

Abstract
Overactivation of the nuclear enzyme poly(ADP-ribose) polymerase (PARP) contributes to the development of cell dysfunction and tissue injury in various pathophysiological conditions associated with oxidative and nitrosative stress, including myocardial reperfusion injury, heart transplantation, diabetic cardiomyopathy and chronic heart failure. In recent studies, we have demonstrated the beneficial effects of a novel ultrapotent PARP inhibitor, INO-1001, on cardiac and endothelial dysfunction and remodeling in rat model of advanced aging-associated chronic heart failure and in a mouse model of heart failure induced by aortic banding. In the current study, we have investigated the effect of INO-1001 on the development of heart failure induced by permanent ligation of the left anterior descending coronary artery, heart failure induced by doxorubicin and acute myocardial dysfunction induced by bacterial endotoxin. In the coronary ligation model, a significantly depressed left ventricular performance and impaired vascular relaxation of aortic rings were found, and PARP inhibition significantly improved both cardiac function and vascular relaxation. In the doxorubicin model, a single injection of doxorubicin induced high mortality and a significant decrease in left ventricular systolic pressure, +dP/dt, −dP/dt, stroke volume, stroke work, ejection fraction and cardiac output. Treatment with the PARP inhibitor reduced doxorubicin-induced mortality and markedly improved cardiac function. PARP inhibition did not interfere with doxorubicin’s antitumor effect. In the endotoxin model of cardiac dysfunction, PARP inhibition attenuated the suppression of myocardial contractility elicited by endotoxin. The current data strengthen the view that PARP inhibition may represent an effective approach for the experimental therapy of various forms of acute and chronic heart failure.
Keywords: doxorubicin, cardiac function, peroxynitrite, heart failure, oxidative stress, ischemia, endotoxin, tumor
Introduction
Reactive oxygen and nitrogen species are overproduced in the cardiovascular system during chronic heart failure. Oxidant-induced cell injury involves the activation of a nuclear enzyme, poly(ADP-ribose) polymerase (PARP), which importantly contributes to the development of cell dysfunction and tissue injury in various pathophysiological conditions associated with oxidative and nitrosative stress, including myocardial reperfusion injury, heart transplantation, diabetic cardiomyopathy and chronic heart failure (overviewed in refs. 1,2).
Recent studies have demonstrated the cardioprotective effect of a novel ultrapotent poly(ADP-ribose) polymerase (PARP) inhibitor, INO-1001 on the development of cardiac hypertrophy and cardiac contractile dysfunction in rat model of advanced aging-associated chronic heart failure and in a murine model of chronic heart failure induced by aortic banding (3,4). In the present study, we investigated the effect of INO-1001 on the cardiac function in three additional models of cardiac dysfunction: a) chronic heart failure induced by chronic ligation of the left anterior descending coronary artery; b) heart failure induced by the cardiotoxic drug doxorubicin; and c) heart failure induced by bacterial lipopolysaccharide (LPS). In the doxorubicin model we have also explored whether INO-1001 interferes with the anticancer effect of doxorubicin.
Materials and methods
The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by US National Institutes of Health (NIH Publication No. 85–23 revised 1985) and was performed with the approval of the local Institutional Animal Care and Use Committee.
Ligation-induced chronic heart failure model
Heart failure was induced in male Wistar rats by chronic ligation of the left anterior descending coronary artery as previously described (5). Seven days after the coronary occlusion, the surviving animals were randomly assigned to receive a minipump containing an ultrapotent PARP inhibitor INO-1001 (3 mg/kg/day) or vehicle. Left ventricular function was measured 8 weeks after the surgery using Millar microtip catheter transducer SPR-524 (Millar Instruments) coupled to a Powerlab/4SP A/D converter (AD Instruments, Mountain View, CA, USA), and then stored and displayed on a computer (5). Vascular function (contractility and relaxation) was studied in isolated aortic rings as previously described (5). The different heart cavities (left and right ventricles, left and right atria) and lungs were dissected and weighed and normalized to the length of the right tibia or body weight. The infarcted (scar) area of the left ventricle (LV) was carefully dissected from the non-infarcted left ventricle, and infarct size was evaluated by computing the ratio of scar tissue weight over the total LV weight.
Doxorubicin-induced heart failure model
Male BALB/c mice were administered a single dose of doxorubicin at 25 mg/kg i.p., and used for functional measurements 5 days later (6). Treatment with the PARP inhibitor INO-1001 (3 mg/kg/day s.c. in ALZET minipumps) started 2 days before the doxorubicin injection and continued until the hemodynamic measurements were made. Five days after DOX administration analysis of left ventricular performance was measured in mice anesthetized with i.p. injections of ketamine (80 mg/kg) and xylazine (10 mg/kg) i.p. The animals were placed on controlled heating pads, and core temperature measured via a rectal probe was maintained at 36–38°C. A microtip pressure-volume catheter (SPR-839; Millar Instruments, Houston, TX) was inserted into the right carotid artery and advanced into the left ventricle (LV) under pressure control as described (6). Polyethylene cannulae (P10) were inserted into the right femoral artery and vein for measurement of mean arterial pressure (MAP) and administration of drugs, respectively. After stabilization for 20 min, the signals were continuously recorded at a sampling rate of 1000/sec using an ARIA pressure-volume conductance system (Millar Instruments) coupled to a Powerlab/4SP A/D converter (AD Instruments), and then stored and displayed on a computer. All pressure-volume loop data were analyzed with a cardiac pressure-volume analysis program (PVAN3.2; Millar Instruments), and the heart rate (HR), maximal left ventricular systolic pressure (LVESP), left ventricular end diastolic pressure (LVEDP), MAP, maximal slope of systolic pressure increment (+dP/dt) and diastolic decrement (−dP/dt), ejection fraction (EF), stroke volume (SV), cardiac output (CO) and stroke work (SW) were computed. After the hemodynamic measurements were made, animals were sacrificed.
