gr-1+CD11b+ cells in tumor metastasis, 4T1 cells were injected into the mammary fat pad of female Balb/c mice, with or without Gr-1+CD11b+ cells sorted from 4T1 tumor tissues or spleens of mice bearing 4T1 tumors. Gr-1+CD11b+ cells from normal spleens were used as a control. We achieved a 95%–98% purity of sorted Gr-1+CD11b+ cells (Figure 2D). The primary tumors were removed after 12 to 14 days, which allows metastasis to develop in lungs avoiding morbidity or mortality caused by large primary tumors. Lungs were harvested and the tumor nodules were counted after the mice showed evidence of distress. We observed a significant increase of tumor nodules in lungs from mice that received 4T1 co-injection with Gr-1+CD11b+ cells derived from tumor tissues or spleens of tumor bearing animals compared with 4T1 alone or 4T1 with normal Gr-1+CD11b+ cells (Figure 2E). These data confirm that Gr-1+CD11b+ cells derived from tumor hosts enhance tumor metastasis.
In further characterization of Gr-1+CD11b+ cells, we found that within Gr-1+CD11b+ cell population, there were approximately 14% of CD34+, 31% VEGFR1+ and 44% F4/80+ cells (Supplementary Figure 1). These data support our hypothesis that Gr-1+CD11b+ cells are constituted of immature myeloid cells at different stages of differentiation.
Gr-1+CD11b+ myeloid cells promote mammary carcinoma cell invasion in vivo and in vitro
Very interestingly, after surgical removal of primary tumors, the recurrent tumors at the primary injection site were significantly larger from 4T1 cells co-injected with tumor derived Gr-1+CD11b+ cells than those from control groups (Figure 3A). In addition, we found no difference in primary tumor growth during the first 10–14 days after injection (data not shown). This indicates that Gr-1+CD11b+ cells derived from tumor tissues or tumor spleens increase tumor cell invasion to surrounding tissues, supporting our earlier observation that Gr-1+CD11b+ cells promote tumor metastasis, as invasion is the first step in the metastatic process.
Figure 3. Gr-1+CD11b+ myeloid cells increased 4T1 tumor invasion in vivo and in vitro.

A: Increased growth of recurrent tumors with 4T1 cells co-injected with Gr-1+CD11b+ cells derived from tumor tissues, or spleens of tumor-bearing mice. Number of animals is indicated in the bar. Results are presented as the mean ± SE. B: Fluorescent microscopy of 4T1 cells that invaded through a matrigel-coated transwell when co-cultured with tumor-derived Gr-1+CD11b+ cells (B-b) compared to normal Gr-1+CD11b+ cells (B-a). 4T1 cells were labeled with a green tracking dye, and Gr-1+CD11b+ cells with a red tracking dye. B-c: Close interaction between 4T1 cells and Gr-1+CD11b+ cells. Scale bar, 50 μM for all figures. C: An MMP inhibitor (GM6001, 1 mM) blocked Gr-1+CD11b+ cell promoted 4T1 cell invasion in vitro. Results are from two experiments with triplicates for each group, are presented as the mean ± SE. tu inf Gr-1+CD11b+ cells: Gr-1+CD11b+ cells from tumor tissues; tu sp Gr-1+CD11b+ cells: Gr-1+CD11b+ cells from spleens of tumor-bearing mice; nor Gr-1+CD11b+ cells: Gr-1+CD11b+ cells from spleens of normal mice. m.g: adjacent mammary gland tissue; tu: tumor tissues.
To further investigate the effects of Gr-1+CD11b+ cells on tumor invasion, we performed in vitro invasion assays. 4T1 cells were labeled with a green cell tracking dye. Sorted Gr-1+CD11b+ cells were labeled with a red cell tracking dye. 4T1 cells with or without Gr-1+CD11b+ cells were seeded into the top chamber of Transwell filters coated with Matrigel. 4T1 cells that had invaded through the membrane were counted (8–10 fields/filter) after overnight incubation. We found that 4T1 cells mixed with Gr-1+CD11b+ cells displayed a significant increase in tumor cell invasion (Figure 3B & 3C). Interestingly, GM6001 (1 uM), a broad spectrum MMP inhibitor, completely attenuated the observed invasion (Figure 3C). These results are in agreement with our in vivo findings that Gr-1+CD11b+ cells directly promote tumor cell invasion and metastasis.
Further, we observed significantly more invasive area in the PyVmT/Tgfbr2MGKO tumors than in the PyVmT/Tgfbr2flox.flox tumors when the two tumor types are at the same stage and with the similar size (Supplementary Figure 2A and 2C). The invasive areas in the PyVmT/Tgfbr2MGKO tumors are often filled with large number of Gr-1+CD11b+ cells (Supplementary Figure 2B). Immunofluorescence staining of E-Cadherin did not show significant changes in E-Cadherin expression between the PyVmT/Tgfbr2MGKO and control tumors, indicating that EMT is unlikely to be involved in PyVmT/Tgfbr2MGKO tumor invasion (Supplementary Figure 2D and 2E). These data suggest that PyVmT/Tgfbr2MGKO tumor invasion may be aided by Gr-1+CD11b+ cells through a collective event between Gr-1+CD11b+ cells and tumor cells.
Tumor residing Gr-1+CD11b+ cells exhibit elevated production and function of MMPs
MMPs contribute to tumor progression and metastasis (Lynch and Matrisian, 2002). Microarray analyses indicated that MMP14 (MT1-MMP), MMP13, and MMP2 are highly elevated in tumor residing Gr-1+CD11b+ cells when compared with those from spleens (data not shown). We further confirmed the results with real time RT-PCR (Figure 4A & B). Since inhibition of MMP activity abolishes Gr-1CD11b+ cell mediated tumor invasion in vitro (Figure 3C), we performed in situ zymography using a quenched fluorogenic substrate that releases fluorescent peptides upon cleavage by gelatinases. This assay allows us to examine the functionality of MMPs in Gr-1+CD11b+ cells directly in tumor tissues. As expected, very strong fluorescence intensity, was localized in the tumor-mammary tissue interface where we had observed significant number of Gr-1+CD11b+ cells (Figure 4C, a-c for 4T1 tumors and 4C-d for MMTV-PyVmT/Tgfbr2MGKO tumors). This was not observed in controls treated with EDTA (Figure 4C-e). Taken together with the in vitro data demonstrating a requirement for metalloproteinase activity, we suggest that tumor residing Gr-1+CD11b+ cells produce multiple MMPs that contribute to breast tumor cell invasion.
Figure 4. Increased production and function of MMPs in tumor residing Gr-1+CD11b+ cells.

Real time RT-PCR of MMP14 and MMP2 (A) as well as MMP13 (B) in Gr-1+CD11b+ cells derived from tumor tissues compared with those from spleens. Results are presented as the mean ± SE. C. In situ zymography of MMPs in 4T1 mammary carcinomas and PyVmT/Tgfbr2MGKO carcinomas. Abundant green fluorescence (indicator of MMP activities) was observed in the invasive front of 4T1 tumors (C-a & c) and PyVmT/Tgfbr2MGKO carcinomas (C-d). Nuclei were labeled with 7AAD (red). Tumor sections treated with EDTA were used as negative controls (C-e). Scale bar, 50 uM for all figures. m.g: adjacent mammary gland tissue; tu: tumor tissues.
