

Abstract
A highly enantioselective and stereocontrolled approach to d-, l- and 8-epi-d-swainsonine has been developed from achiral furan and γ-butyrolactone. A one-pot hydrogenolysis of both azide and benzyl ether followed by an intramolecular reductive amination has been employed as key step to establish the indolizidine ring system. The absolute stereochemistry was installed by the Noyori reduction and the relative stereochemistry by the use of several highly diastereoselective palladium catalyzed glycosylation, Luche reduction, dihydroxylation and palladium catalyzed azide allylation reactions. This practical approach provide multigram quantities of indolizidine natural product d-swainsonine in 13 steps and 25% overall yield.
1. Introduction
Over the past decade, polyhydroxylated indolizidine alkaloids have received considerable attention because of their interesting structures and their diverse biological activities (e.g., antiviral, antitumor, and immunomodulating activity).1 The most well-known member d-swainsonine (1) (Figure 1), a trihydroxyindolizidine alkaloid, was isolated from the fungus Rhizoctonia leguminicola2 and Metarhizium anisopliae,3 as well as found in the legume Swainsona canescens4 and the spotted locoweed Astragalus lentiginosus.5 This natural product has shown to be a potent inhibitor of both lysosomal α-mannosidase6 and mannosidase II7 and has been applied to clinical testing as anticancer drug8 to inhibit tumor growth and metastasis.
Figure 1.
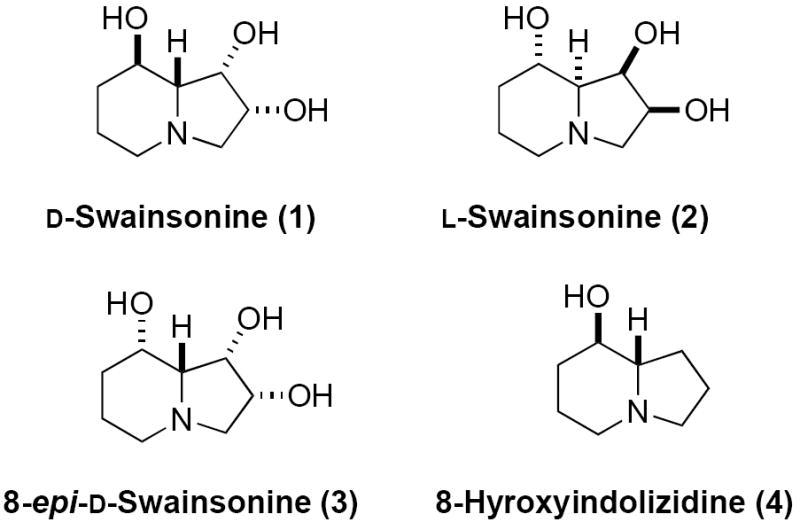
Swainsonine and Analogues
Fleet has shown that the enantiomer of d-swainsonine (1), l-swainsonine (2) selectively inhibited narginase (l-rhamnosidase, Ki = 0.45 μM), whereas the d-swainsonine showed no inhibitory activity toward this enzyme.9 Due to the biological importance, of both d- and l-swainsonine, as well as several epimers and analogues have become attractive targets for syntheses. Thus, over 35 syntheses of both d- and l-swainsonine have been reported since the first syntheses by Richardson and Fleet.10 Most of these syntheses reported to date utilize carbohydrate starting materials to introduce asymmetry; whereas, only eight syntheses used achiral starting materials.10a-d,h Herein we report the full account of a short and efficient de novo syntheses of d- and l-swainsonine (1 and 2) from achiral commercially available starting material.11 In addition to describing our optimised route (multigram scale) to d- and l-swainsonine we disclose for first time the use of this approach for the syntheses of 8-epi-swainsonine (3) and 8-hydroxyindolizidine (4).
Our strategy for the synthesis of d-swainsonine is outlined in Scheme 1, which involves a key one-pot reductive cyclization of azido-sugar 6 into 1. Thus, we envisioned that d-swainsonine could be obtained by hydrogenolytic deprotection and reductive amination of bicyclic amine intermediate 5, which can be derived from azide-sugar 6 by reductive N-alkylation. The manno-stereochemistry of azide 6 could be readily installed by diastereoselective dihydroxylation of 7. The allylic azide 7 in turn could be prepared from palladium catalyzed azide allylation, Luche reduction and stereoselective palladium catalyzed protection of pyranone 8α. A Noyori reduction and Achmatowicz oxidation should convert the furfuryl alcohol 9 into pyranone 8α. Eventually, the furfuryl alcohol could be derived from commercially available furan and γ-butyrolactone 11. In addition to providing either swainsonine enantiomer (1 and 2), we envisioned with route being able to provide the diastereomeric and deoxy-analogues 3 and 4.
Scheme 1.

Retrosynthetic Analysis of (-)-d-Swainsonine (1)
2. Results and Discussion
2.1. Approach to pyranones
The syntheses of d- and l-swainsonine as well as 8-epi-swainsonine began with commercially available achiral γ-butyrolactone 11 and furan (Scheme 2).12 Treatment of γ-butyrolactone 11 with 2-lithiofuran 10 in THF solution afforded furfuryl ketone 12 in 74% yield. Protection of primary alcohol 12 gave TBS-ether 13 (TBSCl/imid. in DMF) in excellent yield (98%). The furfuryl alcohol 9 was obtained from furfuryl ketone 12 by employing a phase transfer variant of the asymmetric Noyori reduction in high enantiomeric excess (> 96% ee) and excellent yield (95%) using an aqueous solution of HCOONa/cetyltrimethylammonium bromide (CTAB).13 Alternatively, the Noyori reduction could be accomplished using a THF solution of HCOOH/Et3N (1:1) in 89% yield, however, this required a 10 fold amount of Noyori catalyst and afforded a slightly lower yield of product. Exposure of furfuryl alcohol 9 to the Achmatowicz conditions14 (NBS in THF/H2O) provided the oxidative rearrangement product hemiacetal 14 (84%), which was converted ((Boc)2O at −78 °C) to a mixture of Boc-protected pyranones 8α and 8β in a good yield (85%) and diastereoselectivity (8:1; α:β).15 The diastereoselectivity of the Boc-acylation to form pyranones 8α and 8β was decreased (8α/8β in 1:1 ratio) when the reaction was performed at elevated temperature (0 °C to rt).