In a separate set of investigations, survival studies were conducted. Animals exposed to an acute dose of doxorubicin (25 mg/kg i.p.) received either INO-1001 (0.03, 0.3, 3 mg/kg s.c. in minipumps) or vehicle starting 2 days (2 days was required for mice to recover following minipump implantation) before doxorubicin injection, respectively. Mortality was monitored and recorded twice daily for 14 days. In a separate set of chronic experiments, doxorubicin was injected in 3 equal doses of 9 mg/kg every 10 days as described previously (6), and survival was followed for 28 days in the presence of INO-1001 (0.03, 0.3 and 3 mg/kg) or vehicle treatment.
In another set of experiments, the potential effect of INO-1001 on tumor growth and the antitumor effect of DOX in a mouse model of breast cancer were investigated in 4T1 mammary adenocarcinoma cells as previously described (6). Cells (n=106) were injected into the mammary fat pad of female BALB/c mice. Fifteen days later, mice were randomized into 4 groups (n=10 per group) and received INO-1001 (3 mg/kg s.c. in minipump), DOX (4 mg/kg i.p. twice a week), DOX + INO-1001, or vehicle. Tumor diameters (x, y, and z) were recorded twice a week, and tumor size was estimated in mm3 and expressed in % of initial size.
Endotoxin-induced heart dysfunction model
Anesthetized male Wistar rats pretreated with vehicle or INO-1001 (3 and 10 mg/kg i.v.) weighing 320–370 g were administered a single dose of E. coli endotoxin (15 mg/kg i.v.) as previously described (7) and hemodynamic parameters were continuously recorded for 90 min.
Statistical analysis
Results are reported as mean ± SEM. Statistical significance between two measurements was determined by the two-tailed unpaired Student’s t-test, and among groups it was determined by analysis of variance with Bonferroni’s correction. In the survival experiments the survival curves of the different groups were compared using log-rank test. Probability values of <0.05 were considered significant.
Results
Effect of INO-1001 on the cardiac dysfunction induced by chronic coronary ligature
Chronic heart failure was characterized by significantly increased left ventricular end-diastolic pressure (LVEDP) and decreased left ventricular systolic pressure (LVSP), mean BP, +dP/dt and −dP/dt and progressive mortality. INO-1001 delayed mortality (Fig. 1) and significantly attenuated the CHF-induced suppression of systolic +dP/dt, LVSP and diastolic −dP/dt and an increase in LVEDP (Fig. 2). INO-1001 significantly improved decreased mean blood pressure in CHF (Fig. 2). The PARP inhibitor exerted no significant effects on hemodynamic parameters in sham rats and the heart rates were not significantly different in all groups studied.
Figure 1.
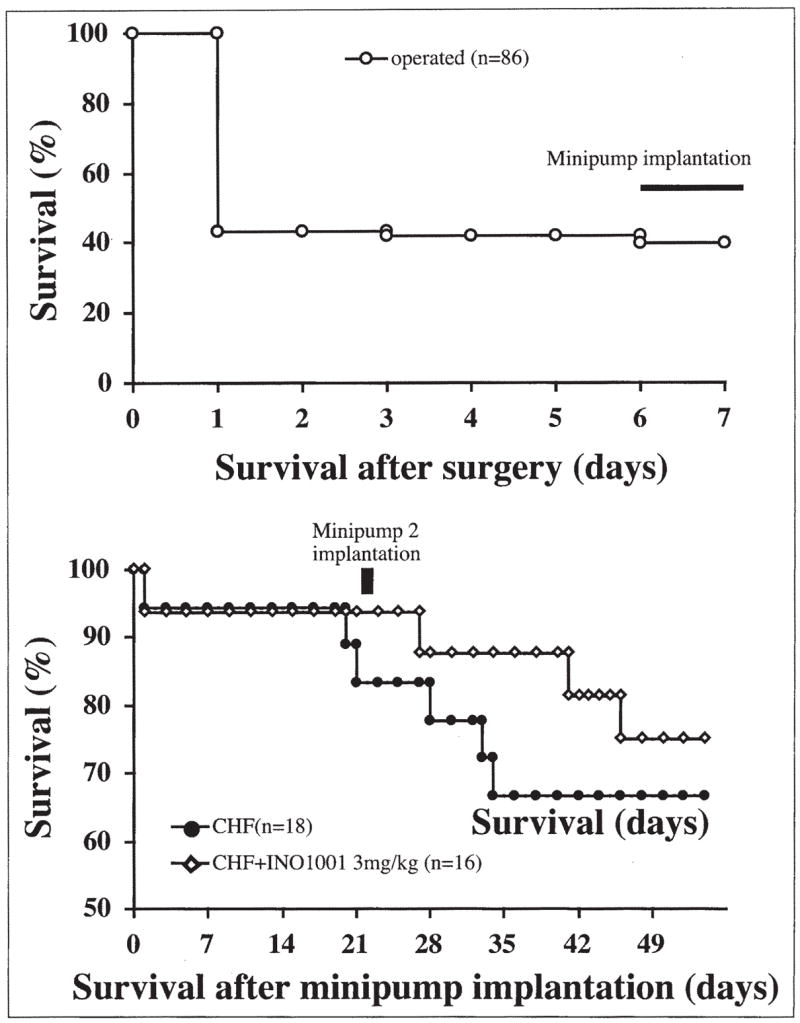
Effect of pharmacological inhibition of PARP on survival in a rat model of chronic heart failure (CHF). Top panel demonstrates mortality prior to randomization of the animals to vehicle and INO-1001 treated groups (n=86 animals). Bottom panel considers surviving animals (n=34) at time of randomization as 100% and shows the effect of INO-1001 on the subsequent mortality.