Increased TGFβ production in mammary carcinomas with genetic deletion of Tgfbr2
In characterizing mammary carcinomas with Tgfbr2 deletion, we found that they contained significantly higher levels of TGFβ1 than the control tumor without Tgfbr2 deletion (Figure 5A). Because significantly more Gr-1+CD11b+ cells infiltrate into tumors with Tgfbr2 deletion, and our preliminary data from microarray analysis indicated that Gr-1+CD11b+ cells produce large amount of TGFβ1, we reasoned that Gr-1+CD11b+ cells may contribute to increased TGFβ1 production in these tumors. We measured TGFβ1 in Gr-1+CD11b+ cells by ELISA. Gr-1+CD11b+ cells derived from tumor spleens produced significantly higher levels of TGFβ1 than those from normal spleens (Figure 5B, left panel). No difference was found in TGFβ1 production between3 tumor cell lines derived from mammary adenocarcinoma with or without Tgfbr2 deletion (Figure 5B, right panel). Immunohistochemistry of tumor sections further demonstrate that TGFβ1 positive cells are mostly stromal cells (Figure 5C), especially in the invasive front where Gr-1+CD11b+ cells were mostly present (Figure 5C). These data support our observation that Gr-1+CD11b+ cells are likely one of the sources for increased TGFβ13 production in mammary carcinomas with genetic deletion of Tgfbr2.
Figure 5. Increased TGFβ1 production in mammary carcinomas with genetic deletion of Tgfbr2.

A: Increased TGFβ1 production in PyVmT/Tgfbr2MGKO tumors compared with PyVmT/Tgfbr2flox/flox tumors. n=4 mice per group. B left panel: Gr-1+CD11b+ cells from spleens of mice bearing large 4T1 tumors exhibited significantly higher TGFβ1 production when compared with Gr-1+CD11b+ cells from normal spleens. n=2 mice per group. B right panel: No difference of TGFβ1 production was found between tumor cell lines derived from PyVmT/Tgfbr2MGKO tumors vs PyVmT/Tgfbr2flox/flox tumors. TGFβ1 in conditioned media was measured by ELISA. Results are presented as the mean ± SE. C: Gr-1+CD11b+ cells are likely the resource for increased TGFβ13 production in mammary carcinomas with genetic deletion of Tgfbr2. IHC of TGFβ1 (left panel) showing TGFβ1 positive cells are mostly in the invasive front where Gr-1+CD11b+ cells were mostly present (right panel with Gr-1 staining). Scale bar, 50 uM for both figures. m.g: adjacent mammary gland tissue; tu: tumor tissues.
Mechanisms of Gr-1+CD11b+ cell recruitment to tumors
Our microarray analyses indicate that CXCL5 was significantly increased in PyVmT/Tgfbr2MGKO tumors compared with the PyVmT/Tgfbr2flox/flox tumors (data not shown). We further confirmed this observation with ELISA assays using cell culture supernatant from established cell lines (Figure 6A, left panel). Interestingly, we also observed elevated CXCL5 production in carcinoma cells with co-expression of a dominant-negative type II TGFβ receptor (DNIIR) with c-Neu under the MMTV promoter compared to c-Neu carcinoma cells (Supplementary Figure 3A). In addition pancreatic cancers with Tgfbr2 deletion also showed increased CXCL5 production (Ijichi and Moses, unpublished data). These data indicate that CXCL5 may be a common mechanism in the tumor microenvironment with deleted or decreased TGFβ signaling. Further analysis confirmed that recombinant CXCL5 increased Gr-1+CD11b+ cell migration in vitro (Figure 6B), and neutralizing CXCL5 using a monoclonal antibody significantly blocked the migration of Gr-1+CD11b+ cells in response to PyVmT/Tgfbr2MGKO cell conditioned medium (Figure 6B). To examine the role of the CXCL5/CXCR2 in Gr-1+CD11b+ cell recruitment to the tumor microenvironment in vivo, we transplanted breast adenocarcinoma with or without Tgfbr2 deletion into the number four mammary gland in syngeneic FVB mice and allowed tumors to grow for 14 days. We then treated mice with equal tumor sizes with a specific CXCR2 antagonist SB-265610 at 2 mg/kg/day for two weeks through I.P. injection. SB-265610 has been shown to have selective inhibition on neutrophil accumulation when used at 1–3 mg/kg/day (Auten et al., 2001). We found that the SB-265610 treatment significantly inhibited the recruitment of Gr-1+CD11b+ cells to the mammary adenocarcinoma with Tgfbr2 deletion but not the control tumors (Figure 6C), with no significant effect on tumor growth (data not shown). Gr-1+CD11b+ cells isolated from PyVmT/Tgfbr2MGKO tumor tissues express CXCR2, in levels that are modestly elevated compared to splenic Gr-1+CD11b+ cells (data not shown). These data indicate that elevated CXCL5 production but not the corresponding CXCR2 receptor regulates the recruitment of Gr-1+CD11b+ cells in the PyVmT/Tgfbr2MGKO tumor microenvironment.
Figure 6. Mechanisms of recruitment of Gr-1+CD11b+ cells to the tumor microenvironment.

A: Elevated production of CXCL5 in cell culture supernatant of PyVmT/Tgfbr2MGKO mammary carcinomas. B: In vitro migration of Gr-1+CD11b+ cells in response to CXCL5. Gr-1+CD11b+ cells migration after 6–8 hr incubation were counted and plotted. Shown is one of the representative experiments of three performed. C: Inhibition of Gr-1+CD11b+ cell recruitment to PyVmT/Tgfbr2MGKO tumors with CXCR2 antagonism. Percentage of Gr-1+CD11b+ cells in all cells from PyVmT/Tgfbr2MGKO and control tumors were plotted. Mice bearing PyVmT/Tgfbr2MGKO and control tumors were treated with a CXCR2 specific antagonist SB-265610 at 2 mg/kg/day for two weeks through I.P. D: Flow cytometry analysis of CXCR4 expression in infiltrating Gr-1+CD11b+ cells from 4T1 tumors as well as PyVmT/Tgfbr2MGKO tumors, compared with those from spleens of the same tumor-bearing mice (as labeled). Histogram of CXCR4 expression was gated on Gr-1+CD11b+ double positive cells. Shown is one of the representative mice analyzed, n=3–5 mice per group. E: In vitro migration of Gr-1+CD11b+ cells in response to SDF-1. Gr-1+CD11b+ cells migration after 6–8 hr incubation were counted and plotted. Shown is one representative experiment of three performed. F: PyVmT/Tgfbr2MGKO tumor metastasis with blockade of CXCR2 or CXCR4 alone or both. Tumor nodules in lung were counted after mice bearing 14-Day tumors were treated with CXCR2 or CXCR4 antagonists or both for 3 weeks. All results are presented as the mean ± SE.