Scheme 2.

Enantioselective Synthesis of Pyranones
2.2. Synthesis of d-Swainsonine 1 via Protected Swainsonine
With the successful synthesis of pyranone 8α, we next installed manno-stereochemistry and C-1 benzyl ether (Scheme 3). Exposure of the pyranone 8α and benzyl alcohol to our palladium glycosylation conditions16 (2.5% Pd(0)/5% PPh3) afforded OBn-protected pyranone 15 as a single diastereomer and in an excellent yield (88%). Diastereoselective reduction of the C-4 ketone in 15 under the Luche condition17 (NaBH4/CeCl3, −78 °C) gave an equatorial allylic alcohol 16 in excellent yield (94%). Dihydroxylation of allylic alcohol 16 using the Upjohn conditions18 (OsO4/NMO) afforded triol 17 by in excellent yield (91%). The C-2/C-3 cis-diol 25 was selectively protected as acetonide 18 by treatment with 2,2-dimethoxypropane and catalytic amount of p-TsOH (96%).
Scheme 3.

Diastereoselective Synthesis of Azide 21
In order to convert the C-4 equatorial hydroxyl group into an equatorial azide, an oxidation/reduction/inversion sequence was employed. Swern oxidation of alcohol 18, provided the C-4 ketone 19, which was reduced with NaBH4 to provide the C-4 axial alcohol 20 in excellent yield for the two steps (93%). Finally, treatment of alcohol 20 with triflic anhydride gave a triflate, which without further purification underwent an SN2 azide displacement (NaN3/DMF) to provide azide 21 in 72% yield from 20.
Alternatively, the acetonide 21 could also be prepared by a short and efficient route that involved a palladium-catalyzed azide installation (Scheme 4),19 this would eliminate the need for the double inversion at C-4 (18 to 20 to 21). To perform the palladium coupling reaction, a C-4 carbonate leaving group was required. Thus, allylic alcohol 16 was treated with methyl chloroformate and catalytic amount of DMAP to produce allylic carbonate 22 in excellent yield (96%). The allylic azide 7 was formed by a regio- and stereoselective Pd-catalyzed allylation of methyl carbonate 22 with TMSN3 in the presence of (allylPdCl)2 and bis(diphenylphosphino)butane in excellent yield (91%). Once again diastereoselective dihydroxylation of allylic azide 7 generated diol 23 (92%), which was protected (2,2- dimethoxypropane/p-TsOH(cat)) to give acetonide 21 in good yield (97%) and two fewer steps.
Scheme 4.

Synthesis of Azide 21 via Palladium-Catalyzed Azide Allylation and Dihydroxylation
To complete the synthesis of d-swainsonine, a one-pot reductive cyclization was employed to furnish indolizidine ring (Scheme 5). TBS-ether 21 was deprotected to yield primary alcohol 24 by treatment with TBAF. Once again mesylation of 24 (MsCl/Et3N) produced mesylate 25 in excellent yield (97%, two steps). The azidosugar 25 was converted into a protected swainsonine 31 via a cascade of transformations (global hydrogenolysis, N-alkylation and reductive amination). Mechanistically, this is believed to begin with an azide reduction to produce aminosugar 26, which undergoes an intramolecular N-alkylation to generate amine 27. Hydrogenolytic removal of Bn-protecting group at the anomeric position should result in hemiacetal 28, which exist in equilibrium with hydroxy aldehyde 29. A reductive amination of 29, via bicyclic iminium ion intermediate 30, provided the protected swainsonine 31. This one-pot reaction was performed with hydrogen (1 atm.) in a THF/ethanol solution and required 7 days for complete conversion (85%). Finally, acidic hydrolysis of the acetonide 31 followed by ion-exchange chromatography (Basic HO− form) gave d-swainsonine (1) in excellent yield (95%).
Scheme 5.

Synthesis of d-Swainsonine (1) via a One-pot Reductive Cyclization
2.3. Alternative Synthesis of Swainsonine
Using the same strategy, we found d-swainsonine (1) could also be efficiently synthesized without acetonide protection in four steps from allylic azide 7 (Scheme 6). Deprotection of TBS-ether 7 with a TBAF solution (99%) afforded the primary alcohol 32, which was after mesylation (MsCl/Et3N) gave mesylate 33 in near quantitative yield (99%). Upjohn dihydroxylation of allylic azide 33 exclusively gave diol 6 (OsO4/NMO, 93%). Finally, the natural product d-swainsonine 1 was obtained by an analogous azide reduction/N-alkylation/reductive amination sequence under hydrogenation condition (100 psi H2, Pd(OH)2/C).
Scheme 6.

Synthesis of d-Swainsonine (1) and 8-Hydroxyindolizidine (4)
In addition, the known indolizidine analogue 8-hydroxyindolizidine (4)20 was also prepared by the same one-pot hydrogenation of allylic azide 33 (68%). It is worth noting this sans-acetonide approach provided 8-hydroxyindolizidine (4) as well as either enantiomer of swainsonine (1 and 2) while using only two protecting groups (Bn and TBS). The route to swainsonine can provide multigram quantities of both enantiomers (3 g of the d-isomer was prepared). The physical and spectral data of our synthetic both d- and l-swainsonine by either way is completely identical to those of the natural materials (1H NMR, 13C NMR, optical rotation and melting point).21
2.4. Synthesis of 8-epi-d-Swainsonine
Encouraged by these results, we next investigated the use of this approach for the synthesis of 8-epi-d-swainsonine (Scheme 7). We envisioned this approach starting with the l-pyranone ent-8β . This enantiomeric β-anomer of 8α was readily prepared in three steps from furfuryl ketone 13 by switching to the use of the (S,S)-Noyori catalyst and performing the Boc-acylation at higher temperature (0°C to rt) to ensure more β-pyranone ent-8β was produced (cf, Scheme 2).
Scheme 7.

