

Abstract
The major components of the cartilage extracellular matrix are type II collagen and aggrecan. Type II collagen provides cartilage with its tensile strength, while the water-binding capacity of aggrecan provides compressibility and elasticity. Aggrecan breakdown leads to an increase in proteolytic susceptibility of articular collagen, hence aggrecan may also have a protective effect on type II collagen. Given their role in aggrecan degradation and differing substrate specificity profiles, the pursuit of inhibitors for both aggrecanase 1 [a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS)-4] and aggrecanase 2 (ADAMTS-5) is desirable. We have previously described collagen-model fluorescence resonance energy transfer (FRET) substrates for aggrecan-degrading members of the ADAMTS family. These FRET substrate assays are also fully compatible with multi-well formats. In the present study, a collagen-model FRET substrate has been examined for inhibitor screening of ADAMTS-4. ADAMTS-4 was screened against a small compound library (n = 960) with known pharmacologic activity. Five compounds were identified that inhibited ADAMTS-4 >60% at a concentration of 1 μM. A secondary screen using RP-HPLC was developed and performed for verification of the five potential inhibitors. Ultimately, piceatannol was confirmed as a novel inhibitor of ADAMTS-4, with an IC50 value of 1 μM. Because the collagen-model FRET substrates have distinct conformational features that may interact with protease secondary substrate sites (exosites), non-active site binding inhibitors can be identified via this approach. Selective inhibitors for ADAMTS-4 would allow for a more definitive evaluation of this protease in osteoarthritis, as well as representing a potential next generation in metalloproteinase therapeutics.
Osteoarthritis (OA)1 is an age-related debilitating disease affecting more than 80% of people over the age of 75, and is caused by the destruction of articular cartilage [1]. Extracellular matrix (ECM) proteins make up approximately 90% of the dry weight of human cartilage [2]. The major components of the cartilage ECM are type II collagen and aggrecan. Type II collagen provides cartilage with its tensile strength, while the water-binding capacity of aggrecan provides compressibility and elasticity [3]. Aggrecan breakdown leads to an increase in proteolytic susceptibility of articular collagen, hence aggrecan may also have a protective effect on type II collagen [4]. Cartilage destruction associated with OA has been shown to be due to increased catabolism rather than decreased synthesis [5]. Therefore, the study of enzymes associated with aggrecan proteolysis compliments those of collagenolytic matrix metalloproteinases (MMPs) in reference to OA.
Several members of the a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) family have been found to catalyze the hydrolysis of aggrecan. Although ADAMTS-1, ADAMTS-4, ADAMTS-5, ADAMTS-8, ADAMTS-9, and ADAMTS-15 are all “aggrecanases,” ADAMTS-4 and ADAMTS-5 are the most robust [6] and have been implicated in OA [7; 8; 9; 10]. The products of ADAMTS-4/ADAMTS-5 aggrecan breakdown have been discovered in the synovial fluid of patients with OA [7; 8]. The processing of aggrecan by ADAMTS-4 and ADAMTS-5 may be complimentary, as ADAMTS-5 is responsible for cleavage within the interglobular domain of aggrecan, while ablation of ADAMTS-5 still results in aggrecan processing in the chondroitin sulfate-rich region, presumably by ADAMTS-4 [11]. Given their role in aggrecan degradation and differing substrate specificity profiles, the pursuit of inhibitors for both ADAMTS-4 and ADAMTS-5 is desirable. However, few inhibitors have been described to date for the aggrecanase members of the ADAMTS family [12; 13; 14].
The discovery of aggrecanase inhibitors could be facilitated by high-throughput screening (HTS) methods. Previously described assays for aggrecanases are not particularly convenient for HTS, as all require antibodies and most are discontinuous [15; 16; 17; 18]. A continuous assay method, such as one that utilizes an increase in fluorescence upon substrate hydrolysis, would allow for rapid and convenient evaluation of aggrecanase inhibitors. To develop an improved HTS assay for aggrecanases, we examined fluorescence resonance energy transfer (FRET) collagen-model substrates recently described by our laboratory [19]. More precisely, ADAMTS-4/ADAMTS-5 FRET substrates had been designed to incorporate the aggrecan 1480–1481 cleavage site within a collagen-model structure [19]. The fluorophore/quencher pair was 7-methoxycoumarin (Mca)/2,4-dinitrophenyl (Dnp), where Mca fluorescence was efficiently quenched by the Dnp group in the intact substrate. Substrate conformation had a significant role in ADAMTS-4 specificity, and these substrates interacted with secondary binding sites (exosites) located outside the enzyme catalytic domain [19]. Thus, HTS with collagen-model aggrecanase substrates may allow for the identification of active site and/or exosite-binding inhibitors.
One of the collagen-model aggrecanase substrates, fSSPa [C6-(Gly-Pro-Hyp-Pro-Hyp-Gly)2-Gly-Pro-Hyp-Gly-Thr-Lys(Mca)-Gly-Glu~Leu~Glu~Gly-Arg~Gly-Thr-Lys(Dnp)-Gly-Ile-Ser-(Gly-Pro-Hyp-Pro-Hyp-Gly)2-Gly-Pro-Hyp-NH2], has been utilized here for screening of a compound library (n = 960) against ADAMTS-4 in a 384-well format. Inhibitory compounds were (a) confirmed by dose-dependence analysis and (b) verified using an RP-HPLC based assay (a “secondary” screen). The overall quality of the screen was also examined.
Materials and methods
All standard chemicals were purchased from Fisher (Atlanta, GA). MMP inhibitor III (a homophenylalanine-hydroxamic acid based broad-spectrum reversible inhibitor) was obtained from EMD Biosciences/Calbiochem (San Diego, CA), while piceatannol, (R,R)-cis-diethyltetrahydro-2,8-chrysenediol, (S)-(+)-camptothecin, N-butanoyl-2-(2-methoxy-6H-isoindolo[2,1-a]indole-11-yl)ethanamine [IIK7], and 8-(p-sulfophenyl)theophylline were purchased from Sigma Chemicals (St. Louis, MO). The synthesis, purification, and characterization of fSSPa have been described [19]. ADAMTS-4 (aggrecanase-1) was expressed as previously described [20]. The amount of active ADAMTS-4 was determined by titration with recombinant N-TIMP-3 [21].