The SDF-1/CXCR4 chemokine axis mediates recruitment of several cell types including hematopoietic progenitors cells (Burger and Kipps, 2006; Mohle et al., 1998; Peled et al., 1999; Urbich and Dimmeler, 2004). Thus, we examined CXCR4 expression in Gr-1+CD11b+ cells using flow cytometry analysis. Tumor infiltrating Gr-1+CD11b+ cells express significantly increased CXCR4 when compared with those derived from spleens of tumor-bearing mice (Figure 6D). This includes Gr-1+CD11b+ cells from 4T1 and PyVmT/Tgfbr2MGKO tumors (Figure 6D), suggesting that upregulated CXCR4 expression is an intrinsic property of these cells regardless of differences in the tumor microenvironment to which they are recruited. Gr-1+CD11b+ cells isolated from tumor tissues migrated in response to recombinant SDF-1 in vitro (Figure 6E). Importantly, neutralizing CXCR4 using an antibody significantly blocked this process (Figure 6E). In addition to CXCR4, CXCR7 has been identified as another signaling receptor for SDF-1(Balabanian et al., 2005; Burns et al., 2006). We found that approximately 19% Gr-1+CD11b+ cells are CXCR7 positive (supplementary Figure 3B). Thus Gr-1+CD11b+ cells could also be recruited through the SDF-1/CXCR7. Together our data suggest that tumor residing Gr-1+CD11b+ cells are recruited to tumor tissues through SDF-1/CXCR4 and potentially through CXCR7 interaction. In addition, PyVmT/Tgfbr2MGKO tumors have increased production of CXCL5 that is also responsible for the recruitment of Gr-1+CD11b+ cells to tumors with deleted or decreased TGFβ signaling.
To further examine the critical roles of Gr-1+CD11b+ cell recruitment in PyVmT/Tgfbr2MGKO tumor invasion and metastases, we transplanted breast adenocarcinoma with Tgfbr2 deletion into the number four mammary gland in syngeneic FVB mice, and allowed tumors to establish for 7 days. We then treated the mice with a CXCR2 specific antagonist SB-265610 at 2 mg/kg/day, through I.P. injection or a CXCR4 antagonist AMD3100 at 1.25 mg/kg/day twice daily through s.c injection, or both for 10 days. The primary tumors were then surgically removed. The mice were allowed to live for another three weeks for tumor metastasis development in the lungs. The effect of the antagonist on tumor metastasis, tumor growth and invasion were evaluated. We found that lung metastasis is significantly inhibited when mice treated with the CXCR2 antagonist, the CXCR4 antagonist or CXCR2 & CXCR4 antagonists when compared with vehicle controls (Figure 6F). No significant difference in primary tumor growth was found between the treatment and the controls (data not shown). H&E staining of the tumor tissues showed the degree of tumor invasion in mice treated with CXCR2, CXCR4 or both was reduced when compared with the vehicle controls (Supplementary Figure 2F-J). These data, together with data from in vitro coculture (Figure 3B and C) and the effect of in vivo coinjection of tumor cells with Gr-1+CD11b+ cells (Figure 3A 2E) strongly suggest that Gr-1+CD11b+ cells are critical players in tumor cell invasion and metastases formation.
Immature myeloid cells in human breast tumors
Finally, we investigated human breast carcinomas for the presence of immature myeloid cells. We chose myeloperoxidase (MPO) as a marker for human immature myeloid cells for IHC. This is based on: 1) A significantly elevated expression of MPO in Gr-1+CD11b+ immature myeloid cells from several mouse models by microarray analysis (data not shown). 2) MPO, a hemoprotein, is abundantly expressed in neutrophils. MPO possesses proinflammatory properties and participate in tumor initiation and progression (Knaapen et al., 2006). We examined eight cases of human breast carcinoma tissues. MPO positive cells were clearly abundant in the invasive front of all samples examined (Figure 7A). These include the front in the collagen-rich area (Figure 7A, a–c) and the front in normal mammary fat tissues (Figure 7A, d–f)). MPO positive cells were found in both situations (Figure 7A, a and d). H&E staining of these tumor tissues further support our observations (Figure 7A, b and e). To further confirm the infiltrating myeloid cells in human breast tumor tissues, we utilized flow cytometry technology. We labeled single cell suspensions from fresh human breast ductal adenocarcinomas (stage 2–3) with antibody cocktails. These include antibodies for lineage identification: CD3 for T cell, CD19 for B cell, CD56 for NK cell, CD40/CD86/HLA-DR for dendritic cells, and CD14 for monocytes, and antibodies for myeloid cell marker including CD33, CD34, and CD15. The myeloid markers are typically used for examining the peripheral blood of patients with myelodysplasia. Tumor infiltrating immature myeloid cells were identified as T, B, NK, monocyte and dendritic cell-lineage negative and positive for CD33, CD34, and CD15 myeloid markers. The percentage of immature myeloid cells was evaluated for each patient, with four patients total. We found an average of 5.82% +/− 2.3 of total cells from tumor tissues are these myeloid cells (Figure 7B). Together, our data suggest that immature myeloid cells not only participate in tumor invasion in mouse models but also likely in human breast cancers. Collectively, our findings point out a new strategy to target tumor metastasis by disrupting immature cell recruitment to tumors.
Figure 7. Immature myeloid cells in human breast tumor tissues.