Figure 2.

Effect of CHF and INO-1001 on left ventricular systolic pressure (LVSP), left ventricular end diastolic pressure (LVEDP), left ventricular +dP/dt, left ventricular −dp/dt in rats. S, sham; CHF, chronic heart failure; S + INO-1001 sham treated with INO-1001 (for 10 weeks); CHF + INO-10001, chronic heart failure treated with PJ34 (for 10 weeks). Results are mean ± SEM of 8–10 experiments in each group. *P<0.05 vs. S; #P<0.05 vs. CHF.
There was no difference in the infarct size among the two groups, consistent with the fact that the start of the PARP inhibition treatment was delayed to a time point subsequent to the coronary ligation. Chronic heart failure led to a significant increase in the weights of right and left ventricles and atria (Fig. 3). Heart failure was also characterized by increased lung weights, indicative of marked pulmonary edema secondary to cardiac decompensation (Fig. 3). Treatment with INO-1001 significantly reduced the heart failure-induced increase in ventricular, atrial and lung weights (Fig. 3). Heart failure was characterized by loss of endothelial function in isolated aortic rings of rats (maximal relaxation to 10 μM acetylcholine was 95±4% in sham group and 30±4% in rats with heart failure) (Fig. 4). Inhibition of PARP with INO-1001 improved vascular function in rats with heart failure (maximal relaxation to 10 μM acetylcholine was 30±4% in heart failure vs. 70±5% in heart failure + INO-1001 groups) (Fig. 4). Contractile response to epinephrine and endothelium-independent relaxant responses to sodium nitroprusside were unchanged in all groups studied (Fig. 4).
Figure 3.
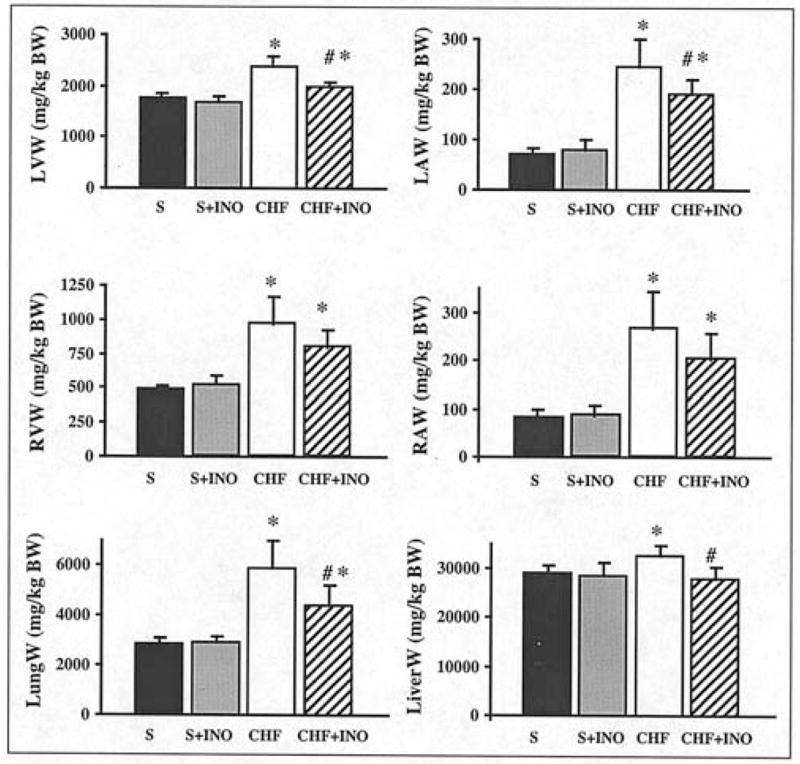
Effect of pharmacological inhibition of PARP on cardiac remodeling and lung weight in rat model of chronic heart failure (CHF) induced by permanent ligation of left anterior descending (LAD) coronary artery. Left ventricular (LVW), right ventricular (RVW), left atrial (LAW), right atrial (RAW) and lung (LungW) weights in sham (S), INO-1001-treated (for 10 weeks) sham (S + INO-1001), chronic heart failure (CHF) and INO-1001-treated (for 10 weeks) chronic heart failure (CHF + INO-1001) rats. Results are mean ± SEM of 8–10 experiments in each group. The weight of each cavity and lung was normalized to the length of the right tibia. *P<0.05 vs. S; #P<0.05 vs. CHF.
Figure 4.