FRET Substrate Assays
Initial evaluation of the aggrecanase assay was performed in a 384-well format as follows. fSSPa was prepared as a stock solution in Tris salt buffer (TSB; 50 mM Tris•HCl, pH 7.5, 100 mM NaCl, 10 mM CaCl2, 0.05% Brij-35, 0.02% NaN3). All reagents were brought to 25 °C. TSB was added to each well as needed. Inhibitor was added to each well. Enzyme and substrate were added to final concentrations of 1–20 nM and 2–10 μM, respectively, and the plate was incubated 4–24 h at 25 °C (the plate was sealed or kept in a humidified environment to prevent evaporative losses). Enzyme activity was monitored as a change in fluorescence (end points – beginning points) for each well. Fluorescence was measured on a Molecular Devices SPECTRAmax Gemini EM Dual-Scanning Microplate Spectrofluorometer using λexcitation = 324 nm and λemission = 393 nm.
Library Screening
Inhibitor screening was performed in 384-well format with a subset (n = 960) of the Library of Pharmacologically Active Compounds1280 (LOPAC1280) (Sigma, St. Louis, MO, catalog # LO1280) as follows. First, 20 μL per well of 40 nM ADAMTS-4 was added to test wells using a peristaltic bulk dispenser (Wellmate, Matrix Technologies Corporation, Hudson, NH) followed by addition of 18 nL of compound or controls to the test wells via a 384-well pintool (Biomek FX, Beckman Coulter Inc., Fullerton, CA). Then 20 μL of 20 μM substrate was added to the appropriate wells. The assay was run in a 40 μL total volume, resulting in a final test compound concentration of 1 μM. Plates were spun at 200 × g for 1 min, then read immediately on a Spectramax M2 (Molecular Devices, Sunnyvale, CA) at λexcitation = 324 nm and λemission = 393 nm, followed by an 8 h read. Data was analyzed via HTS database software (MDL Assay Explorer, Elsevier, San Ramon, CA).
Primary Screen Data Normalization, Quality Control, and Hit Selection Criteria
Prior to normalization of screening data, the change in relative fluorescence units (ΔRFU) for each well was determined by the following equation:
where (I393nm)X h is the intensity measured at λemission = 393 nm from the test well at the respective time point. The percent inhibition for each well was then calculated as follows:
where the “high control” was measured from wells containing an IC100 of MMP inhibitor III (750 nM) and the “low control” was measured from wells containing DMSO only.
To determine the quality of the screen results, three parameters were calculated on a per-plate basis: (a) the signal-to-background ratio (S/B); (b) the coefficient for variation [CV; CV = (standard deviation/mean) × 100] for all compound test wells; and (c) Z- or Z′-factor [Z′-factor = 1 − [[3 × (σp + σn)]/(μp − μn)], where σ is the standard deviation and μ the mean for positive (p) and negative (n) controls] [22]. A mathematical algorithm was used to determine nominally inhibitory compounds (“hits”) in the primary screen. Two values were calculated: (a) the average percent inhibition of all compounds tested; and (b) three times their standard deviation. The sum of these two values was used as a cutoff parameter, i.e., any compound that exhibited greater % inhibition than the cutoff parameter was declared active [23].
Ki Determination
Varying concentrations of inhibitor diluted in TSB were added to the plate. The ADAMTS-4 and fSSPa were added. Since Ki(app) = Ki (1 + [S]/KM), the peptide was used at <5–10% KM. This insures that Ki(app) was a close approximation of Ki. Fluorescence was monitored for a time in which <10% of the substrate was hydrolyzed, thus the increases in fluorescence versus time was linear. Initial velocities (Vi) were expressed as relative fluorescence/time and monitored with increasing concentration of inhibitor. Ki values were calculated using SigmaPlot software. Data was collected at enzyme concentrations <10Ki.
Secondary Screen
Stock solutions of fSSPa were prepared at 100 μM concentration in TSB. Inhibitors were prepared as 2 mM solutions in DMSO and then further diluted with TSB. ADAMTS-4 assays were conducted in TSB by incubating 10 μM substrate with 10 nM enzyme for 24 h in the presence and absence of inhibitors. Final concentrations of inhibitors were 0.1, 1.0, and 10 μM. Fluorescence readings (λexcitation = 324 nm and λemission = 393 nm) were obtained at 0, 5, and 24 h. The change in relative fluorescence units (ΔRFU) was calculated by ΔRFU = RFU (S + E + I)24 h − RFU (S + E + I)0 h. After the final reading at 24 h, the reaction solution was analyzed by RP-HPLC. Analytical RP-HPLC was performed on a Hewlett Packard 1100 Liquid Chromatograph equipped with a Vydac 208TP5415 protein and peptide C8 column (15–10 μm particle size, 300 Å pore size, 150 × 4.1 mm). Eluants were 0.1% TFA in water (A) and 0.1% TFA in acetonitrile (B). The elution gradient was 0–50% B in 20 min with a flow of 1 ml/min. Detection was at λ = 220, 324, and 363 nm. Reaction yields in the presence of inhibitors were evaluated by the integration of the HPLC peaks formed compared to the enzyme reaction without inhibitor present. Integrations were averaged from two injections. Product identification was achieved by MALDI-TOF-MS on an ABD DE-STR Voyager mass spectrometer using α-cyano-4-hydroxycinnamic acid matrix.
Results
Screening of ADAMTS-4 Inhibition by MMP Inhibitor III
We have previously described two FRET substrates, fTHPa and fSSPa, for ADAMTS-4 and ADAMTS-5, and used these substrates to quantify ADAMTS-4 activity [19; 24]. For the present study, we have examined fSSPa in a screening assay for ADAMTS-4. Recombinant ADAMTS-4-2 was the enzyme variant screened. ADAMTS-4-2 contains a typical reprolysin-type zinc-binding motif, followed by a disintegrin-like domain, a single thrombospondin type I module, and the Cys-rich domain (Figure 1). This form is found during C-terminal processing of ADAMTS-4 [20; 25], and has reasonable activity towards fSSPa [19]. Inhibition of ADAMTS-4-2 by MMP inhibitor III was examined over an inhibitor range of 1.6–5000 nM, and produced a dose-dependent response with Ki = 187.5 ± 0.02 nM (Figure 2). At inhibitor concentrations near the Ki value, the signal-to-background ratio was 26.2 (>5 is desirable) and the CV was 4% (<10% is desirable). Z′ (Z-factor) was 0.882 (>0.5 is desirable). Thus, the fSSPa assay, in 384-well format, was of sufficient quality to proceed with a more robust screening approach.