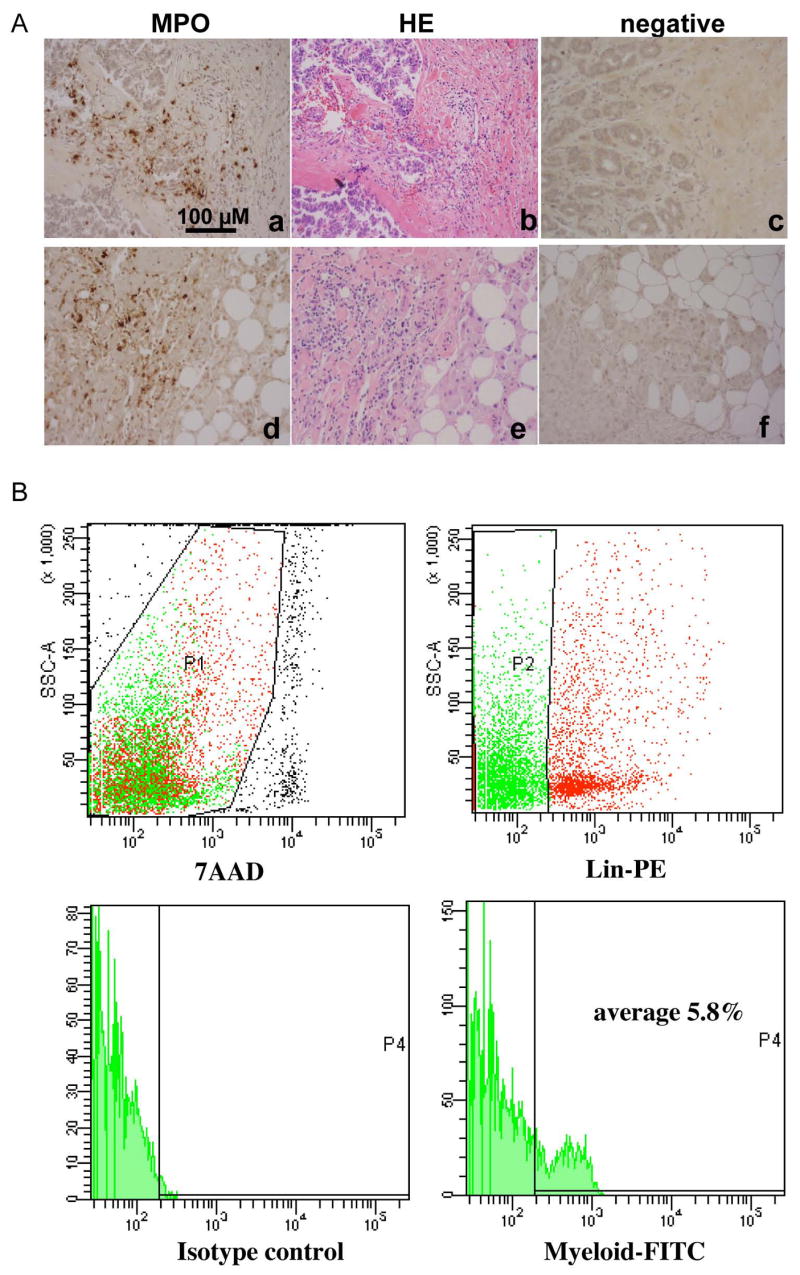
A: IHC staining of myeloperoxidase to identify bone marrow-derived immature myeloid cells at the invasive front of human breast ductal adenocarcinomas (a & d). H&E staining of sequential tumor sections figure b to a, and e to d, respectively. Figure c and f are negative controls. Scale bar, 100 uM for all figures. B: Flow cytometry analysis of immature myeloid cells from single cell suspension of human breast ductal adenocarcinomas (stage 2–3). The analyzed cells were gated as 7AAD negative (top left panel, P1), negative for lineage markers including CD3 for T cell, CD19 for B cell, CD56 for NK cell, CD40/CD86/HLA-DR for dendritic cells, and CD14 for monocytes, and CD33, CD34 and CD15 positive (top right panel, P2). A histogram for FITC positive myeloid cells is shown in the lower right pane, with isotype control in the lower left panel.
Discussion
Using the MMTV PyVmT mouse model we demonstrate that deletion of Tgfbr2 in mammary carcinomas specifically enhances recruitment of Gr-1+CD11b+ myeloid cells to the invasive front. We further demonstrate that Gr-1+CD11b+ cells promote tumor invasion and metastasis through increased MMP production. TGFβ functions as a tumor suppressor or a tumor promoter depending on the context and stage of tumor progression. We and others have shown that abrogation of TGFβ signaling with deletion of Tgfbr2 resulted in aggressive tumor progression in several mouse models (Biswas et al., 2004; Forrester et al., 2005; Ijichi et al., 2006; Munoz et al., 2006; Lu et al., 2006). However, other reports show that the enhancement of TGFβ signaling in mammary epithelial cells in conjunction with c-Neu or PyVmT increases pulmonary metastases (Muraoka et al., 2003; Muraoka-Cook et al., 2004; Muraoka-Cook et al., 2006). These seem to be contradicting observations, which now could be explained by the recruitment of Gr-1+CD11b+ cells into the tumor microenvironment. Our data suggest that autologous TGFβ signaling in mammary epithelial cells act as a tumor suppressor; when it is deleted or altered, it results in Gr-1+CD11b+ cell recruitment. This leads to increased MMP and TGFβ production that enhances tumor invasion and metastasis. The switch of TGFβ signaling as tumor-suppressor to tumor promoter thus involves an additional component, which is the recruitment of Gr-1+CD11b+ cells in the tumor microenvironment. Indeed, inflammatory cells (CD45 and BM8 positive cells) have been observed in head-and-neck tumors lacking TGFβ signaling (Lu et al., 2006). In addition, inflammation causes precancerous lesions in mice with TGFβ1 deficiency that can progress to colon cancer (Engle et al., 2002). Very recently, CCR1+ myeloid cells (CD34+) have been show to be recruited to colon cancers with deletion of Smad4, down stream signaling molecules of TGFβ signaling, promote tumor invasion (Kitamura et al., 2007). These CCR1+ cells seem to be different Gr-1+CD11b+ cells we described here as they do not express high levels of Gr-1.
Several lines of evidence make the contribution of Gr-1+CD11b+ to tumor metastasis particularly interesting. 1) These cells are overproduced in tumor hosts that include cancer patients with a variety of tumors. 2) Gr-1+CD11b+ cells are composed of immature myeloid cells at the early stages of differentiation (Almand et al., 2001; Yang et al., 2004). They are different from terminally differentiated tumor associated macrophages (TAM), identified as Mac-1 (CD11b) and F4/80+, which have been shown to promote tumor progression and metastasis through elevated CSF-1 production and enhanced EGF signaling in cancer cells (Condeelis and Pollard, 2006). Similarities between TAM and these immature myeloid cells were noticed from profiling work (Biswas et al., 2006), however, differences between the two populations were also evident. For example, myeloid suppressor cells produce high level of TGF-β1, whereas TGF-β1 expression in TAMs was restricted to unstimulated TAMs and was not further increased by M2-biasing cytokines (Biswas et al., 2006). Gr-1+CD11b+ cells are also different from two other cell types in the tumor microenvironment, neutrophils and mast cells, which express Gr-1 but not CD11b. 3) Gr-1+CD11b+ cells interact with other host immune cells including T, B, and NK cells. These immune cells may dictate the tumor microenvironment that a shift from inflammation/immune response to anti-inflammatory/immune-suppressive responses (Th1/Th2-like cytokine shift) may be responsible in the metastatic liver milieu (Budhu et al., 2006). It is unclear whether systemic immune suppression and direct participation in tumor progression are two different properties or different manifestations of the same process.
Our data demonstrate that at least two distinct chemokine axes regulate the recruitment of Gr-1+CD11b+ cells in tumor tissues. One is SDF-1/CXCR4. Interestingly, tumor residing Gr-1+CD11b+ cells, from both mammary tumors with Tgfbr2 deletion or 4T1 tumors, express high level of CXCR4 when compared with controls (Figure 6D). This indicates that upregulation of CXCR4 in Gr-1+CD11b+ cells is an intrinsic property of Gr-1+CD11b+ cells regardless the difference of tumor microenvironment into which they are recruited. SDF-1/CXCR4 has been implicated in the recruitment of hematopoietic stem and progenitor cells (Balkwill and Coussens, 2004; Jin et al., 2006; Mohle et al., 1998; Peled et al., 1999; Urbich and Dimmeler, 2004), supporting our observation that Gr-1+CD11b+ cells are hemaetopoietic progenitors. In addition to the SDF-1/CXCR4 chemokine axis, mammary carcinomas with Tgfbr2 deletion showed an increased production of CXCL5 (Figure 6A). The role of CXCL5 in cancer associated inflammation is indicated in the prostate (Hochreiter et al., 2000). CXCL5 also has profound angiogenic potential (Wente et al., 2006). Interestingly, elevated CXCL5 production was also observed in dominant negative type II TGFβ receptor/MMTV-C-Neu cells (Supplementary Figure 3), indicating CXCL5 may be a result of diminished TGFβ signaling in the tumor microenvironment. These data suggested two distinct mechanisms for Gr-1+CD11b+ cell recruitment: increased expression of CXCR4 in Gr-1+CD11b+ cells as an intrinsic property and elevated CXCL5 level in PyVmT/Tgfbr2MGKO tumors as tumor microenvironment cues. When PyVmT/Tgfbr2MGKO tumors were treated with a CXCR2 specific antagonist or a CXCR4 antagonist, there was a significant decreased tumor metastasis, strongly suggesting a critical role of Gr-1+CD11b+ cells in PyVmT/Tgfbr2MGKO tumor invasion and metastases.