Effect of pharmacological inhibition of PARP on endothelium-dependent and endothelium-independent relaxations of rat thoracic aortae ex vivo. Data shown represent sham (S), INO-1001-treated (for 10 weeks) sham (S + INO-1001), chronic heart failure (CHF) and INO-1001-treated (for 10 weeks) chronic heart failure (CHF + INO-1001) aortic relaxant responses to acetylcholine (top) and sodium nitroprusside (bottom) in rings precontracted with epinephrine. Middle panel shows that the contractile ability of the rings was not affected by CHF or PARP inhibition. Results are mean ± SEM of 8–10 experiments in each group. *P<0.05 vs. S; #P<0.05 vs. CHF.
Effect of INO-1001 on the cardiac dysfunction induced by doxorubicin injection
Doxorubicin induced a severe depression of left ventricular function indicated by rightward shift and decreased amplitude of LV pressure-volume loops (Fig. 5). Doxorubicin decreased heart rate, mean blood pressure, left ventricular systolic pressure, +dP/dt, −dP/dt, stroke volume, stroke work, ejection fraction and cardiac output in BALB/c mice (Fig. 6). DOX also induced increase in left ventricular end-diastolic pressure.
Figure 5.
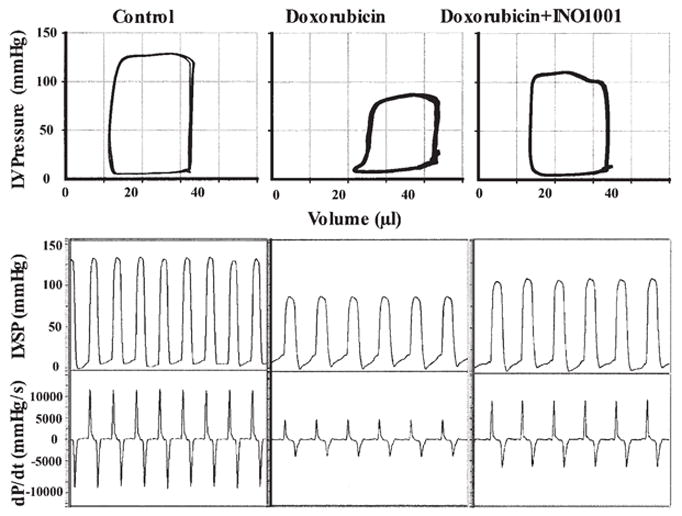
Pharmacological inhibition of PARP improves doxorubicin (DOX)-induced left ventricular dysfunction. Representative PV loops (top part) and left ventricular pressure signal (bottom part) from control, DOX and DOX + INO-1001 treated mice. Please note that the rightward shift of PV loops in doxorubicin treated animals, the decrease of maximal left ventricular pressure, and +dP/dt indicate depressed cardiac contractility. INO-1001 improves baseline contractile function.
Figure 6.
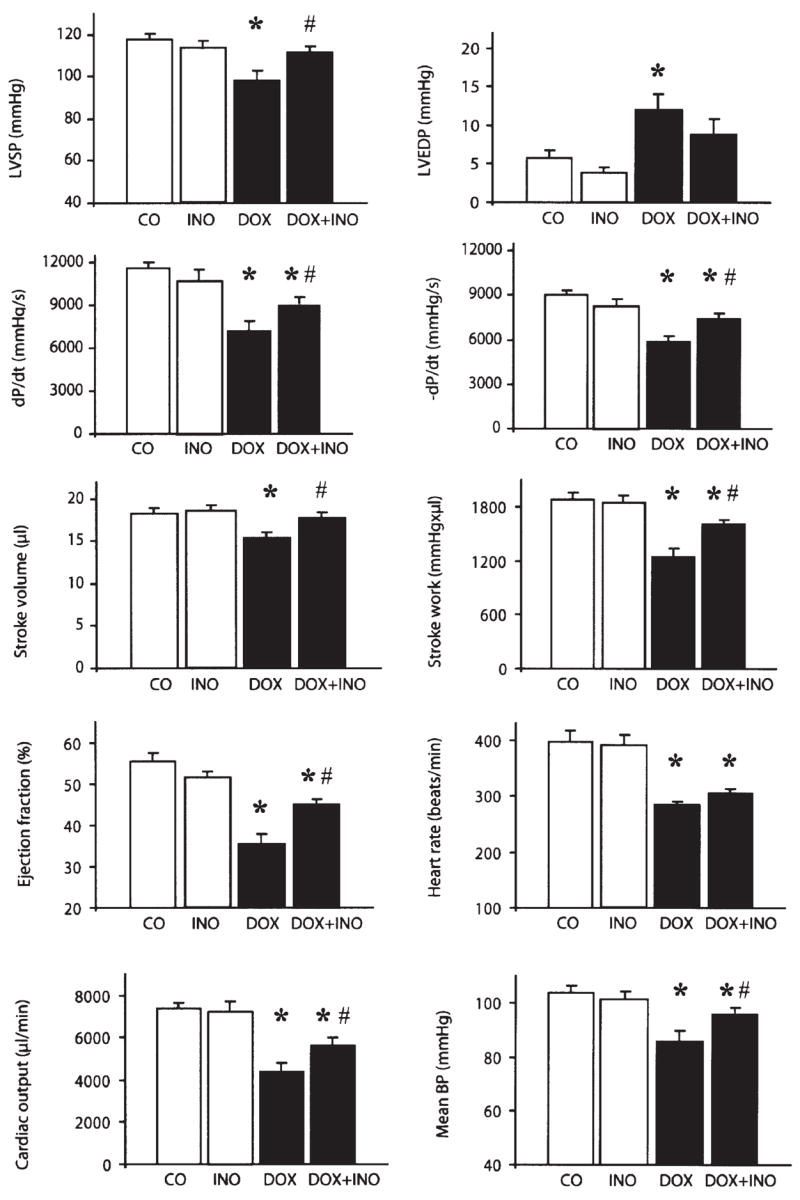
Pharmacological inhibition of PARP improves doxorubicin (DOX)-induced cardiac dysfunction. Effect of DOX and INO-1001 on left ventricular systolic pressure (LVSP), left ventricular end diastolic pressure (LVEDP), left ventricular +dP/dt, left ventricular −dP/dt, stroke volume, stroke work, ejection fraction, mean blood pressure (mean BP), cardiac output and hear rate in BALB/c mice. CO, control; DOX, doxorubicin treated (a single dose of 25 mg/kg); CO + INO-1001, control treated with INO-1001 (3 mg/kg/day s.c.); DOX + INO-1001, doxorubicin (a single dose of 25 mg/kg) and INO-1001 (3 mg/kg/day s.c.) treated. Hemodynamic parameters were measured 5 days after DOX administration. Results are mean ± SEM of 7–10 experiments in each group. *P<0.05 vs. CO; #P<0.05 vs. DOX.