Figure 1.

Domain structures of ADAMTS-4 and ADAMTS-5. The catalytic domain contains a typical reprolysin-type zinc-binding motif, and is followed by a disintegrin-like domain, a single thrombospondin type I module, and a Cys-rich domain. The C-terminal portion of the proteins consists of a spacer domain, which shows little similarity between ADAMTS family members or other protein domain structures [49]. ADAMTS-5 has an additional thrombospondin type I repeat after the spacer domain [49].
Figure 2.

Inhibition of ADAMTS-4-2 by MMP inhibitor III. The change in relative fluorescence units (ΔRFU) for 10 nM ADAMTS-4-2 hydrolysis of 10 μM fSSPa was monitored over an MMP inhibitor III concentration range of 1 nM to 10μM as described in Materials and Methods. Assays were performed in triplicate; bars indicate standard deviations.
Screening a Library to Identify Inhibitors of ADAMTS-4
ADAMTS-4 underwent screening using a 960 compound library (Figure 3). The library contained small organic compounds of known pharmacologic activity, such as interaction with G protein-coupled receptors, designed to target various pathways of therapeutic interest, including cell signaling pathways, apoptotic pathways, and several different enzyme classes [26]. Primary actives for ADAMTS-4 were selected based upon a standard “average plus 3 standard deviations” inhibition cutoff algorithm, described in detail in the Materials and Methods section. This resulted in a cut-off of >59.5% inhibition. Five compounds (Figure 4) inhibited ADAMTS-4 at this level: (a) piceatannol [(E)-4-[2-(3,5-dihydroxyphenyl)ethenyl]1,2-benzenediol] (100% inhibition); (b) (R,R)-cis-diethyl tetrahydro-2,8-chrysenediol (100% inhibition); (c) (S)-(+)-camptothecin (77% inhibition); (d) N-butanoyl 2-(9-methoxy-6H-isoindolo[2,1-a]indol-11-yl)ethanamine (70% inhibition); and (e) 8-(p-sulfophenyl)theophylline (69% inhibition). Dose-dependent inhibition of ADAMTS-4-2 was then compared for piceatannol versus (S)-(+)-camptothecin. Both compounds were found to inhibit ADAMTS-4-2, with IC50 values of 1.0 and 4.0 μM for piceatannol and (S)-(+)-camptothecin, respectively (Figure 5). These results confirmed the behavior of piceatannol and (S)-(+)-camptothecin in the screen, where the former compound was found to be more active than the latter.
Figure 3.
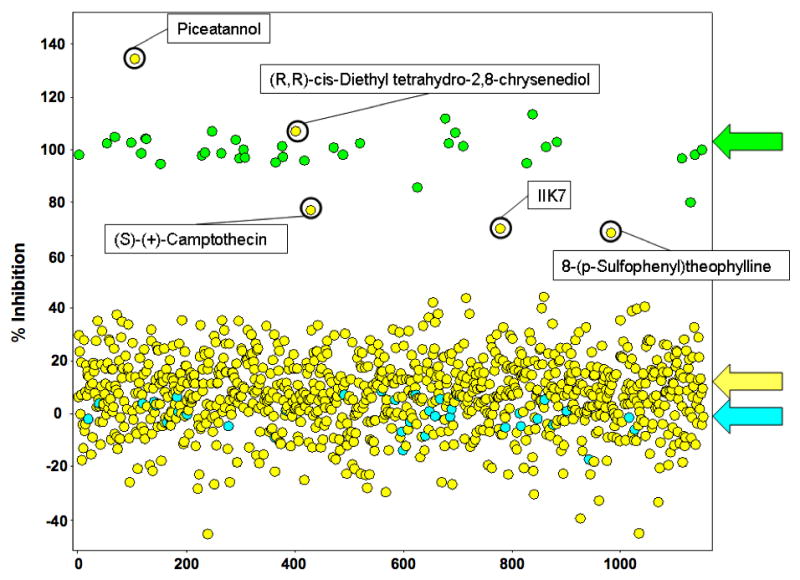
Results of the 384-well screen for inhibitors of ADAMTS-4-2. Displayed are the results of test compounds (n = 960; yellow arrow) as well as positive (100% inhibition; green arrow) and negative (0% inhibition; blue arrow) controls. Circled are the inhibition results for the five active compounds (“hits”) identified in the screen.
Figure 4.

Structures of (a) piceatannol [(E)-4-[2-(3,5-dihydroxyphenyl)ethenyl] 1,2-benzenediol; trans-3,3′,4,5′-tetrahydroxystilbene]; (b) (R,R)-cis-diethyltetrahydro-2,8-chrysenediol; (c) (S)-(+)-camptothecin (4-ethyl-4-hydroxy-1H-pyrano[3′,4′:6,7] indolizino[1,2-b] quinoline-3,14(4H,12H)dione); (d) N-butanoyl-2-(2-methoxy-6H-isoindolo[2,1-a] indole-11-yl)ethanamine [IIK7]; and (e) 8-(p-sulfophenyl)theophylline.
Figure 5.

Inhibition of ADAMTS-4-2 by piceatannol (closed triangles) or (S)-(+)-camptothecin (closed circles). The change in relative fluorescence units (ΔRFU) for 10 nM ADAMTS-4-2 hydrolysis of 10 μM fSSPa was monitored over an inhibitor concentration range of 1 nM to 10 μM as described in Materials and Methods. IC50 = 1.0 and 4.0 μM for piceatannol and (S)-(+)-camptothecin, respectively. Assays were performed in triplicate; bars indicate standard deviations.