Gr-1+CD11b+ cells promote tumor metastasis likely through enhanced production and function of MMPs including MT1-MMP (MMP14), MMP13 and MMP2. Treatment with a pan MMP inhibitor significantly attenuated tumor invasion in vitro. MT1-MMP plays a crucial role in degrading extracellular matrix and allows tumor cells to escape the matrix-enforced growth control effect (Hotary et al., 2003). MT1-MMP is known to be able to activate MMP2 and MMP13, both of which are associated with tumor invasion (d’Ortho et al., 1997; English et al., 2001; Leeman et al., 2002; Nelson et al., 2000). Results from our in vitro coculture (Figure 3B and C) and in vivo coinjection of tumor cells with Gr-1+CD11b+ cells (Figure 3A 2E) strongly suggest a role for these cells in tumor invasion and metastasis. In addition, PyVmT/Tgfbr2MGKO tumors, with a large number of Gr-1+CD11b+ cells, showed significantly more invasive area compared with PyVmT/Tgfbr2flox.flox tumors. Further, tumors treated with CXCR2 or CXCR4 antagonists showed reduced invasiveness. These data suggest that the increased invasion of PyVmT/Tgfbr2MGKO tumor may be aided by Gr-1+CD11b+ cells through a collective event between Gr-1+CD11b+ cells and tumor cells with the myeloid cells providing MMPs. We cannot rule out the contribution of other factors produced by Gr-1+CD11b+ cells to tumor cell invasion, but our results strongly suggest that an induction of proteolytic activity is a major contributing factor. In addition, the observed increased tumor growth and metastasis-promoting effects of Gr-1+CD11b+ cells could also be due to their effect on angiogenesis as we previously reported (Yang et al., 2004).
Tumor-infiltrating Gr-1+CD11b+ myeloid cells are likely one of the major sources for increased TGFβ13 production observed in mammary carcinomas with genetic3 deletion of Tgfbr2 (Figure 5). This is demonstrated by the IHC of tumor sections showing that TGFβ1 positive cells are mostly stromal cells (Figure 5C), especially in the invasive front where Gr-1+CD11b+ cells were most abundant. Our observation is supported by a recent publication showing that in response to Tgfbr2 deletion, there was increased TGFβ1 expression in the stroma (Lu et al., 2006). We have now shown that Gr-1+CD11b+ cells are components of these stromal cells. In addition, as Gr-1+CD11b+ cells produce high levels of TGFβ1, they are likely to have a major effect on tumor evasion from host immune detection.
In summary, we have demonstrated that deletion of the type II TGFβ receptor gene in mammary carcinomas results in the recruitment of Gr-1+CD11b+ myeloid cells through two distinct mechanisms: SDF-1/CXCR4 as an intrinsic mechanism for Gr-1+CD11b+ cells and CXCL5/CXCR2. Gr-1+CD11b+ cells directly promote tumor metastasis through enhanced MMP and TGFβ production (Figure 8). Cellular and molecular targeting of Gr-1+CD11b+ cell production/recruitment should not only improve host immunosurveillance but also inhibit tumor metastasis, having the effect of “killing two birds with one stone” and provide additional therapeutic options.
Figure 8. Mechanisms for enhanced tumor progression resulting from loss of TGFβ signaling.

Deletion of the type II TGFβ receptor gene in mammary carcinomas results in elevated production of CXCL5, which interact with CXCR2 on Gr-1+CD11b+ cells thus recruiting the cells into the tumor microenvironment. Gr-1+CD11b+ cells, as hemaetopoietic cells, express high level of CXCR4 that interact with SDF-1 in the tumor microenvironment, and are also recruited. Gr-1+CD11b+ cells promote tumor invasion through high expression of MMP14, MMP13 and MMP2. In addition, Gr-1+CD11b+ cells produce high levels of TGFβ1 that inhibit host immune surveillance.
Experimental procedures
Cell lines and mice
4T1 breast cancer cell line was obtained and maintained per standard cell culture techniques. MMTV-PyVmT/Tgfbr2MGKO and floxed control cell lines were established as described (Forrester et al., 2005). Eight to ten week-old female Balb/c mice were purchased from Harlan Inc. (Indianapolis, IN.). Tgfbr2flox/flox, Tgfbr2MGKO, MMTV-PyVmT/Tgfbr2flox/flox, and MMTV-PyVmT/Tgfbr2MGKO mice were established and maintained as described (Forrester et al., 2005). The studies were approved by IACUC at Vanderbilt University Medical Center.
Flow cytometry analysis
Single cell suspensions were made from spleens and BM of normal and tumor-bearing mice (Yang et al., 2004), and tumor tissues (Ljung et al., 1989). These cells were labeled with fluorescence-conjugated antibodies (BD PharMingen), and isotype-matched IgG controls. The cells were analyzed on a FACScan flow cytometer (Becton Dickinson).
Single-cell sorting
Splenocytes from normal or tumor-bearing mice were stained with fluorescence-labeled antibodies, and sorted with a FACStarPlus® flow cytometer (Becton Dickinson). Gr-1+CD11b+ cells were collected for tumor growth, invasion and migration experiments.
4T1 breast tumor model for metastasis
4T1 (5 × 105 cells), without or with Gr-1+CD11b+ cells (0.5 × 105), were injected into the mammary fat pad of female Balb/c mice. Primary tumors were completely removed and weighed 12–14 days after tumor inoculation. The mice were sacrificed 4 weeks later and the lung metastasis was examined Whole Lung Mounting procedures. In addition, the weight of recurrent tumors at the primary injection sites was also recorded.
Whole lung mounting
Mice were sacrificed by anesthetic overdose. Lungs were processed as described (Jessen et al., 2004). The tumor nodules in lung were then counted.
Immunohistochemistry and immunofluorescence
Paraffin embedded tumor sections were fixed in 4% paraformaldehyde and incubated with a rat polyclonal anti-Gr-1 antibody (BD Pharmingen), rabbit polyclonal anti-myeloperioxidase (Abcam) or anti TGFβ1 (Pharmingen). A biotinylated 2nd antibody was applied, followed by incubation with streptavidin-conjugated HRP. Peroxidase activity was localized with diaminobenzidine (Vectastain ABC kit, Vector Laboratories). For immunofluorescence, anti E-cadherin antibody (BD transduction laboratories, 1:500 dilution) was applied to paraffin embedded tumor sections, Alexa flour 488 conjugated goat anti mouse secondary antibody (Molecular Probes, 10 ug/ml) was used to detect E-cadherin expression.