Synthesis of Allylic Alcohol 36
Palladium catalyzed glycosylation of pyranone ent-8β with benzyl alcohol diastereoselectively afforded the C-1 BnO-pyranone 34 in 88% yield (Scheme 7). Unfortunately, the Luche reduction (NaBH4/CeCl3) of pyranone 34 provided a 1:1.3 mixture of allylic alcohols 35 and 36 in 86% yield. Improved diastereoselectivity was obtained (35/36 in a 1:2.5 ratio) when DIBAL-H was used as the reducing agent (89%). The easily separated minor diastereomer, equatorial allylic alcohol 35, could be easily converted into axial allylic alcohol 36 via a 2-step Mitsunobu/ester hydrolysis sequence. Thus, exposure of allylic alcohol 35 to typical Mitsunobu reaction conditions (BzOH, DIAD and PPh3) yielded benzoate ester 37 with inverted stereochemistry at C-4. Methanolysis of ester 37 (K2CO3/MeOH) provided allylic alcohol 35 in good yield (82%, two steps).
With a practical route to allylic alcohol 36 (the C-5 diastereomer of 16) in hand, we explored the analogous application of this approach for the synthesis of 8-epi-d-swainsonine (3) (Scheme 8). Treatment of allylic alcohol 36 with methyl chloroformate formed carbonate 38 (Pyr/DMAP) in 94% yield. The carbonate 38 was coupled with TMSN3 to give allylic azide ((allylPdCl)2/dppb) in excellent yield (92%). Stereoselective dihydroxylation of allylic azide 38 under Upjohn conditions (OsO4/NMO) afforded diol 40 as a single talo-diastereomer. Diol 40 was protected as acetonide 41 in excellent yield (95% two steps). Deprotection of TBS-ether 41 provided primary alcohol 42 (TBAF, 99% yield), which was followed by mesylation to afford mesylate 43 in 98% yield. A one-pot global hydrogenation/hydrogenolysis of azide 43 produced protected 8-epi-d-swainsonine (3) in excellent yield (83%). Eventually, 8-epi-d-swainsonine (3) was obtained by treatment with HCl in 94% yield. The spectral and physical data of 8-epi-d-swainsonine (3) by our approach match those of references reported (1H NMR, 13C NMR, optical rotation and melting point).22
Scheme 8.

Synthesis of 8-epi-d-Swainsonine (3)
3. Conclusions
In conclusion, a de novo asymmetric approach to d-swainsonine (1), l-swainsonine (2), 8-epi-d-swainsonine (3) as well as 8-hydroxylindolizidine (4) has been developed. Both d- and l-swainsonine were achieved in 13 steps and 25% overall yield from achiral furan 10. These practical syntheses are comparable to the previous carbohydrate-based routes.10 Key to the successful approach is the use of the Noyori reduction, palladium-catalyzed glycosylation, diastereoselective dihydroxylation, palladium-catalyzed azide allylation and one-pot reductive cyclization. The 8-epi-d-swainsonine (4) was also synthesized in 10 steps in 34% overall yield from Boc-pyranone ent-8β (15 steps from furan 10 in 8% overall yield). Further application of this approach to the synthesis of various analogues is ongoing.
4. Experimental Section23
4.1. General
1H and 13C spectra were recorded on 270 and 600 spectrometers. Chemical shifts were reported relative to internal tetramethylsilane (δ 0.00 ppm) or CDCl3 (δ 7.26 ppm) or CD3OD (δ 4.89 ppm) for 1H and CDCl3 (δ 77.1 ppm) or CD3OD (δ 49.15 ppm) for 13C. Optical rotations were measured with a digital polarimeter in the solvent specified. Infrared (IR) spectra were obtained on a FT-IR spectrometer. Flash column chromatography was performed on ICN reagent 60 (60-200 mesh) silica gel. Analytical thin-layer chromatography was performed with precoated glass-backed plates (K6F 60Å, F254) and visualized by quenching of fluorescence and by charring after treatment with p-anisaldehyde or phosphomolybdic acid or potassium permanganate stain. Rf values were obtained by elution in the stated solvent ratios (v/v). Ether, THF, methylene chloride and triethylamine were dried by passing through activated alumina (8 × 14 mesh) column with nitrogen gas pressure. Commercial reagents were used without purification unless otherwise noted. Air and/or moisture-sensitive reactions were carried out under an atmosphere of argon/nitrogen using oven/flamed-dried glassware and standard syringe/septa techniques.
4.1.1. (2S,3S,4S,5S,6R)-2-(benzyloxy)-tetrahydro-6-(3-tert-butyldimethylsilyloxyprop-yl)-2H-pyran-3,4,5-triol (17)
To a t-butanol/acetone (0.53 mL, 1:1, 1 M) solution of allylic alcohol 16 (200 mg, 0.53 mmol) at 0 °C was added a solution of (50% w/v) of N-methyl morpholine N-oxide/water (0.25 mL). Crystalline OsO4 (1.4 mg, 1 mol %) was added and the reaction was stirred for 24 h. The reaction mixture was concentrated with a small silica gel under reduced pressure, purified using silica gel flash chromatography eluting with 100% EtOAc to give triol 17 (199 mg, 0.48 mmol, 91%); Rf = 0.29 (70% EtOAc/hexane); (c = 3.11, MeOH); IR (thin film, cm-1) 3397, 2928, 2857, 1455, 1389, 1253, 1061, 833; 1H NMR (600 MHz, CD3OD)δ 7.36-7.43 (m, 5H), 4.91 (s, 1H), 4.78 (d, J = 11.4 Hz, 1H), 4.57 (d, J = 11.4 Hz, 1H), 3.97 (m, 1H), 3.83 (dd, J = 9.0, 3.0 Hz, 1H), 3.77 (m, 2H), 3.64 (dd, J = 9.0, 9.0 Hz, 1H), 3.59 (m, 1H), 2.00-2.11 (m, 1H), 1.90-1.97 (m, 1H), 1.66-1.73 (m, 1H), 1.58-1.64 (m, 1H), 1.01 (s, 9H), 0.16 (s, 6H); 13C NMR (150 MHz, CD3OD) δ 138.9, 129.5, 129.0, 128.8, 100.5, 73.6, 72.7, 72.4, 72.1, 64.5, 30.3, 29.0, 26.6, 19.2, -4.9; CIHRMS: [C21H36O6SiNa+]: 435.2176, found: 435.2179.