Treatment with INO-1001 significantly attenuated the doxorubicin-induced changes in left ventricular systolic pressure, mean blood pressure, systolic +dP/dt, stroke volume, stroke work, ejection fraction, cardiac output and diastolic −dP/dt (Fig. 6). The PARP inhibitor exerted no significant effects on hemodynamic parameters in control mice (Fig. 6).
The results of the survival experiments are shown in Fig. 7. Treatment with INO-1001 significantly decreased the DOX-induced mortality both in acute and chronic models.
Figure 7.
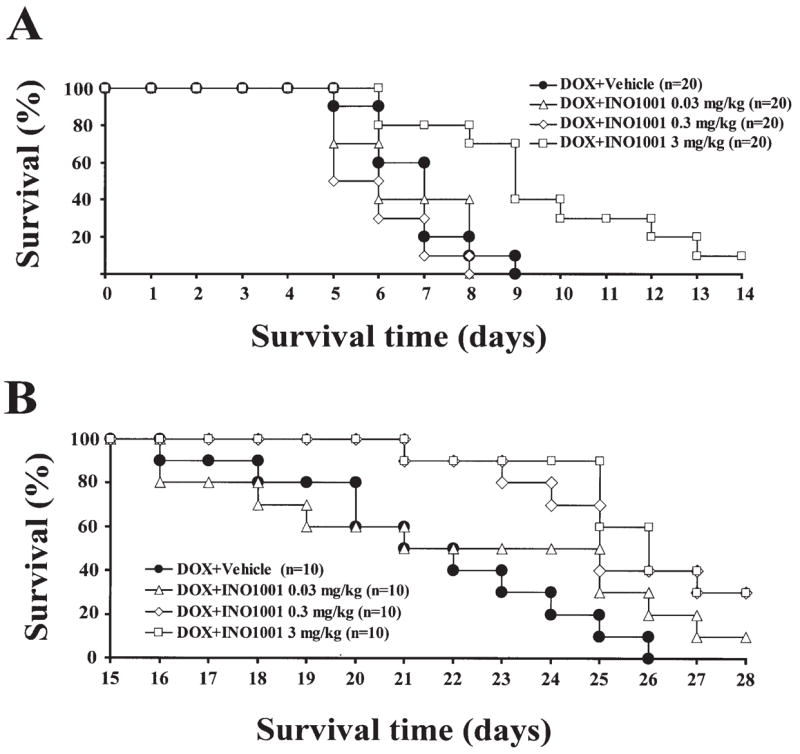
Pharmacological inhibition of PARP with INO-1001 improves survival of mice treated with DOX. Effects of INO-1001 on survival in a DOX-induced acute (A) or chronic (B) heart failure models in mice. (A) Effect of various doses of INO-1001 (0.03, 0.3, and 3 mg/kg/day s.c.) on DOX-induced mortality (25 mg/kg IP) in mice. (B) Effect of various doses of INO-1001 (0.03, 0.3, and 3 mg/kg/day s.c.) on DOX-induced chronic mortality (3 doses of 9 mg/kg IP every 10 days) in mice. Treatment with INO-1001 (3 mg/kg) significantly decreased the DOX-induced mortality.
INO-1001, at the highest dose used (3 mg/kg/day s.c. in minipumps), was tested on tumor growth and on the antineo-plastic activity of DOX, and it failed to affect these parameters, indicating that PARP inhibition with INO-1001 does not interfere with tumor growth and antitumor activity of DOX in the present experimental model (Fig. 8).
Figure 8.
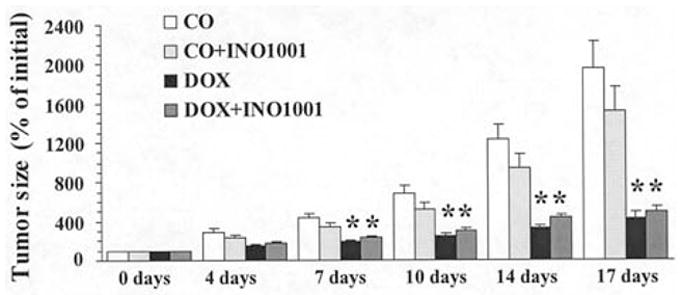
Effects of INO-1001 on mouse breast carcinoma growth and antineoplastic effect of DOX. CO, controls; CO + INO-1001, control treated with INO-1001 (3 mg/kg/day s.c. minipump); DOX, DOX-treated (twice 4 mg/kg/week PO); DOX + INO-1001, treated with DOX (twice 4 mg/kg/week) and INO-1001 (3 mg/kg/day s.c. minipump). Tumor growth was individually followed in all mice, and tumor diameters (x, y, and z) were measured twice a week after initiation of treatments. Tumor size was calculated in mm3 and expressed as percentage of increase over time compared with initial size at start of treatment (day 0). Results are mean ± SEM of 10 mice in each group. *P<0.05 vs CO.