Immunoassay for chemokines
Conditioned media and tissue lysates were analyzed for CXCL5, TGFβ1 expression by ELISA using commercial kits (R&D Systems).
In vitro cell migration and invasion assays
For cell migration, Gr-1+CD11b+ cells (50,000) were seeded onto the top chamber of transwell filters (8 uM, VWR Scientific). The filters were placed in a 24-well plate that contains medium with recombinant mouse SDF-1 or isotype control at 100 ng/ml. To neutralize CXCR4, the CXCR4 monoclonal ab at 10 ug/ml was added to the top chamber of the transwell. Migrated Gr-1+CD11b+ cells were counted (5–7 fields/well, triplicate for each experimental groups) 6–8 hours after incubation. For in vitro invasion assays, 4T1 cells were labeled with green fluorescence dye CMFDA (Molecular Probes). Gr-1+CD11b+ cells were labeled with red fluorescence dye CMTMR. 4T1 cells (30,000) alone or with Gr-1+CD11b+ cells (30,000) were seeded into the top chamber of transwell filters coated with Matrigel (Becton-Dickinson). The filters were placed in a 24-well plate that contains culture media of 10% FCS RPMI. Invaded 4T1 cells were counted (3 fields/filter, triplicate for each experimental groups) 6–8 hours after incubation.
In vivo inhibition of Gr-1+CD11b+ cell recruitment to breast adenocarcinomas
MMTV-PyVmT/Tgfbr2MGKO and MMTV-PyVmT/Tgfbr2flox/flox tumors from donor mice were cut into approximately 1 mm3 and planted into #4 mammary fat pad of syngeneic FVB recipient mice. Tumor size was determined two weeks later as described (Yang et al., 2004). Mice with similar tumor size were treated with CXCR2 specific antagonist SB-265610 (GSK Pharmaceuticals) at 2 mg/kg/day through I.P. for two weeks (DMSO as vehicle). Tumor size was measured again at the end of the treatment. For the effect of CXCR2 and CXCR4 blockade on PyVmT/Tgfbr2MGKO tumor metastasis, mice were implanted tumors for seven days and then were treated with SB-265610 as described above, or a CXCR4 antagonist (AMD3100, Sigma, at 1.25 mg/kg/day twice daily through s.c, PBS as vehicle) or both for 10 days. The primary tumors were then surgically removed. The mice were allowed to live for another three weeks for lung metastasis development without further treatment. Metastasis nodules were counted. Primary tumors were removed, weighed and processed for HE staining.
In situ Zymography of MMPs
Substrate (DQ-gelatin, Molecular Probes) was dissolved to a final concentration of 25 ug/ml in a mixture of 2% gelatin and 2% sucrose in PBS + 0.02% sodium azide. 100 ul of this solution was then coated on a slide, set on ice for 10 minutes, then stored at RT until next day. Protease inhibitors including Aprotinin (2 ug/ml), Leupeptin (100 uM) and pepstatin were included in the substrate solution to exclude other protease activities in addition to MMPs. EDTA (25 mM), an inhibitor of metalloproteinases, was used in negative controls. Frozen tumor sections (5–10 um) were cut directly onto the coated slides and the slides were incubated in dark at 30° C for 24–36 hrs. The slides were then mounted using an aqueous fluorescence-saving mountant with 50 nM 7AAD, a nucleic acid stain (Molecular Probes). The slides were then examined under a fluorescence microscope.
Quantitative RT-PCR
Total RNA was extracted from sorted Gr-1+CD11b+ cells as described using an RNeasy Mini Kit (QIAGEN Inc.). cDNA was synthesized using In Vitrogen superscripttm First-strand synthesis system for RT-PCR. Primers specific for MMP2, MMP13 and MT1-MMP were used and relative gene expression was determined using BioRad iCycler-iQ SYBR Green PCR kit (BioRad Laboratories). The comparative threshold cycle method was used to calculate gene expression normalized to β-actin as a gene reference. Primers were designed using Beacon Designer 4 software and synthesized by Operon (a Qiagen Company). Primer sequences are available upon request.
Human breast tumor tissues
Human breast ductal adenocarcinomas (stage 2–3) were collected from patients under surgery in Tianjing Cancer Institute, China, with written consent. The samples were rinsed in cold PBS and put in RPMI supplemented with FCS. The samples were then processed to obtain single cell suspension as described earlier. The cells were then labeled with fluorescence-conjugated antibodies (BD PharMingen), and isotype-matched IgG controls. The cells were analyzed on a FACSaria flow cytometer (Becton Dickinson, Mountain).
Statistical analysis
All data was analyzed by the Student t-test, and were expressed as means±SE, and differences were considered statistically significant when the p-value < 0.05.
Supplementary Material
Acknowledgments
We thank Dr. Carlos Arteaga for critical reading of the manuscript. We thank Leigh Moberly and Mergan Jackson from Berea College, Kentucky, for their contribution in these studies during the summer internship. We appreciate the technical assistance from the VA FACS Core, Vanderbilt FACS Core and Mouse Pathology and Immunohistochemistry Core (funded in part by grant CA068485). We thank Dr Barbara Fingleton for advise for in situ zymography. This work was supported by grants CA085492, CA102162 and U54 CA126505 to Harold Moses, CA108856 to Charles Lin and CA126505 to Lynn Matrisian, as well as P30 DK58404 (Brent Polk) pilot grant and Breast SPORE 5P50CA098131 (Carlos Arteaga) career development award to Li Yang.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahuja SK, Murphy PM. The CXC chemokines growth-regulated oncogene (GRO) alpha, GRObeta, GROgamma, neutrophil-activating peptide-2, and epithelial cell-derived neutrophil-activating peptide-78 are potent agonists for the type B, but not the type A, human interleukin-8 receptor. J Biol Chem. 1996;271:20545–20550. doi: 10.1074/jbc.271.34.20545. [DOI] [PubMed] [Google Scholar]
- Akhurst RJ, Derynck R. TGF-beta signaling in cancer--a double-edged sword. Trends Cell Biol. 2001;11:S44–51. doi: 10.1016/s0962-8924(01)02130-4. [DOI] [PubMed] [Google Scholar]
- Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- Auten RL, Richardson RM, White JR, Mason SN, Vozzelli MA, Whorton MH. Nonpeptide CXCR2 antagonist prevents neutrophil accumulation in hyperoxia-exposed newborn rats. J Pharmacol Exp Ther. 2001;299:90–95. [PubMed] [Google Scholar]
- Bagadi SA, Prasad CP, Srivastava A, Prashad R, Gupta SD, Ralhan R. Frequent loss of Dab2 protein and infrequent promoter hypermethylation in breast cancer. Breast Cancer Res Treat. 2006 doi: 10.1007/s10549-006-9422-6. [DOI] [PubMed] [Google Scholar]