4.1.2. (3aS,4S,6R,7R,7aS)-4-(benzyloxy)-tetrahydro-6-(3-tert-butyldimethylsilyloxypropyl)-2,2-dimethyl-3aH-[1,3]dioxolo[4,5-c]pyran-7-ol (18)
Para-toluenesulfonic acid monohydrate (19.4 mg, 5 mol%) was added to a stirred solution of diol 17 (840 mg, 2.08 mmol) and 2,2-dimethoxypropane (5.87 mL) in acetone (31.2 mL) for 0.5 h at 0 °C. The reaction mixture was quenched with sodium bicarbonate solution (5 mL), removed acetone in vacuo, extracted with Et2O (3 × 25 mL), dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified using silica gel flash chromatography eluting with 15% EtOAc/hexane to give 904 mg (2.0 mmol, 96%) of colorless oil acetonide 18: Rf (20% EtOAc/hexane) = 0.40; (c = 1.52, CH2Cl2); IR (thin film, cm-1) 3467, 2930, 2857, 1382, 1248, 1087, 835; 1H NMR (600 MHz, CDCl3)δ 7.34 (m, 5H), 5.06 (s, 1H), 4.74 (d, J = 11.4 Hz, 1H), 4.51 (d, J = 11.4 Hz, 1H), 4.18 (d, J = 6.0 Hz, 1H), 4.11 (dd, J = 6.0, 7.2 Hz, 1H), 3.67 (m, 2H), 3.56 (ddd, J = 9.0, 9.0, 3.0 Hz, 1H), 3.48 (m, 1H), 2.15 (d, J = 3.0, 1H), 1.83-1.95 (m, 2H), 1.52-1.65 (m, 2H), 1.51 (s, 3H), 1.31 (s, 3H), 0.90 (s, 9H), 0.06 (s, 6H); 13C NMR (150 MHz, CDCl3) δ 137.0, 128.6, 128.3, 128.1, 109.5, 96.2, 78.6, 75.9, 73.2, 69.8, 69.1, 63.3, 28.8, 28.1, 27.9, 26.3, 26.1, 18.5, -5.2; CIHRMS: Calculated for [C24H40O6SiNa+]: 475.2491, found: 475.2492.
4.1.3. (3aS,4S,6R,7aR)-4-(benzyloxy)-dihydro-6-(3-tert-butyldimethylsilyloxypropyl)-2,2-dimethyl-6H-[1,3]dioxolo[4,5-c]pyran-7(7aH)-one (19)
To a solution of (COCl)2 (0.15 mL, 1.76 mmol) in CH2Cl2 (3.5 mL) was added DMSO (0.51 mL, 3.52 mmol) at −78 °C, and the mixture was stirred for 10 min. A solution of alcohol 18 (400 mg, 0.88 mmol) in 1.76 mL CH2Cl2 was added to the resulting mixture, stirring for 0.5 h at −78 °C. Then triethylamine (0.50 mL, 3.52 mmol) was added at −78 °C and the reaction mixture was warmed to 0 °C for 1 h. 50 mL water was added to quench the mixture, and the aqueous mixture was extracted (3 × 80 mL) with Et2O, dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified using silica gel flash chromatography eluting with 15% EtOAc/hexane to give ketone 19 (392 mg, 0.87 mmol, 99%) as a colorless oil: Rf (20% EtOAc/hexane) = 0.61; (c = 2.0, CH2Cl2); IR (thin film, cm-1) 2954, 2929, 2857, 1741, 1472, 1253, 1077, 1023, 834; 1H NMR (600 MHz, CDCl3.)δ 7.35 (m, 5H), 5.03 (d, J = 1.2 Hz, 1H), 4.76 (d, J = 11.4 Hz, 1H), 4.57 (d, J = 11.4 Hz, 1H), 4.47 (dd, J = 6.6, 1.2 Hz, 1H), 4.46 (d, J = 6.6, 1H), 4.18 (dd, J = 4.8, 8.4 Hz, 1H), 3.65 (m, 2H), 1.90-1.96 (m, 1H), 1.72-1.84 (m, 2H), 1.58-1.65 (m, 1H), 1.48 (s, 3H), 1.35 (s, 3H) 0.90 (s, 9H), 0.05 (s, 6H); 13C NMR (67.5 MHz, CDCl3) δ 196.5, 143.3, 137.1, 128.6, 128.2 (two carbon), 127.9, 92.1, 74.0, 70.6, 62.9, 28.5, 26.0, 18.4, -5.2; CIHRMS: Calculated for [C24H38O6Si+H+]: 451.2514, found: 451.2516.
4.1.4. (3aS,4S,6R,7S,7aS)-4-(benzyloxy)-tetrahydro-6-(3-tert-butyldimethylsilyloxypropyl)-2,2-dimethyl-3aH-[1,3]dioxolo[4,5-c]pyran-7-ol (20)
A CH2Cl2 (1 mL) solution of ketone 19 (370 mg, 0.82 mmol) and MeOH (1 mL, 1 M) was cooled to 0 °C. NaBH4 (37 mg, 0.98 mmol) was added and the reaction mixture was stirred at 0 °C for 20 min. The reaction mixture was diluted with ether (20 mL) and quenched with 15 mL of saturated NaHCO3, extracted (3 × 30 mL) with Et2O, dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified using silica gel flash chromatography eluting with 15% EtOAc/hexane to give colorless oil 323 mg (0.71 mmol, 87%) of alcohol 20: Rf (20% EtOAc/hexane) = 0.41; (c = 1.6, CH2Cl2); IR (thin film, cm-1) 3568, 2928, 2857, 1381, 1251, 1070, 833; 1H NMR (600 MHz, CDCl3) δ 7.35 (m, 5H), 5.15 (s, 1H), 4.76 (d, J = 11.4 Hz, 1H), 4.55 (d, J = 11.4 Hz, 1H), 4.22 (dd, J = 4.8, 6.0 Hz, 1H), 4.11 (d, J = 6.0 Hz, 1H), 3.76 (dd, J = 4.8, 9.0 Hz, 1H), 3.63-3.70 (m, 2H), 3.61 (d, J = 6.0, 6.0 Hz, 1H), 3.48 (m, 1H), 2.21 (d, J = 6.0, 1H), 1.84-1.90 (m, 1H), 1.73-1.80 (m, 1H), 1.65-1.71 (m, 1H),1.59-1.64 (m, 1H), 1.58 (s, 3H), 1.38 (s, 3H), 0.90 (s, 9H), 0.05 (s, 6H); 13C NMR (150 MHz, CDCl3) δ 137.1, 128.6, 128.3, 128.1, 109.3, 96.7, 73.6, 72.9, 69.4, 68.5, 66.1, 63.1, 29.4, 27.6, 26.1, 25.9, 25.4, 18.4, -5.2; CIHRMS: Calculated for [C24H40O6SiNa+]: 475.2491, found: 475.2490.