Secondary Screen of ADAMTS-4 Inhibitors
An RP-HPLC based secondary screen was performed for the 5 putative ADAMTS-4 inhibitors. The assay was performed in 384-well plate format and fluorescence intensity of the wells was read at 0, 5, and 24 h. After 24 h the samples were analyzed by RP-HPLC and the yield of products was determined by integration of the peaks. The results obtained by the two methods were compared. Piceatanol showed inhibition by both methods (Figures 6 and 7). However, the other four compounds did not inhibit ADAMTS-4 activity when product formation was analyzed by RP-HPLC (Figure 8).
Figure 6.

RP-HPLC elution profiles of ADAMTS-4-2 hydrolysis of fSSPa in the absence (blue) and presence (magenta) of picetannol. ADAMTS-4-2 (10 nM) hydrolysis of 10 μM fSSPa was examined after 24 h following addition of 0 or 10 μM piceatannol as described in Materials and Methods. The intact substrate eluted at 14.667 min, while cleavage products were observed at 12.069, 12.361, and 13.410 min. The identities of the cleavage products have been described previously [19].
Figure 7.

Inhibition of ADAMTS-4-2 by piceatannol, as monitored by RP-HPLC and fluorescence. The change in RP-HPLC peak areas (closed circles) or relative fluorescence units (ΔRFU) (closed triangles) for 10 nM ADAMTS-4-2 hydrolysis of 10 μM fSSPa was monitored over a piceatannol concentration range of 0.1 to 10 μM as described in Materials and Methods. Assays were performed in duplicate.
Figure 8.

Inhibition of ADAMTS-4-2 by piceatannol (open circles), (R,R)-cis-diethyltetrahydro-2,8-chrysenediol (closed squares), (S)-(+)-camptothecin (closed diamonds), N-butanoyl-2-(2-methoxy-6H-isoindolo[2,1-a] indole-11-yl)ethanamine (closed circles), and 8-(p-sulfophenyl)theophylline (closed triangles), as monitored by RP-HPLC. The change in RP-HPLC peak areas for 10 nM ADAMTS-4-3 hydrolysis of 10 μM fSSPa was monitored over an inhibitor concentration range of 0.1 to 10 μM as described in Materials and Methods. Assays were performed in duplicate.
Quality of the Inhibitor Screen Results
The quality of the screen was examined on a per-plate basis utilizing the ΔRFU value for wells that contained positive (high inhibition) and negative (low inhibition) controls. Over 5 plates assayed, Z′-factor was 0.64 ± 0.5 (>0.5 is desirable), the signal-to-background (S/B) ratio was 2.6 ± 0.1 (>5 is desirable), and the average CV was 4.32% (<10% is desirable). In an attempt to improve the S/B ratio, changes in velocity (ΔRFU) were examined as a function of enzyme concentration for different plate assay volumes (10, 20, 40, or 80 μL). RFUs were measured at 0 and 18 h. For all volumes, a linear relationship was observed for ΔRFU versus [ADAMTS-4] (Figure 9), and thus there were no significant fluorescence quenching effects. Interestingly, greater signal change was seen for larger assay volumes; since the same microtiter plates were used for all experiments, this increase was attributed to the longer sample pathlength being interrogated by the plate reader.
Figure 9.

Hydrolysis of fSSPa as a function of ADAMTS-4-2 concentration in different assay volumes. The change in relative fluorescence units (ΔRFU) for ADAMTS-4-2 hydrolysis of 10 μM fSSPa was monitored using total reaction volumes of 10 (closed circles), 20 (closed triangles), 40 (closed squares), or 80 (closed diamonds) μL as described in Materials and Methods. Assays were performed in triplicate; bars indicate standard deviations.
Discussion
The use of FRET substrates for HTS requires several design considerations. First, the fluorophore should have a high quantum yield, yet be efficiently quenched by a resonance energy transfer mechanism. Substantial quantum yields are found for Mca derivatives, which can be readily incorporated into peptides [27]. Mca fluorescence is also efficiently quenched by the Dnp group [28]. Second, one would like to incorporate the fluorophore and quencher internally in the peptide sequence. Since efficient quenching of the Mca fluorescence occurs when the Dnp group is within 8 residues, internal incorporation of these groups would allow for quenching regardless of substrate size. Third, the substrate should be sufficiently soluble such that assays can be performed in an aqueous environment at 25 or 37 °C. The incorporation of 4-5 Gly, Pro, and Hyp residues on the N- and C-termini of the substrate increase the solubility of these molecules [19]. Our collagen-model FRET substrates satisfy the above three criteria, making them applicable for HTS.
The present screening protocol initially profiled 960 compounds at a 1 μM concentration for each compound. Hits were selected based upon an average of % inhibition for all samples plus 3 standard deviations, which turned out to be >59.5% inhibition. The quality of a hit was evaluated by dose-dependence. Finally, a secondary screen was performed to eliminate compounds that inhibit non-specifically (e.g., interact with the substrate) or interfere with fluorescence of the Mca-containing peptide fragment. Ultimately, piceatannol [(E)-4-[2-(3,5-dihydroxyphenyl)ethenyl]1,2-benzenediol] (Figure 4a) was revealed as a novel inhibitor of ADAMTS-4. Piceatannol, a red wine polyphenolic compound, has been most prominently recognized as a non-receptor Tyr kinase inhibitor [29]. Unlike most aggrecanase inhibitors [12; 13], this compound does not possess a zinc-chelating hydroxamic acid. Piceatannol has structural similarities to (−)-epigallocatechin gallate (EGCG), which inhibits ADAMTS-4 and ADAMTS-5 with IC50 values of 100–150 nM and ADAMTS-1 with an IC50 value of 200–250 nM [14]. EGCG inhibition of aggrecanases is not due to zinc chelation [14]. Future experiments will evaluate the mode of ADAMTS-4 inhibition by piceatannol and determine the key functional groups. It should be noted that piceatannol itself has a wide range of activities, including downregulation of the transcription factor STAT-3 [30] which has been associated with cytotoxic effects primarily in tumor cells [31] but potentially in other cell types as well. Piceatannol also stimulates osteogenesis through effects on bone morphogenetic protein-2 production [32] and upregulates endothelial heme oxygenase-1 expression [33].