- Balabanian K, Lagane B, Infantino S, Chow KY, Harriague J, Moepps B, Arenzana-Seisdedos F, Thelen M, Bachelerie F. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem. 2005;280:35760–35766. doi: 10.1074/jbc.M508234200. [DOI] [PubMed] [Google Scholar]
- Balkwill F, Coussens LM. Cancer: an inflammatory link. Nature. 2004;431:405–406. doi: 10.1038/431405a. [DOI] [PubMed] [Google Scholar]
- Bierie B, Moses HL. TGF-beta and cancer. Cytokine Growth Factor Rev. 2006;17:29–40. doi: 10.1016/j.cytogfr.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, Bottazzi B, Doni A, Vincenzo B, Pasqualini F, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation) Blood. 2006;107:2112–2122. doi: 10.1182/blood-2005-01-0428. [DOI] [PubMed] [Google Scholar]
- Bozic CR, Gerard NP, Gerard C. Receptor binding specificity and pulmonary gene expression of the neutrophil-activating peptide ENA-78. Am J Respir Cell Mol Biol. 1996;14:302–308. doi: 10.1165/ajrcmb.14.3.8845182. [DOI] [PubMed] [Google Scholar]
- Bronte V, Apolloni E, Cabrelle A, Ronca R, Serafini P, Zamboni P, Restifo NP, Zanovello P. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood. 2000;96:3838–3846. [PMC free article] [PubMed] [Google Scholar]
- Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107:1761–1767. doi: 10.1182/blood-2005-08-3182. [DOI] [PubMed] [Google Scholar]
- Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, Penfold ME, Sunshine MJ, Littman DR, Kuo CJ, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med. 2006;203:2201–2213. doi: 10.1084/jem.20052144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Carter D, Garrigue-Antar L, Reiss M. Transforming growth factor beta type I receptor kinase mutant associated with metastatic breast cancer. Cancer Res. 1998;58:4805–4810. [PubMed] [Google Scholar]
- Chen T, Jackson CR, Link A, Markey MP, Colligan BM, Douglass LE, Pemberton JO, Deddens JA, Graff JR, Carter JH. Int7G24A variant of transforming growth factor-beta receptor type I is associated with invasive breast cancer. Clin Cancer Res. 2006;12:392–397. doi: 10.1158/1078-0432.CCR-05-1518. [DOI] [PubMed] [Google Scholar]
- Chytil A, Magnuson MA, Wright CV, Moses HL. Conditional inactivation of the TGF-beta type II receptor using Cre:Lox. Genesis. 2002;32:73–75. doi: 10.1002/gene.10046. [DOI] [PubMed] [Google Scholar]
- Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Ortho MP, Will H, Atkinson S, Butler G, Messent A, Gavrilovic J, Smith B, Timpl R, Zardi L, Murphy G. Membrane-type matrix metalloproteinases 1 and 2 exhibit broad-spectrum proteolytic capacities comparable to many matrix metalloproteinases. Eur J Biochem. 1997;250:751–757. doi: 10.1111/j.1432-1033.1997.00751.x. [DOI] [PubMed] [Google Scholar]
- de Jong JS, van Diest PJ, van der Valk P, Baak JP. Expression of growth factors, growth inhibiting factors, and their receptors in invasive breast cancer. I: An inventory in search of autocrine and paracrine loops. J Pathol. 1998;184:44–52. doi: 10.1002/(SICI)1096-9896(199801)184:1<44::AID-PATH984>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Engle SJ, Ormsby I, Pawlowski S, Boivin GP, Croft J, Balish E, Doetschman T. Elimination of colon cancer in germ-free transforming growth factor beta 1-deficient mice. Cancer Res. 2002;62:6362–6366. [PubMed] [Google Scholar]
- English WR, Holtz B, Vogt G, Knauper V, Murphy G. Characterization of the role of the “MT-loop”: an eight-amino acid insertion specific to progelatinase A (MMP2) activating membrane-type matrix metalloproteinases. J Biol Chem. 2001;276:42018–42026. doi: 10.1074/jbc.M107783200. [DOI] [PubMed] [Google Scholar]
- Forrester E, Chytil A, Bierie B, Aakre M, Gorska AE, Sharif-Afshar AR, Muller WJ, Moses HL. Effect of conditional knockout of the type II TGF-beta receptor gene in mammary epithelia on mammary gland development and polyomavirus middle T antigen induced tumor formation and metastasis. Cancer Res. 2005;65:2296–2302. doi: 10.1158/0008-5472.CAN-04-3272. [DOI] [PubMed] [Google Scholar]
- Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, Kavanaugh D, Carbone DP. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2:1096–1103. doi: 10.1038/nm1096-1096. [DOI] [PubMed] [Google Scholar]
- Gobbi H, Dupont WD, Simpson JF, Plummer WD, Jr, Schuyler PA, Olson SJ, Arteaga CL, Page DL. Transforming growth factor-beta and breast cancer risk in women with mammary epithelial hyperplasia. J Natl Cancer Inst. 1999;91:2096–2101. doi: 10.1093/jnci/91.24.2096. [DOI] [PubMed] [Google Scholar]
- Grady WM, Markowitz SD. Genetic and epigenetic alterations in colon cancer. Annu Rev Genomics Hum Genet. 2002;3:101–128. doi: 10.1146/annurev.genom.3.022502.103043. [DOI] [PubMed] [Google Scholar]
- Hochreiter WW, Nadler RB, Koch AE, Campbell PL, Ludwig M, Weidner W, Schaeffer AJ. Evaluation of the cytokines interleukin 8 and epithelial neutrophil activating peptide 78 as indicators of inflammation in prostatic secretions. Urology. 2000;56:1025–1029. doi: 10.1016/s0090-4295(00)00844-x. [DOI] [PubMed] [Google Scholar]
- Hotary KB, Allen ED, Brooks PC, Datta NS, Long MW, Weiss SJ. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell. 2003;114:33–45. doi: 10.1016/s0092-8674(03)00513-0. [DOI] [PubMed] [Google Scholar]
- Ijichi H, Chytil A, Gorska AE, Aakre ME, Fujitani Y, Fujitani S, Wright CV, Moses HL. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006;20:3147–3160. doi: 10.1101/gad.1475506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KA, Liu SY, Tepper CG, Karrim J, McGoldrick ET, Rosner A, Munn RJ, Young LJ, Borowsky AD, Cardiff RD, Gregg JP. Molecular analysis of metastasis in a polyomavirus middle T mouse model: the role of osteopontin. Breast Cancer Res. 2004;6:R157–169. doi: 10.1186/bcr768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyaseelan S, Manzer R, Young SK, Yamamoto M, Akira S, Mason RJ, Worthen GS. Induction of CXCL5 during inflammation in the rodent lung involves activation of alveolar epithelium. Am J Respir Cell Mol Biol. 2005;32:531–539. doi: 10.1165/rcmb.2005-0063OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin DK, Shido K, Kopp HG, Petit I, Shmelkov SV, Young LM, Hooper AT, Amano H, Avecilla ST, Heissig B, et al. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med. 2006;12:557–567. doi: 10.1038/nm1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Kometani K, Hashida H, Matsunaga A, Miyoshi H, Hosogi H, Aoki M, Oshima M, Hattori M, Takabayashi A, et al. SMAD4-deficient intestinal tumors recruit CCR1(+) myeloid cells that promote invasion. Nat Genet. 2007;39:467–475. doi: 10.1038/ng1997. [DOI] [PubMed] [Google Scholar]