4.1.5. (3aR, 9R, 9aR, 9bS)-octahydro-2,2-dimethyl-[1,3]dioxolo[4, 5-a]indolizin-9-ol (31)
To a solution of acetonide 25 (7.79 g, 17.6 mmol) in dry EtOH/THF (76 mL, v/v = 1:1) was added Pd(OH)2/C (1.9 g) and the mixture was stirred under H2 at an 100 psi pressure for 3 days at room temperature. The catalyst was filtered off through a short pad of celite, concentrated under reduced pressure. The resulting crude was purified using silica gel flash chromatography eluting with 30% MeOH/EtOAc to give protected swainsonine 31 (3.2 g, 15.0 mmol, 85%); a colorless needles, Rf = 0.44 (10% MeOH/EtOAc); (c = 1.0, CH2Cl2); mp 102-104; IR (neat, cm-1) 3194, 2942, 2793, 1466, 1371, 1260, 1209, 1113, 1068; 1H NMR (600 MHz, CDCl3) δ 4.67 (dd, J = 6.2, 4.8, 1H), δ 4.24 (dd, J = 6.0, 4.2, 1H), 3.79 (ddd, J = 10.9, 8.9, 4.4, 1H), 3.11 (d, J = 10.8, 1H), 2.96 (dt, J = 10.8, 3.0 Hz, 1H), 2.52 (br s, 1H), 2.09 (dd, J = 10.8, 4.8 Hz, 1H), 2.00-2.03 (m, 1H), 1.82 (ddd, J =10.8, 10.8, 3.6 Hz, 1H), 1.61-1.65 (m, 1H), 1.59 (dd, J = 9.0, 4.8 Hz, 1H), 1.48 (s, 3H) , 1.31 (s, 3H), 1.25(m, 1H); 13C NMR (150 MHz, CDCl3) δ 111.3, 79.2, 78.2, 73.8, 67.3, 60.0, 51.6, 33.0, 26.0, 24.9, 24.1.
4.1.6. (2S, 6S)-6-(benzyloxy)-2-(3-tert-butyldimethylsilyloxypropyl)-2H-pyran-3(6H)-one (34)
To a solution of Boc-protected pyranone 8α (5.86 g, 15.8 mmol) and benzyl alcohol (3.28 g, 30.4 mmol) in dry CH2Cl2 (8 mL), was added Pd2(DBA)3•CHCl3 (97.7 mg, 0.9 mol% Pd) and PPh3 (101.5 mg, 3.6 mol%) at 0 °C under argon atmosphere. After stirring for 2 h at 0 °C to room temperature, the reaction mixture was quenched with 200 mL of saturated NaHCO3, extracted (3 × 200 mL) with Et2O, dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified using silica gel flash chromatography eluting with 6% EtOAc/hexane to give benzyl ether 34 (5.02 g, 13.30 mmol, 88%) as a colorless oil: Rf (10% EtOAc/hexane) = 0.75; (c = 1.4, CH2Cl2); IR (thin film, cm-1) 2954, 2929, 2856, 1695, 1471, 1254, 1092, 1024, 832; 1H NMR (600 MHz, CDCl3) δ 7.36 (m, 5H), 6.89 (dd, J = 10.2, 1.8 Hz, 1H), 6.10 (dd, J = 10.2, 1.2 Hz, 1H), 5.37 (d, J = 1.2 Hz, 1H), 4.95 (d, J = 11.4 Hz, 1H), 4.71 (d, J = 11.4 Hz, 1H), 4.09 (dd, J = 7.8, 3.6 Hz, 1H), 3.66 (m, 2H), 2.03-2.09 (m, 1H), 1.85-1.91 (m, 1H), 1.75-1.82 (m, 1H), 1.67-1.74 (m, 1H), 0.91 (s, 9H), 0.06 (s, 6H); 13C NMR (150 MHz, CDCl3) δ 200.0, 146.5, 137.0, 128.6, 128.15 (two carbon), 128.1, 94.6, 78.9, 70.3, 62.9, 28.5, 28.0, 26.0, 18.4, -5.2; CIHRMS: Calculated for [C21H32O4SiNa+]: 399.1962, found: 399.1970.