Four false positives were observed in the screening protocol. One problem with FRET-based assays is that compounds being screened may have absorption maxima that coincide with the emission wavelength of the fluorophore. This results in quenching of fluorescence by the compound and an incorrect designation as an inhibitor. Alternatively, fluorescent compounds that have similar excitation and emission maxima as the fluorophore will fluoresce during the assay, and may not be recognized as inhibitors. As noted by George et al. for HTS of MMP-3, the CyDye pair of Cy3/Cy5Q was much less susceptible to false results than the Mca/Dnp pair, as <1% of a random library were auto-fluorescent at Cy3 wavelengths while >10% of the same library could not be screened using Mca/Dnp due to auto-fluorescence and interference [34]. One could create complimentary substrates differing only by their respective fluorophore/quencher pairs, and use these different substrates to screen potential inhibitors. Compounds would need to exhibit activity in both assays to be classified as inhibitor hits, and thus those that interfered with fluorescence or quenching for one substrate would be inactive in the other assay. The ADAMTS-4 substrate fSSPa could be easily modified to incorporate other donor/quencher pairs, and we are presently examining these options. However, it should also be noted that the use of 6-carboxyfluorescein and Qsy-9 for an 18 residue ADAMTS-4 FRET substrate lead to a significant inner filter effect and subsequent difficulties in kinetic evaluation of the substrate, limiting its use as a HTS tool [35].
For the screening approach using fSSPa, the CV and more importantly Z′-factor [22] were found to be acceptable. The S/B ratio was less than desirable. Since longer pathlengths resulted in improved S/B ratios (Figure 9), the S/B may be improved by either increasing the assay volume in the existing plate format or running the assay in different plate types that have a higher well height/volume aspect, in 384- or even 1536-well format. Longer pathlengths would then be obtained with possibly lower reagent volumes, conserving assay reagents in the process. These applications will be studied for future full-scale HTS endeavors.
The present ADAMTS-4 screening assay is convenient, in that it is continuous and requires fluorescent monitoring of substrate hydrolysis. Prior HTS assays for aggrecanases utilized aggrecan, the aggrecan interglobular domain (IGD), or a 41 amino acid peptide that includes the aggrecanase cleavage site in the aggrecan IGD [15; 16; 17; 18]. None are as convenient as the fluorogenic assay described herein, as all require antibodies and most are discontinuous. For example, the assay described by Thomas et al. utilizes cleavage of the aggrecan IGD between Glu373 and Ala374 to reveal a neoepitope, which is bound by an antibody to the neoepitope followed by a fluorescent anti-IgG binding to the neoepitope antibody [18]. The optimal conditions utilized 0.25 nM enzyme (ADAMTS-4 ~40 kDa form) and 0.25 μM substrate. Miller et al. used the same neoepitope approach (except that the secondary antibody was HRP-conjugated) to monitor hydrolysis of the 41 residue aggrecan sequence described above [15]. The full-length peptide was required to detect activity. Will et al. used the same neoepitope/secondary antibody approach as Miller et al. for monitoring ADAMTS-1, ADAMTS-4, and ADAMTS-5 (~41 kDa forms of each) hydrolysis of the aggrecan IGD and a site-specific mutated IGD [17]. Lastly, Peppard et al. developed an aggrecanase assay based on antibodies to aggrecan chondroitin and keratin sulfate [16]. The antibodies were linked to beads, which generated a fluorescent signal via luminescent oxygen signaling when intact aggrecan was present. Hydrolysis of aggrecan resulted in decreased fluorescence. This approach was used for HTS of 400,000 compounds against ADAMTS-4. Although the HTS assay was of high quality, the assay technology is proprietary and prone to photobleaching. Further, the identity and confirmation of any hits from that screening effort was not reported.
ADAMTS-4 has modest activity towards fSSPa, with KM = 279.0 μM and kcat = 0.0720 sec−1 [19]. In the present screening study, [S] = 10 μM, resulting in [S]/KM = 0.036. These conditions favor the discovery of competitive inhibitors [36]. However, our other collagen-model aggrecanase substrate, fTHPa, is hydrolyzed by ADAMTS-4 with KM = 47.1 μM and kcat = 0.0120 sec−1 [19]. The lower KM value of fTHPa would allow for screening at [S]/KM ~ 1, representing near balanced assay conditions that allows for equivalent evaluation of all inhibition mechanisms [36]. For the 41 residue aggrecan IGD peptide substrate, with aggrecanase from bovine cartilage culture medium, KM = 480 ± 83 μM and Vmax = 10.9 ± 1.2 pmol product/h/μg total protein [15]. Thus, due to the high KM value, the bias towards competitive inhibitors is great when screening with this substrate. Conversely, KM was < 20 nM, while Vmax = 10.3 ± 5.1 and 151.5 ± 93.5 nmol substrate/min•mg for aggrecanase hydrolysis of wild type and mutant IGDs, respectively [17]. In this case, 1.5 nM enzyme and 0.1 μM substrate could be used for screening. Another group found optimal conditions for screening ADAMTS-4 (~40 kDa form) with the aggrecan IGD to be 0.25 nM enzyme and 0.25 μM substrate [18]. Given the low KM values observed during aggrecanase hydrolysis of the aggrecan IGD, [S] will be much greater than KM during screening, favoring the discovery of uncompetitive inhibitors [36]. In fact, when [S] ≫ KM, competitive inhibition can be completely overcome due to the competition between the substrate and inhibitor [37].
HTS for MMPs has been previously established using synthetic FRET substrates with Mca as fluorophore and Dnp as quencher [38; 39; 40; 41; 42]. We have demonstrated that rapid inhibitor screening for ADAMTS family members using our collagen-model FRET substrates can be achieved. The collagen-model substrates have distinct conformational features that interact with secondary binding sites (exosites) found within ADAMTSs [19]. Thus, use of substrates such as fSSPa might allow for identification of ADAMTS-4 exosite inhibitors. Exosite inhibitors could be covalently linked to active site inhibitors, creating high affinity and selective lead compounds. Exosites/allosteric sites have been shown to represent unique opportunities for the design of selective inhibitors [43; 44; 45]. Recently described ADAMTS-4 FRET substrates obtained from phage display screening [35; 46] are most likely too small to use for identifying exosite inhibitors [47]. In contrast, the 73-residue ADAMTS-13 FRET substrate, based on the ADAMTS-13 substrate von Willebrand factor, may be useful for screening exosite inhibitors [48]. Selective inhibitors for ADAMTS-4 and ADAMTS-5, as well as MMP-13, would allow for a more definitive evaluation of these proteases in OA, as well as representing a potential next generation in metalloproteinase therapeutics.