- Knaapen AM, Gungor N, Schins RP, Borm PJ, Van Schooten FJ. Neutrophils and respiratory tract DNA damage and mutagenesis: a review. Mutagenesis. 2006;21:225–236. doi: 10.1093/mutage/gel032. [DOI] [PubMed] [Google Scholar]
- Leeman MF, Curran S, Murray GI. The structure, regulation, and function of human matrix metalloproteinase-13. Crit Rev Biochem Mol Biol. 2002;37:149–166. doi: 10.1080/10409230290771483. [DOI] [PubMed] [Google Scholar]
- Ljung BM, Mayall B, Lottich C, Boyer C, Sylvester SS, Leight GS, Siegler HF, Smith HS. Cell dissociation techniques in human breast cancer--variations in tumor cell viability and DNA ploidy. Breast Cancer Res Treat. 1989;13:153–159. doi: 10.1007/BF01806527. [DOI] [PubMed] [Google Scholar]
- Lu SL, Herrington H, Reh D, Weber S, Bornstein S, Wang D, Li AG, Tang CF, Siddiqui Y, Nord J, et al. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 2006;20:1331–1342. doi: 10.1101/gad.1413306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch CC, Matrisian LM. Matrix metalloproteinases in tumor-host cell communication. Differentiation. 2002;70:561–573. doi: 10.1046/j.1432-0436.2002.700909.x. [DOI] [PubMed] [Google Scholar]
- Melani C, Chiodoni C, Forni G, Colombo MP. Myeloid cell expansion elicited by the progression of spontaneous mammary carcinomas in c-erbB-2 transgenic BALB/c mice suppresses immune reactivity. Blood. 2003;102:2138–2145. doi: 10.1182/blood-2003-01-0190. [DOI] [PubMed] [Google Scholar]
- Mohle R, Bautz F, Rafii S, Moore MA, Brugger W, Kanz L. The chemokine receptor CXCR-4 is expressed on CD34+ hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1. Blood. 1998;91:4523–4530. [PubMed] [Google Scholar]
- Munoz NM, Upton M, Rojas A, Washington MK, Lin L, Chytil A, Sozmen EG, Madison BB, Pozzi A, Moon RT, et al. Transforming growth factor beta receptor type II inactivation induces the malignant transformation of intestinal neoplasms initiated by Apc mutation. Cancer Res. 2006;66:9837–9844. doi: 10.1158/0008-5472.CAN-06-0890. [DOI] [PubMed] [Google Scholar]
- Muraoka RS, Koh Y, Roebuck LR, Sanders ME, Brantley-Sieders D, Gorska AE, Moses HL, Arteaga CL. Increased malignancy of Neu-induced mammary tumors overexpressing active transforming growth factor beta1. Mol Cell Biol. 2003;23:8691–8703. doi: 10.1128/MCB.23.23.8691-8703.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraoka-Cook RS, Kurokawa H, Koh Y, Forbes JT, Roebuck LR, Barcellos-Hoff MH, Moody SE, Chodosh LA, Arteaga CL. Conditional overexpression of active transforming growth factor beta1 in vivo accelerates metastases of transgenic mammary tumors. Cancer Res. 2004;64:9002–9011. doi: 10.1158/0008-5472.CAN-04-2111. [DOI] [PubMed] [Google Scholar]
- Muraoka-Cook RS, Shin I, Yi JY, Easterly E, Barcellos-Hoff MH, Yingling JM, Zent R, Arteaga CL. Activated type I TGFbeta receptor kinase enhances the survival of mammary epithelial cells and accelerates tumor progression. Oncogene. 2006;25:3408–3423. doi: 10.1038/sj.onc.1208964. [DOI] [PubMed] [Google Scholar]
- Nelson AR, Fingleton B, Rothenberg ML, Matrisian LM. Matrix metalloproteinases: biologic activity and clinical implications. J Clin Oncol. 2000;18:1135–1149. doi: 10.1200/JCO.2000.18.5.1135. [DOI] [PubMed] [Google Scholar]
- Pasche B, Knobloch TJ, Bian Y, Liu J, Phukan S, Rosman D, Kaklamani V, Baddi L, Siddiqui FS, Frankel W, et al. Somatic acquisition and signaling of TGFBR1*6A in cancer. Jama. 2005;294:1634–1646. doi: 10.1001/jama.294.13.1634. [DOI] [PubMed] [Google Scholar]
- Peled A, Petit I, Kollet O, Magid M, Ponomaryov T, Byk T, Nagler A, Ben-Hur H, Many A, Shultz L, et al. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science. 1999;283:845–848. doi: 10.1126/science.283.5403.845. [DOI] [PubMed] [Google Scholar]
- Seitz S, Wassmuth P, Plaschke J, Schackert HK, Karsten U, Santibanez-Koref MF, Schlag PM, Scherneck S. Identification of microsatellite instability and mismatch repair gene mutations in breast cancer cell lines. Genes Chromosomes Cancer. 2003;37:29–35. doi: 10.1002/gcc.10196. [DOI] [PubMed] [Google Scholar]
- Serafini P, Borrello I, Bronte V. Myeloid suppressor cells in cancer: Recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol. 2006;16:53–65. doi: 10.1016/j.semcancer.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004;64:6337–6343. doi: 10.1158/0008-5472.CAN-04-0757. [DOI] [PubMed] [Google Scholar]
- Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, Fuh G, Gerber HP, Ferrara N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b(+)Gr1(+) myeloid cells. Nat Biotechnol. 2007;25:911–920. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- Siegel PM, Shu W, Cardiff RD, Muller WJ, Massague J. Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci U S A. 2003;100:8430–8435. doi: 10.1073/pnas.0932636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Bottinger EP, Jakowlew SB, Bagnall KM, Mariano J, Anver MR, Letterio JJ, Wakefield LM. Transforming growth factor-beta1 is a new form of tumor suppressor with true haploid insufficiency. Nat Med. 1998;4:802–807. doi: 10.1038/nm0798-802. [DOI] [PubMed] [Google Scholar]
- Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res. 2004;95:343–353. doi: 10.1161/01.RES.0000137877.89448.78. [DOI] [PubMed] [Google Scholar]
- Wente MN, Keane MP, Burdick MD, Friess H, Buchler MW, Ceyhan GO, Reber HA, Strieter RM, Hines OJ. Blockade of the chemokine receptor CXCR2 inhibits pancreatic cancer cell-induced angiogenesis. Cancer Lett. 2006 doi: 10.1016/j.canlet.2005.10.041. [DOI] [PubMed] [Google Scholar]
- Yang L, Debusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, Matrisian LM, Carbone DP, Lin PC. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–421. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Young MR, Lathers DM. Myeloid progenitor cells mediate immune suppression in patients with head and neck cancers. Int J Immunopharmacol. 1999;21:241–252. doi: 10.1016/s0192-0561(99)00008-9. [DOI] [PubMed] [Google Scholar]
- Zineh I, Aquilante CL, Langaee TY, Beitelshees AL, Arant CB, Wessel TR, Schofield RS. CXCL5 gene polymorphisms are related to systemic concentrations and leukocyte production of epithelial neutrophil-activating peptide (ENA-78) Cytokine. 2006 doi: 10.1016/j.cyto.2006.02.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.