Effect of INO-1001 on the cardiac dysfunction induced by endotoxin
Endotoxin induced a significant decrease in mean BP, LVSP, +dP/dt and −dP/dt at 30 and 60 min following the i.v. injection, and all these parameters were improved by INO-1001 (10 mg/kg i.v.) pretreatment (Fig. 9).
Figure 9.

Pharmacological inhibition of PARP with INO-1001 (10 mg/kg i.v.) improves endotoxin (LPS 15 mg/kg i.v.)-induced acute cardiac dysfunction. Results are mean ± SEM of 9 experiments in each group. *P<0.05 vs. baseline; #P<0.05 vs. LPS.
Discussion
A multitude of new pharmacological approaches have been introduced during the past decade for experimental therapy of heart failure. Although many of these approaches have been successful in animal models and clinical trials, morbidity and mortality from CHF remain high and research into patho-mechanisms of disease and new disease-modifying pharmacological targets continues.
Experimental and clinical studies have suggested an increased production of reactive oxygen and nitrogen species (superoxide, hydrogen peroxide, hydroxyl radical, peroxynitrite) in the failing myocardium (overviewed in refs. 1,2,8). These oxidant and free radical species trigger the activation of a recently emerging pathway of myocardial dysfunction, which involves the activation of the nuclear enzyme PARP by DNA single strand breaks generated by oxidative and nitrosative stress. Evidence for the importance of this pathway has been demonstrated in hearts subjected to regional or global ischemia and reperfusion. The mode of PARP inhibitors’ cardioprotective action involves a conservation of myocardial energetics, as well as a prevention of the up-regulation of various pro-inflammatory pathways (cytokines, adhesion receptors, mononuclear cell infiltration) triggered by ischemia and reperfusion (overviewed in refs. 1,9).
Recent work began to demonstrate the importance of PARP activation in various forms of heart failure. There is evidence for activation of PARP in animal and human hearts with heart failure, and there is evidence for cardioprotective effects of PARP inhibitors of various classes in these models (overviewed in ref. 1; 3–5,10–15).
The PARP inhibitor INO-1001 has emerged in recent years as a clinically applicable drug development candidate PARP inhibitor. Preclinical studies have shown its efficacy in a variety of models of myocardial reperfusion, cardiopulmonary bypass, as well as other models that involve oxidative and nitrosative stress and PARP activation, including stroke and acute lung failure (overviewed in ref. 9; 15–25). INO-1001 has been tested in an advanced aging-associated heart failure model and in a mouse model of aortic banding-induced heart failure, where it reduced myocardial hypertrophy and improved the contractility of the heart (3,4). In the current study, we extended these studies, and tested the effect of the compound in three different models of heart failure, one induced by chronic coronary ligation, one induced by doxorubicin-induced heart failure and one induced by endotoxin. The results of the current studies demonstrate that INO-1001 exerts significant protective effects in all three models tested, thereby further strengthening the view that PARP inhibition may be a valid approach for cardioprotection. Similarly to a previous study with a PARP inhibitor of an earlier class (5), we have also demonstrated remote protective effects (i.e. effects outside the myocardium). The current results obtained with INO-1001 further support the view that the impairment of the cardiac and endothelial function in CHF -at least in the current experimental model - are dependent on PARP activity. Although the exact mechanism of this impairment is not known, it is conceivable that following the myocardial infarction the compensatory increased sympathetic activity, activated renin-angiotensin system and increased workload of the non-infracted regions leads to myocardial hypertrophy, myocardial ischemia/functional hypoxia, increased oxidative stress and PARP activation, which in turn results in vascular dysfunction. In this respect, it is noteworthy that angiotensin II induces PARP-dependent endothelial dysfunction in vitro and in chronic angiotensin-infusion models (25). It is also conceivable that enhanced catecholamine auto-oxidation may lead to reactive species that may induce PARP activation in endothelial cells (although this hypothesis remains to be tested).
In the doxorubicin model there are multiple effector pathways leading to heart failure including oxidative and nitrosative stress, metalloproteinase activation, calcium dyshandling and PARP activation (6,11,12,26). The relationship of these factors is not completely understood at present, but according to a likely scenario, oxidative and nitrosative stress induces/activates matrix metalloproteinases and upregulates and/or catalytically activates PARP. The calcium dyshandling may be related to an interrelated action of some or all the above-listed factors.
PARP inhibitors have been shown to exert anticancer effects in some models (overviewed in refs. 9,27), including an enhancement of doxorubicin-induced cell killing in P53 deficient cell lines in vitro (28). The current study investigated whether PARP inhibition may interfere or affect the anti-cancer effect of doxorubicin in a murine breast cancer line. The results demonstrate that INO-1001 does not interfere with the antitumor effects (even though we failed to demonstrate any enhancement). It is possible that the effect of PARP inhibitors are cell-type selective and future work is needed to test whether PARP inhibitors enhance the effect of doxorubicin in P53 deficient tumor cell lines in vivo.