4.1.7. (2S, 3S, 6S)-6-(benzyloxy)-3,6-dihydro-2-(3-tert-butyldimethylsilyloxypropyl)-2H-pyran-3-ol (36)
To a solution of pyranone 34 (6.7 g, 17.7 mmol) in 130 mL THF was added dropwise 1.0 M DIBAL-H in hexane (17.8 mL, 17.8 mmol) at −78 °C under nitrogen atmosphere. The reaction mixture was kept stirring for 3 h at −78 °C, then treated with 1.0 M Na/K tartaric solution (50 mL) and allowed to warm to room temperature. The resulting mixture was extracted with diethyl ether (3 × 200 mL) and the combined organic phases were dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified using silica gel flash chromatography eluting with 20 % EtOAc/hexane to afford colorless oil 6.0 g (15.8 mmol, 89%) of allylic alcohol 35 and 36 in 1:2.5. 36: Rf (20% EtOAc/hexane) = 0.61; (c = 1.0, CH2Cl2); IR (thin film, cm-1) 3417, 2952, 2928, 2856, 1471, 1254, 1094, 1056, 834; 1H NMR (600 MHz, CDCl3) δ 7.27-7.38 (m, 5H), 6.10 (dd, J = 9.6, 4.8 Hz, 1H), 5.86 (d, J = 9.6, 1H), 5.09 (d, J = 1.2 Hz, 1H), 4.92 (d, J = 11.4 Hz, 1H), 4.67 (d, J = 11.4 Hz, 1H), 3.70 (m, 1H), 3.69 (m, 2H), 3.54 (ddd, J = 7.2, 4.8, 2.4 Hz, 1H), 1.97 (d, J = 2.4 Hz, 1H), 1.78-1.84 (m, 1H), 1.69-1.77 (m, 2H), 1.62-1.67 (m, 1H), 0.91 (s, 9H), 0.07 (s, 6H); 13C NMR (150 MHz, CDCl3) δ 137.6, 131.4, 130.9, 128.4, 128.1, 127.8, 97.2, 75.6, 70.0, 63.9, 63.2, 28.7, 27.3, 26.0, 18.4, -5.2; CIHRMS: Calculated for [C21H34O4SiNa+]: 401.2118, found: 401.2132. 35: Rf (20% EtOAc/hexane) = 0.58; (c = 1.0, CH2Cl2); IR (thin film, cm-1) 3426, 2932, 2928, 2856, 1471, 1254, 1093, 1056, 834; 1H NMR (600 MHz, CDCl3) δ 7.26-7.37 (m, 5H), 5.95 (ddd, J = 9.6, 3.0, 1.8 Hz, 1H), 5.81 (ddd, J = 9.6, 3.6, 1.2 Hz, 1H), 5.16 (dd, J =3.6 1.8 Hz, 1H), 4.87 (d, J = 11.4 Hz, 1H), 4.64 (d, J = 11.4 Hz, 1H), 3.99 (ddd, J = 7.8, 7.2, 2.4 Hz, 1H), 3.68 (m, 2H), 3.47 (ddd, J = 7.8, 7.2, 3.6 Hz, 1H), 2.29 (d, J = 7.2 Hz, 1H), 1.77-1.92 (m, 2H), 1.61-1.71 (m, 2H), 0.92 (s, 9H), 0.08 (s, 6H); 13C NMR (150 MHz, CDCl3) δ 137.7, 132.8, 128.8, 128.4, 128.0, 127.7, 96.0, 78.2, 69.5, 67.2, 63.2, 28.9, 28.8, 26.0, 18.4, -5.2; CIHRMS: Calculated for [C21H34O4SiNa+]: 401.2118, found: 401.2132.
4.1.8. (2S,3S,6S)-6-(benzyloxy)-3,6-dihydro-2-(3-tert-butyldimethylsilyloxypropyl)-2H-pyran-3-yl benzoate (37)
Allylic alcohol 35 (1.1 g, 3.38 mmol) was dissolved in THF (5 mL). The solution was cooled to 0 °C and triphenylphosphine (1.77 g, 6.75 mmol), benzoic acid (0.82 g, 6.75 mmol), and diisopropyl azodicarboxylate (1.33 mL, 6.75 mmol) were added to the solution. The solution was stirred overnight, quenched with saturated aqueous sodium bicarbonate (50 mL), and extracted with ether (3 × 50 mL). The organic fractions were combined, dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified using silica gel flash chromatography eluting with 5% EtOAc/hexane to afford colorless oil 1.38 g (2.85 mmol, 84%) of allylic ester 37: Rf (20% EtOAc/hexane) = 0.64; (c = 1.0, CH2Cl2); IR (thin film, cm-1) 2953, 2927, 2857, 1790, 1717, 1452, 1268, 1108, 1061, 835; 1H NMR (600 MHz, CDCl3) δ 8.09-8.10 (m, 2H), 7.52-7.57 (m, 2H), 7.28-7.45 (m, 6H), 6.19 (ddd, J = 10.2, 4.8, 1.8 Hz, 1H), 6.05 (d, J = 10.2 Hz, 1H), 5.28 (ddd, J = 7.2, 4.8, 2.4 Hz, 1H), 5.20 (d, J = 1.2 Hz, 1H), 4.94 (d, J = 11.4 Hz, 1H), 4.74 (d, J = 11.6 Hz, 1H), 3.78 (ddd, J = 7.8, 4.8, 3.0 Hz, 1H), 3.60-3.68 (m, 2H), 1.84-1.90 (m, 1H), 1.75-1.82 (m, 1H), 1.67-1.73 (m, 1H), 1.58-1.65 (m, 1H), 0.08 (s, 9H), 0.02 (s, 3H), 0.01 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 166.3, 137.8, 134.6, 130.6, 129.9, 128.9, 128.5, 128.4, 128.1, 127.8, 127.1, 96.7, 74.0, 69.6, 66.0, 63.1, 28.9, 27.5, 26.0, 18.4, -5.3; CIHRMS: Calculated for [C28H38O5SiNa+]: 505.2381, found: 505.2386.