Acknowledgments
We gratefully acknowledge support of this work from the National Institutes of Health (MH 078948 and CA 98799 to G.B.F., AR 40994 to H.N.), the Wellcome Trust (reference number 057508 to H.N.), a Glenn/American Federation for Aging Research (AFAR) Scholarship (to J.L.L.-F.), and the FAU Center of Excellence in Biomedical and Marine Biotechnology. The National Institutes of Health Molecular Library Screening Center Network (5U54MH074404, Professor H. Rosen, Principal Investigator) subsidized the research efforts of P.C., T.S., and P.H.
Footnotes
Abbreviations: ADAMTS, a disintegrin and metalloproteinase with thrombospondin motifs; Dnp, 2,4-dinitrophenyl; ECM, extracellular matrix; Fmoc, 9-fluorenylmethoxycarbonyl; fSSP, fluorogenic single-stranded peptide; fTHP, fluorogenic triple-helical peptide; HTS, high-throughput screening; Hyp, 4-hydroxy-L-proline; LOPAC, Library of Pharmacologically Active Compounds; MALDI-TOF-MS, matrix-assisted laser desorption ionization time-of-flight mass spectrometry; Mca, (7-methoxycoumarin-4-yl)acetyl; MMP, matrix metalloproteinase; OA, osteoarthritis; RP-HPLC, reversed-phase high-performance liquid chromatography; TFA, trifluoroacetic acid; THP, triple-helical peptide; TIMP, tissue inhibitor of metalloproteinases; TSB, Tris salt buffer.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Felson DT. The epidemiology of OA: Results from the Framingham OA study. Seminars Arthritis Rheum. 1990;20:42–50. doi: 10.1016/0049-0172(90)90046-i. [DOI] [PubMed] [Google Scholar]
- 2.Nagase H, Kashiwagi M. Aggrecanases and cartilage matrix degradation. Arthritis Res Ther. 2003;5:94–103. doi: 10.1186/ar630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knudson CB, Knudson W. Cartilage proteoglycans. Seminars Cell Dev Biol. 2001;12:69–78. doi: 10.1006/scdb.2000.0243. [DOI] [PubMed] [Google Scholar]
- 4.Pratta M, Yao W, Decicco C, Tortorella MD, Liu RW, Copeland RA, Magolda R, Newton RC, Trzaskos JM, Arner EC. Aggrecan protects cartilage collagen from proteolytic cleavage. J Biol Chem. 2003;278:45539–45545. doi: 10.1074/jbc.M303737200. [DOI] [PubMed] [Google Scholar]
- 5.Wang J, Verdonk P, Elewaut D, Yveys EM, Verbruggen G. Homeostasis of the ECM of normal and OA human articular cartilage chondrocytes in vitro. Osteoarthritis Cartilage. 2003;11:801–809. doi: 10.1016/s1063-4584(03)00168-7. [DOI] [PubMed] [Google Scholar]
- 6.Porter S, Clark IM, Kevorkian L, Edwards DR. The ADAMTS metalloproteinases. Biochem J. 2005;386:15–27. doi: 10.1042/BJ20040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sandy JD, Flannery CR, Neame PJ, Lohmander LS. The structure of aggrecan fragments in human synovial fluid: Evidence for the involvement in osteoarthritis of a novel proteinase which cleaves the Glu 373-Ala 374 bond of the interglobular domain. J Clin Invest. 1992;89:1512–1516. doi: 10.1172/JCI115742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lohmander LS, Neame PJ, Sandy JD. The structure of aggrecan fragments in human synovial fluid: Evidence that aggrecanase mediates cartilage degradation in inflammatory joint disease, joint injury, and osteoarthritis. Arthritis Rheum. 1993;36:1214–1222. doi: 10.1002/art.1780360906. [DOI] [PubMed] [Google Scholar]
- 9.Stanton H, Rogerson FM, East CJ, Golub SB, Lawlor KE, Meeker CT, Little CB, Last K, Farmer PJ, Campbell IK, Fourie AM, Fossang AJ. ADAMTS4 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature. 2005;434:648–652. doi: 10.1038/nature03417. [DOI] [PubMed] [Google Scholar]
- 10.Song RH, Tortorella MD, Malfait AM, Alston JT, Yang Z, Arner EC, Griggs DW. Aggrecan degradation in human articular cartilage explants is mediated by both ADAMTS-4 and ADAMTS-5. Arthritis Rheum. 2007;56:575–585. doi: 10.1002/art.22334. [DOI] [PubMed] [Google Scholar]
- 11.East CJ, Stanton H, Golub SB, Rogerson FM, Fosang AJ. ADAMTS-5 deficiency does not block aggrecanolysis at preferred cleavage sites in the chondroitin sulfate-rich region of aggrecan. J Biol Chem. 2007;282:8632–8640. doi: 10.1074/jbc.M605750200. [DOI] [PubMed] [Google Scholar]
- 12.Yao W, Wasserman ZR, Chao M, Reddy G, Shi E, Liu RQ, Covington MB, Arner EC, Pratta MA, Tortorella M, Magolda RL, Newton R, Qian M, Ribadeneira MD, Christ D, Wexler RR, Decicco CP. Design and synthesis of a series of (2R)-N4-hydroxy-2-(3-hydroxybenzyl)-N1-[(1S,2R)-2-hydroxy-2,3-dihydro-1H-inden-1-yl] butanediamide derivatives as potent, selective, and orally bioavailable aggrecanase inhibitors. J Med Chem. 2001;44:3347–3350. doi: 10.1021/jm015533c. [DOI] [PubMed] [Google Scholar]
- 13.Yao W, Chao M, Wasserman ZR, Liu RQ, Covington MB, Newton R, Christ D, Wexler RR, Decicco CP. Potent P1’ biphenylmethyl substituted aggrecanase inhibitors. Bioorg Med Chem Lett. 2002;12:101–104. doi: 10.1016/s0960-894x(01)00704-1. [DOI] [PubMed] [Google Scholar]