In conclusion, our study provides evidence that the PARP activation importantly contributes to the pathogenesis of cardiovascular dysfunction in several distinct experimental model of CHF. The current results strengthen the view that PARP inhibition may represent a novel approach for the experimental therapy of CHF.
Acknowledgments
This work was supported by grants from the National Institutes of Health: R01 HL59266 and R01 GM66189, and by the Hungarian Research Fund (OTKA). L.L. is supported by Swiss National Fund grant Nr PPOOB/68882/1.
Abbreviations
- PARP
poly(ADP-ribose) polymerase
- DOX
doxorubicin
- BP
blood pressure
- LVEDP
left ventricular end-diastolic pressure
- LVSP
left ventricular systolic pressure
- +dp/dt
maximal slope of systolic pressure increment
- −dp/dt
maximal slope of diastolic pressure decrement
References
- 1.Pacher P, Schulz R, Liaudet L, Szabo C. Nitrosative stress and pharmacological modulation of heart failure. Trends Pharmacol Sci. 2005;26:302–310. doi: 10.1016/j.tips.2005.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ungvari Z, Gupte SA, Recchia FA, Batkai S, Pacher P. Role of oxidative-nitrosative stress and downstream pathways in various forms of cardiomyopathy and heart failure. Curr Vasc Pharmacol. 2005;3:221–229. doi: 10.2174/1570161054368607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pacher P, Vaslin A, Benko R, Mabley JG, Liaudet L, Hasko G, Marton A, Batkai S, Kollai M, Szabo C. A new, potent poly(ADP-ribose) polymerase inhibitor improves cardiac and vascular dysfunction associated with advanced aging. J Pharmacol Exp Ther. 2004;311:485–491. doi: 10.1124/jpet.104.069658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao CY, Chen M, Zsengeller Z, Li H, Kiss L, Kollai M, Szabo C. Poly(ADP-ribose) polymerase promotes cardiac remodeling, contractile failure, and translocation of apoptosis-inducing factor in a murine experimental model of aortic banding and heart failure. J Pharmacol Exp Ther. 2005;312:891–898. doi: 10.1124/jpet.104.077164. [DOI] [PubMed] [Google Scholar]
- 5.Pacher P, Liaudet L, Mabley J, Komjati K, Szabo C. Pharmacologic inhibition of poly(adenosine diphosphate-ribose) polymerase may represent a novel therapeutic approach in chronic heart failure. J Am Coll Cardiol. 2002;40:1006–1016. doi: 10.1016/s0735-1097(02)02062-4. [DOI] [PubMed] [Google Scholar]
- 6.Pacher P, Liaudet L, Bai P, Mabley JG, Kaminski PM, Virag L, Deb A, Szabo E, Ungvari Z, Wolin MS, Groves JT, Szabo C. Potent metalloporphyrin peroxynitrite decomposition catalyst protects against the development of doxorubicin-induced cardiac dysfunction. Circulation. 2003;107:896–904. doi: 10.1161/01.cir.0000048192.52098.dd. [DOI] [PubMed] [Google Scholar]
- 7.Batkai S, Pacher P, Jarai Z, Wagner JA, Kunos G. Cannabinoid antagonist SR-141716 inhibits endotoxic hypotension by a cardiac mechanism not involving CB1 or CB2 receptors. Am J Physiol. 2004;287:H595–H600. doi: 10.1152/ajpheart.00184.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrari R, Guardigli G, Mele D, Percoco GF, Ceconi C, Curello S. Oxidative stress during myocardial ischaemia and heart failure. Curr Pharm Des. 2004;10:1699–1711. doi: 10.2174/1381612043384718. [DOI] [PubMed] [Google Scholar]
- 9.Jagtap P, Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- 10.Pacher P, Cziraki A, Mabley JG, Liaudet L, Papp L, Szabo C. Role of poly(ADP-ribose) polymerase activation in endotoxin-induced cardiac collapse in rodents. Biochem Pharmacol. 2002;64:1785–1791. doi: 10.1016/s0006-2952(02)01421-1. [DOI] [PubMed] [Google Scholar]
- 11.Szenczi O, Kemecsei P, Holthuijsen MF, van Riel NA, van der Vusse GJ, Pacher P, Szabo C, Kollai M, Ligeti L, Ivanics T. Poly(ADP-ribose) polymerase regulates myocardial calcium handling in doxorubicin-induced heart failure. Biochem Pharmacol. 2005;69:725–732. doi: 10.1016/j.bcp.2004.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pacher P, Liaudet L, Bai P, Virag L, Mabley JG, Hasko G, Szabo C. Activation of poly(ADP-ribose) polymerase contributes to development of doxorubicin-induced heart failure. J Pharmacol Exp Ther. 2002;300:862–867. doi: 10.1124/jpet.300.3.862. [DOI] [PubMed] [Google Scholar]
- 13.Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabo E, Szabo C. The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002;51:514–521. doi: 10.2337/diabetes.51.2.514. [DOI] [PubMed] [Google Scholar]