4.1.9. (2S,3S,6S)-6-(benzyloxy)-3,6-dihydro-2-(3-tert-butyldimethylsilyloxypropyl)-2H-pyran-3-yl-methyl carbonate (38)
To a solution of allylic alcohol 36 (4.0 g, 10.6 mmol) and pyridine (2.62 g, 31.6 mmol) in dry CH2Cl2 (53 mL) at 0 °C, was added DMAP (260 mg), and added dropwise methyl chloroformate (6.24 g, 63.6 mmol). After stirring 1 h at 0 °C, a saturated Cu2SO4 solution (500 mL) was added and then the mixture was extracted with CH2Cl2 (3 × 300 mL), dried (Na2SO4), concentrated under reduced pressure. The crude product was purified using silica gel flash chromatography eluting with 5% EtOAc/hexane to give 4.34 g (9.96 mmol, 94%) of colorless oil carbonate 38: Rf (20% EtOAc/hexane) = 0.62; (c = 1.4, CH2Cl2); IR (thin film, cm-1) 2955, 2929, 2886, 2857, 1745, 1442, 1264, 1098, 1059, 833; 1H NMR (600 MHz, CDCl3) δ 7.35 (m, 5H), 6.12 (ddd, J = 10.2, 4.8, 1.2 Hz, 1H), 6.03 (d, J = 10.2 Hz, 1H), 5.13 (d, J =1.2 Hz, 1H), 4.88 (d, J = 11.4 Hz, 1H), 4.87 (dd, J = 4.8, 1.8 Hz, 1H), 4.68 (d, J = 11.4 Hz, 1H), 3.78 (s, 3H), 3.62-3.73 (m, 3H), 1.73-1.84 (m, 2H), 1.64-1.70 (m, 1H), 1.57-1.63 (m, 1H), 0.90 (s, 9H), 0.06 (s, 6H); 13C NMR (150 MHz, CDCl3) δ 155.8, 137.7, 133.7, 128.4, 128.1, 127.8, 126.5, 96.5, 73.6, 69.4, 69.2, 63.1, 55.0, 28.9, 27.1, 26.0, 18.4, -5.2; CIHRMS: Calculated for [C23H36O6SiNa+]: 459.2173, found: 459.2179.
4.1.10. (2S,3S,6S)-3-azido-6-(benzyloxy)-3,6-dihydro-2-(3-tert-butyldimethylsilyloxyprop-yl)-2H-pyran (39)
To a mixture of carbonate 38 (2.2 g, 5.45 mmol), allylpalladium chloride dimer (43.5 mg, 0.11 mmol) and 1,4-bis(diphenylphosphino)butane (189 mg, 0.44 mmol) in dry THF (1.2 mL) was added TMSN3 (684 mg, 5.95 mmol) under argon atmosphere. The solution was stirred at room temperature for 0.5 h. Then the reaction mixture was passed Celite pad, concentrated under reduced pressure, and then purified using silica gel flash chromatography eluting with 4% EtOAc/hexane to give 2.0 g (4.96 mmol, 91%) allylic azide 39 as colorless oil: Rf (20% EtOAc/hexane) = 0.64; (c = 1.9, CH2Cl2); IR (thin film, cm-1) 2953, 2929, 2857, 2099, 1472, 1253, 1098, 835; 1H NMR (600 MHz, CDCl3) δ 7.36 (m, 5H), 6.10 (d, J = 10.2 Hz, 1H), 5.92 (ddd, J = 10.2, 4.8, 1.2 Hz, 1H), 5.17 (d, J = 1.2 Hz, 1H), 4.90 (d, J = 11.4 Hz, 1H), 4.68 (d, J = 11.4 Hz, 1H), 3.72 (ddd, J = 7.2, 4.8, 1.8 Hz, 1H), 3.68 (m, 2H), 3.63 (dd, J = 4.8, 3.0, 1.8 Hz, 1H), 1.81-1.89 (m, 1H), 1.69-1.75 (m, 2H), 1.59-1.64 (m, 1H), 0.91 (s, 9H), 0.07 (s, 6H); 13C NMR (150 MHz, CDCl3) δ 137.8, 133.3, 128.4, 128.1, 127.8, 126.1, 96.8, 75.5, 69.3, 63.1, 55.3, 28.7, 28.6, 26.0, 18.4, -5.2; CIHRMS: Calculated for [C21H33N3O6SiNa+]: 426.2183, found: 426.2189.
4.1.11. (2S,3S,4S,5S,6S)-5-azido-2-(benzyloxy)-tetrahydro-6-(3-tert-butyldimethylsilyl-oxypropyl)-2H-pyran-3, 4-diol (40)
To a t-butanol/acetone (13.4 mL, 1:1, 1 M) solution of allylic azide 39 (2.7 g, 6.7 mmol) at 0 °C was added a solution of (50% w/v) of N-methyl morpholine N-oxide/water (4 mL). Crystalline OsO4 (17 mg, 1 mol %) was added and the reaction was stirred for 24 h. The reaction mixture was quenched with 20 mL saturated Na2S2O3 solution, extracted with ether (3 × 200), dried (Na2SO4), concentrated under reduced pressure, and then purified using silica gel flash chromatography eluting with 30% EtOAc/hexane to give diol 40 (2.81 g, 6.43 mmol, 96%); colorless oil, Rf = 0.47 (40% EtOAc/hexane); (c = 1.0, CH2Cl2); IR (thin film, cm-1) 3426, 2953, 2929, 2858, 2102, 1471, 1077, 835; 1H NMR (600 MHz, CDCl3) δ 7.34 (m, 5H), 4.93 (d, J = 11.4 Hz, 1H), 4.63 (d, J = 7.8 Hz, 1H), 4.56 (d, J = 11.4 Hz, 1H), 4.23 (dd, J = 4.2, 2.4, 1.2 Hz, 1H), 3.95 (ddd, J = 7.8, 4.2, 1.2 Hz, 1H), 3.62-3.72 (m, 3H), 3.56 (dd, J = 3.0, 1.2 Hz, 1H), 2.73 (d, J = 1.2, 1H), 2.53 (d, J = 2.4 Hz, 1H), 1.80-1.87 (m, 1H), 1.71-1.77 (m, 1H), 1.57-1.68 (m, 2H), 0.91 (s, 9H), 0.07 (s, 6H); 13C NMR (150 MHz, CDCl3) δ 137.1, 128.6, 128.3, 128.2, 99.6, 72.5, 70.8, 70.1, 68.9, 63.6, 62.9, 29.3, 27.7, 26.1, 18.4, -5.2; CIHRMS: Calculated for [C21H35N3O5SiNa+]: 460.2238, found: 460.2243.