- 14.Vankemmelbeke MN, Jones GC, Fowles C, Ilic MZ, Handley CJ, Day AJ, Knight CG, Mort JS, Buttle DJ. Selective inhibition of ADAMTS-1, -4 and -5 by catechin gallate esters. Eur J Biochem. 2003;270:2394–2403. doi: 10.1046/j.1432-1033.2003.03607.x. [DOI] [PubMed] [Google Scholar]
- 15.Miller JA, Liu RQ, Davis GL, Pratta MA, Trzaskos JM, Copeland RA. A microplate assay specific for the enzyme aggrecanase. Anal Biochem. 2003;314:260–265. doi: 10.1016/s0003-2697(02)00638-3. [DOI] [PubMed] [Google Scholar]
- 16.Peppard J, Glickman F, He Y, Hu SI, Doughty J, Goldberg R. Development of a high-throughput screening assay for inhibitors of aggrecan cleavage using luminescent oxygen channelling (AlphaScreen) J Biomol Screen. 2003;8:149–156. doi: 10.1177/1087057103252308. [DOI] [PubMed] [Google Scholar]
- 17.Will H, Dettloff M, Bendzko P, Sveshnikov P. A quantitative assay for aggrecanase activity. J Biomol Tech. 2005;16:459–472. [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas M, Sabatini M, Bensaude F, Mignard B, Ortuno JC, Caron I, Boutin JA, Ferry G. A microplate assay for the screening of ADAMTS-4 inhibitors. Matrix Biol. 2006;25:261–267. doi: 10.1016/j.matbio.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 19.Lauer-Fields JL, Sritharan T, Kashiwagi M, Nagase H, Fields GB. Substrate Conformation Modulates Aggrecanase (ADAMTS-4) Affinity and Sequence Specificity: Suggestion of a Common Topological Specificity of Functionally Diverse Proteases. J Biol Chem. 2007;282:142–150. doi: 10.1074/jbc.M605236200. [DOI] [PubMed] [Google Scholar]
- 20.Kashiwagi M, Enghild JJ, Gendron C, Hughes C, Caterson B, Itoh Y, Nagase H. Altered proteolytic activities of ADAMTS-4 expressed by C-terminal processing. J Biol Chem. 2004;279:10109–10119. doi: 10.1074/jbc.M312123200. [DOI] [PubMed] [Google Scholar]
- 21.Kashiwagi M, Tortorella M, Nagase H, Brew K. TIMP-3 is a potent inhibitor of aggrecanase 1 (ADAM-TS4) and aggrecanase 2 (ADAM-TS5) J Biol Chem. 2001;276:12501–12504. doi: 10.1074/jbc.C000848200. [DOI] [PubMed] [Google Scholar]
- 22.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 23.Hodder PS, Cassaday J, Peltier R, Berry K, Inglese J, Feuston B, Culberson C, Bleicher L, Cosford NDP, Bayly C, Suto C, Varney M, Strulovici B. Identification of Metabotropic Glutamate Receptor Antagonists Using an Automated High Throughput Screening System. Anal Biochem. 2003;313:246–254. doi: 10.1016/s0003-2697(02)00608-5. [DOI] [PubMed] [Google Scholar]
- 24.Lauer-Fields JL, Minond D, Brew K, Fields GB. Application of Topologically Constrained Mini-Proteins as Ligands, Substrates, and Inhibitors. Methods Mol Biol. 2007;386:125–166. doi: 10.1007/978-1-59745-430-8_5. [DOI] [PubMed] [Google Scholar]
- 25.Gao G, Westling J, Thompson VP, Howell TD, Gottschall PE, Sandy JD. Activation of the proteolytic activity of ADAMTS4 (aggrecanase-1) by C-terminal truncation. J Biol Chem. 2002;277:11034–11041. doi: 10.1074/jbc.M107443200. [DOI] [PubMed] [Google Scholar]
- 26.Darvas F, Dorman G, Karancsi T, Nagy T, Bágyi I. Estimation of stability and shelf life for compounds, libraries and repositories in combination with systematic discovery of new rearrangment pathways. In: Nicolaou KC, Hanko R, Hartwig W, editors. Handbook of Combinatorial Chemistry. Wiley-VCH; Weinheim: 2002. pp. 806–828. [Google Scholar]
- 27.Fields GB. Using fluorogenic peptide substrates to assay matrix metalloproteinases. In: Clark IM, editor. Methods in Molecular Biology 151: Matrix Metalloproteinase Protocols. Humana Press; Totowa, NJ: 2001. pp. 495–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knight CG, Willenbrock F, Murphy G. A novel coumarin-labelled peptide for sensitive continuous assays of the matrix metalloproteinases. FEBS Lett. 1992;296:263–266. doi: 10.1016/0014-5793(92)80300-6. [DOI] [PubMed] [Google Scholar]
- 29.Ashikawa K, Majumdar S, Banerjee S, Bharti AC, Shishodia S, Aggarwal BB. Piceatannol inhibits TNF-induced NF-κB activation and NF-κB-mediated gene expression through suppression of IκBα kinase and p65 phosphorylation. J Immunol. 2002;169:6490–6497. doi: 10.4049/jimmunol.169.11.6490. [DOI] [PubMed] [Google Scholar]
- 30.Kumari AL, Ali AM, Das S, Pardhasaradhi BVV, Varalakshmi C, Khar A. Role of STAT3 and NFκB signaling in the serum factor-induced apoptosis in AK-5 cells. Biochem Biophys Res Commun. 2005;336:860–867. doi: 10.1016/j.bbrc.2005.08.185. [DOI] [PubMed] [Google Scholar]
- 31.Chowdhury SA, Kishino K, Satoh R, Hashimoto K, Kikuchi H, Nishikawa H, Shirataki Y, Sakagami H. Tumor-specificity and apoptosis-inducing activity of stilbenes and flavonoids. Anticancer Res. 2005;25:2055–2063. [PubMed] [Google Scholar]