- 14.Pillai JB, Russell HM, Raman J, Jeevanandam V, Gupta MP. Increased expression of poly(ADP-ribose) polymerase-1 contributes to caspase-independent myocyte cell death during heart failure. Am J Physiol. 2005;288:H486–H496. doi: 10.1152/ajpheart.00437.2004. [DOI] [PubMed] [Google Scholar]
- 15.Xiao CY, Chen M, Zsengeller Z, Szabo C. Poly(ADP-ribose) polymerase contributes to the development of myocardial infarction in diabetic rats and regulates the nuclear translocation of apoptosis-inducing factor. J Pharmacol Exp Ther. 2004;310:498–504. doi: 10.1124/jpet.104.066803. [DOI] [PubMed] [Google Scholar]
- 16.Shimoda K, Murakami K, Enkhbaatar P, Traber LD, Cox RA, Hawkins HK, Schmalstieg FC, Komjati K, Mabley JG, Szabo C, Salzman AL, Traber DL. Effect of poly(ADP ribose) synthetase inhibition on burn and smoke inhalation injury in sheep. Am J Physiol. 2003;285:L240–L249. doi: 10.1152/ajplung.00319.2002. [DOI] [PubMed] [Google Scholar]
- 17.Khan TA, Ruel M, Bianchi C, Voisine P, Komjati K, Szabo C, Sellke FW. Poly(ADP-ribose) polymerase inhibition improves postischemic myocardial function after cardioplegia-cardiopulmonary bypass. J Am Coll Surg. 2003;197:270–277. doi: 10.1016/S1072-7515(03)00538-6. [DOI] [PubMed] [Google Scholar]
- 18.Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, Brownlee M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest. 2003;112:1049–1057. doi: 10.1172/JCI18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Komjati K, Mabley JG, Virag L, Southan GJ, Salzman AL, Szabo C. Poly(ADP-ribose) polymerase inhibition protect neurons and the white matter and regulates the translocation of apoptosis-inducing factor in stroke. Int J Mol Med. 2004;13:373–382. [PubMed] [Google Scholar]
- 20.Murakami K, Enkhbaatar P, Shimoda K, Cox RA, Burke AS, Hawkins HK, Traber LD, Schmalstieg FC, Salzman AL, Mabley JG, Komjati K, Pacher P, Zsengeller Z, Szabo C, Traber DL. Inhibition of poly (ADP-ribose) polymerase attenuates acute lung injury in an ovine model of sepsis. Shock. 2004;21:126–133. doi: 10.1097/01.shk.0000108397.56565.4a. [DOI] [PubMed] [Google Scholar]
- 21.Murthy KG, Xiao CY, Mabley JG, Chen M, Szabo C. Activation of poly(ADP-ribose) polymerase in circulating leukocytes during myocardial infarction. Shock. 2004;21:230–234. doi: 10.1097/01.shk.0000110621.42625.10. [DOI] [PubMed] [Google Scholar]
- 22.Farivar AS, Woolley SM, Fraga CH, Thomas R, Salzman AL, Szabo C, Mulligan MS. Intratracheal poly (ADP) ribose synthetase inhibition ameliorates lung ischemia reperfusion injury. Ann Thorac Surg. 2004;77:1938–1943. doi: 10.1016/j.athoracsur.2003.10.120. [DOI] [PubMed] [Google Scholar]
- 23.Szabo G, Soos P, Mandera S, Heger U, Flechtenmacher C, Seres L, Zsengeller Z, Sack FU, Szabo C, Hagl S. Mesenteric injury after cardiopulmonary bypass: role of poly(adenosine 5′-diphosphate-ribose) polymerase. Crit Care Med. 2004;32:2392–2397. doi: 10.1097/01.ccm.0000148009.48919.6a. [DOI] [PubMed] [Google Scholar]
- 24.Szabo G, Soos P, Mandera S, Heger U, Flechtenmacher C, Bahrle S, Seres L, Cziraki A, Gries A, Zsengeller Z, Vahl CF, Hagl S, Szabo C. INO-1001 a novel poly(ADP-ribose) polymerase (PARP) inhibitor improves cardiac and pulmonary function after crystalloid cardioplegia and extracorporal circulation. Shock. 2004;21:426–432. doi: 10.1097/00024382-200405000-00005. [DOI] [PubMed] [Google Scholar]
- 25.Szabo C, Pacher P, Zsengeller Z, Vaslin A, Komjati K, Benko R, Chen M, Mabley JG, Kollai M. Angiotensin II-mediated endothelial dysfunction: role of poly(ADP-ribose) polymerase activation. Mol Med. 2004;10:28–35. doi: 10.2119/2004-00001.szabo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bai P, Mabley JG, Liaudet L, Virag L, Szabo C, Pacher P. Matrix metalloproteinase activation is an early event in doxorubicin-induced cardiotoxicity. Oncol Rep. 2004;11:505–508. [PubMed] [Google Scholar]
- 27.Masutani M, Nakagama H, Sugimura T. Poly(ADP-ribosyl)ation in relation to cancer and autoimmune disease. Cell Mol Life Sci. 2005;62:769–783. doi: 10.1007/s00018-004-4509-x. [DOI] [PubMed] [Google Scholar]
- 28.Munoz-Gamez JA, Martin-Oliva D, Aguilar-Quesada R, Canuelo A, Nunez MI, Valenzuela MT, Ruiz de Almodovar JM, De Murcia G, Oliver FJ. PARP inhibition sensitizes p53-deficient breast cancer cells to doxorubicin-induced apoptosis. Biochem J. 2005;386:119–125. doi: 10.1042/BJ20040776. [DOI] [PMC free article] [PubMed] [Google Scholar]