- 32.Chang JK, Hsu YL, Teng IC, Kuo PL. Piceatannol stimulates osteoblast differentiation that may be mediated by increased bone morphogenetic protein-2 production. Eur J Pharmacol. 2006;551:1–9. doi: 10.1016/j.ejphar.2006.08.073. [DOI] [PubMed] [Google Scholar]
- 33.Wung BS, Hsu MC, Wu CC, Hsieh CW. Piceatannol upregulates endothelial heme oxygenase-1 expression via novel protein kinase C and tyrosine kinase pathways. Pharmacol Res. 2006;53:113–122. doi: 10.1016/j.phrs.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 34.George J, Teear ML, Norey CG, Burns DD. Evaluation fo an imaging platform during the development of a FRET protease assay. J Biomol Screen. 2003;8:72–80. doi: 10.1177/1087057102239778. [DOI] [PubMed] [Google Scholar]
- 35.Hills R, Mazzarella R, Fok K, Liu M, Nemirovskiy O, Leone J, Zack MD, Arner EC, Viswanathan M, Abujoub A, Muruganandam A, Sexton DJ, Bassill GJ, Sato AK, Malfait AM, Tortorella MD. Identification of an ADAMTS-4 cleavage motif using phase display leads to the development of fluorogenic peptide substrates and reveals matrillin-3 as a novel substrate. J Biol Chem. 2007 doi: 10.1074/jbc.M611588200. in press. [DOI] [PubMed] [Google Scholar]
- 36.Copeland RA. Assay considerations for compound library screening. In: Copeland RA, editor. Evaluation of Enzyme Inhibitors in Drug Discovery. John Wiley & Sons, Inc; Hoboken, NJ: 2005. pp. 82–110. [Google Scholar]
- 37.Copeland RA. Reversible inhibitors. In: Copeland RA, editor. Enzymes. 2. Wiley-VCH; New York: 2000. pp. 266–304. [Google Scholar]
- 38.Schullek JR, Butler JH, Zhi-Jie N, Chen D, Yuan Z. A high-density screening format for encoded combinatorial libraries: Assay miniturization and its application to enzymatic reactions. Anal Biochem. 1997;246:20–29. doi: 10.1006/abio.1996.9958. [DOI] [PubMed] [Google Scholar]
- 39.Szardenings AK, Antonenko V, Campbell DA, DeFrancisco N, Ida S, Shi L, Sharkov N, Tien D, Wang Y, Navre M. Identification of highly selective inhibitors of collagenase-1 from combinatorial libraries of diketopiperazines. J Med Chem. 1999;42:1348–1357. doi: 10.1021/jm980475p. [DOI] [PubMed] [Google Scholar]
- 40.Vassiliou S, Mucha A, Cuniasse P, Georgiadis D, Lucet-Levannier K, Beau F, Kannan R, Murphy G, Knauper V, Rio MC, Basset P, Yiotakis A, Dive V. Phosphinic pseudo-tripeptides as potent inhibitors of matrix metalloproteinases: A structure-activity study. J Med Chem. 1999;42:2610–2620. doi: 10.1021/jm9900164. [DOI] [PubMed] [Google Scholar]
- 41.Buchardt J, Schiodt CB, Krog-Jensen C, Delaissé JM, Foged NT, Meldal M. Solid phase combinatorial library of phosphinic peptides for discovery of matrix metalloproteinase inhibitors. J Comb Chem. 2000;2:624–638. doi: 10.1021/cc000031q. [DOI] [PubMed] [Google Scholar]
- 42.Schiodt CB, Buchardt J, Terp GE, Christensen U, Brink M, Larsen YB, Meldal M, Foged NT. Phosphinic peptide inhibitors of macrophage metalloelastase (MMP-12): Selectivity and mechanism of binding. Current Med Chem. 2001;8:967–976. doi: 10.2174/0929867013372670. [DOI] [PubMed] [Google Scholar]
- 43.Dennis MS, Eigenbrot C, Skelton NJ, Ultsch MH, Santell L, Dwyer MA, O’Connell MP, Lazarus RA. Peptide exosite inhibitors of factor VIIa as anticoagulants. Nature. 2000;404:465–470. doi: 10.1038/35006574. [DOI] [PubMed] [Google Scholar]
- 44.Roberge M, Santell L, Dennis MS, Eigenbrot C, Dwyer MA, Lazarus RA. A novel exosite on coagulation factor VIIa and its molecular interactions with a new class of peptide inhibitors. Biochemistry. 2001;40:9522–9531. doi: 10.1021/bi010592d. [DOI] [PubMed] [Google Scholar]
- 45.Scheer JM, Romanowski MJ, Wells JA. A common allosteric site and mechanism in caspases. Proc Natl Acad Sci USA. 2006;103:7595–7600. doi: 10.1073/pnas.0602571103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wayne GJ, Deng SJ, Amour A, Borman S, Matico R, Carter HL, Murphy G. TIMP-3 inhibition of ADAMTS-4 (aggrecanase-1) is modulated by interactions between aggrecan and the C-terminal domain of ADAMTS-4. J Biol Chem. 2007 doi: 10.1074/jbc.M610721200. in press. [DOI] [PubMed] [Google Scholar]
- 47.Wittwer AJ, Hills RL, Keith RH, Munie GE, Arner EC, Anglin CP, Malfait AM, Tortorella MD. Substrate-dependent inhibition kinetics of an active site-directed inhibitor of ADAMTS-4 (aggrecanase 1) Biochemistry. 2007 doi: 10.1021/bi7000642. in press. [DOI] [PubMed] [Google Scholar]
- 48.Kokame K, Nobe Y, Kokubo Y, Okayama A, Miyata T. FRETS-VWF73, a first fluorogenic substrate for ADAMTS13 assay. Br J Haematol. 2005;129:93–100. doi: 10.1111/j.1365-2141.2005.05420.x. [DOI] [PubMed] [Google Scholar]
- 49.Hurskainen TL, Hirohata S, Seldin MF, Apte SS. ADAM-TS5, ADAM-TS6, and ADAM-TS7, novel members of a new family of zinc metalloproteases. J Biol Chem. 1999;274:25555–25563. doi: 10.1074/jbc.274.36.25555. [DOI] [PubMed] [Google Scholar]