4.1.12. (3-((3aS,4S,6S,7R,7aS)-7-azido-4-(benzyloxy)-tetrahydro-2,2-dimethyl-3aH-[1,3]dioxolo[4,5-c]pyran-6-yl)propoxy)(tert-butyl)dimethylsilane (41)
Para-toluenesulfonic acid monohydrate (61.5 mg, 5 mol%) was added to a stirred solution of diol 40 (2.9 g, 6.6 mmol) and 2,2-dimethoxypropane (18.6 mL) in acetone (99 mL) for 0.5 h at 0 °C. The reaction mixture was quenched with sodium bicarbonate solution (100 mL), removed acetone in vacuo, extracted with Et2O (3 × 200 mL), dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified using silica gel flash chromatography eluting with 5% EtOAc/hexane to give 3.12 g (6.53 mmol, 99%) of colorless oil acetonide 41: Rf (20% EtOAc/hexane) = 0.67; (c = 1.0, CH2Cl2); IR (thin film, cm-1) 2952, 2930, 2857, 2102, 1249, 1049, 835; 1H NMR (600 MHz, CDCl3) δ 7.27-7.37 (m, 5H), 4.91 (d, J = 11.4 Hz, 1H), 4.66 (d, J = 11.4 Hz, 1H), 4.42 (dd, J = 5.4, 1.8 Hz, 1H), 4.41 (d, J = 7.8 Hz, 1H), 4.05 (dd, J = 7.2, 5.4 Hz, 1H), 3.72 (ddd, J = 8.4, 4.8, 1.8 Hz, 1H), 3.67 (m, 2H), 3.55 (dd, J = 1.8, 1.8 Hz, 1H), 1.84-1.90 (m, 1H), 1.65-1.78 (m, 2H), 1.57-1.64 (m, 1H), 1.37 (s, 3H), 1.31 (s, 3H), 0.91 (s, 9H), 0.06 (s, 6H); 13C NMR (150 MHz, CDCl3) δ 137.2, 128.4, 128.0, 127.8, 109.6, 101.0, 75.8, 74.4, 73.0, 70.1, 62.9, 60.2, 28.7, 29.1, 27.8, 26.3, 26.0, 18.3, -5.2; CIHRMS: Calculated for [C24H39N3O5SiNa+]: 500.2551, found: 500.2556.
4.1.13. 3-((3aS,4S,6S,7R,7aS)-7-azido-4-(benzyloxy)-tetrahydro-2,2-dimethyl-3aH-[1,3]dioxolo[4,5-c]pyran-6-yl)propan-1-ol (42)
To a solution of TBS-ether 41 (540 mg, 1.13 mmol) in dry THF (1.1 mL), TBAF (1.3 mL, 1.3 mmol) was added at room temperature under the argon atmosphere. After 12 h, the reaction mixture was quenched with sodium bicarbonate solution (500 mL), extracted with Et2O (3 × 100 mL), dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified using silica gel flash chromatography eluting with 40% EtOAc/hexane to give alcohol 42 (401 mg, 1.11 mmol, 98%); as colorless oil, Rf = 0.45 (40% EtOAc/hexane); (c = 0.8, CH2Cl2); IR (thin film, cm-1) 3434, 2988, 2941, 2875, 2102, 1383, 1221, 1046, 854; 1H NMR (600 MHz, CDCl3) δ 7.26-7.38 (m, 5H), 4.90 (d, J = 11.4 Hz, 1H), 4.66 (d, J = 11.4 Hz, 1H), 4.42 (dd, J = 5.4, 1.8 Hz, 1H), 4.41 (d, J = 7.8 Hz, 1H), 4.06 (dd, J = 7.8, 5.4 Hz, 1H), 3.73 (ddd, J = 8.4, 4.8, 1.8 Hz, 1H), 3.67 (m, 2H), 3.55 (dd, J = 1.8, 1.8 Hz, 1H), 1.88-1.95 (m, 1H), 1.73-1.81 (m, 1H), 1.65-1.71 (m, 2H), 1.37 (s, 3H), 1.32 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 137.2, 128.4, 128.2, 127.8, 109.7, 101.1, 75.7, 74.3, 73.1, 70.2, 62.5, 60.3, 29.1, 27.8, 26.3; CIHRMS: Calculated for [C18H25N3O5Na+]: 386.1686, found: 386.1680.
4.1.14. 3-((3aS,4S,6S,7R,7aS)-7-azido-4-(benzyloxy)-tetrahydro-2,2-dimethyl-3aH-[1,3]dioxolo[4,5-c]pyran-6-yl)propyl methanesulfonate (43)
To a stirred solution of alcohol 42 (1.8 g, 4.95 mmol) and Et3N (1.5 g, 14.9 mmol) in dry CH2Cl2 (4.95 mL) was added dropwise CH3SO2Cl (1.7 g, 1.49 mmol) at 0 °C. The reaction mixture was allowed to keep stirring for 0.5 h, and then water 30 mL was added, extracted with Et2O (3 × 200 mL), dried (Na2SO4), and concentrated under reduced pressure. The resulting crude product was purified using silica gel flash chromatography eluting with 45% EtOAc/hexane to give mesylate 43 (2.14 g, 4.85 mmol, 98%); colorless solid, mp: 85-87 °C; Rf = 0.61 (50% EtOAc/hexane); (c = 1.2, CH2Cl2); IR (thin film, cm-1) 2988, 2940, 2870, 2104, 1354, 1173, 1051, 832; 1H NMR (600 MHz, CDCl3) δ 7.26-7.37 (m, 5H), 4.90 (d, J = 11.4 Hz, 1H), 4.67 (d, J = 11.4 Hz, 1H), 4.43 (d, J = 7.8 Hz, 1H), 4.42 (dd, J = 5.4, 1.8 Hz, 1H), 4.30 (m, 1H), 4.25 (m, 1H), 4.06 (dd, J = 7.8, 5.4 Hz, 1H), 3.73 (ddd, J = 8.4, 4.8, 1.8 Hz, 1H), 3.53 (dd, J = 1.8, 1.8 Hz, 1H), 3.01 (s, 3H), 1.92-2.01 (m, 2H), 1.81-1.89 (m, 1H), 1.67-1.72 (m, 1H), 1.37 (s, 3H), 1.33 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 137.1, 128.4, 128.0, 127.8, 109.8, 101.2, 75.6, 74.3, 72.5, 70.4, 69.5, 60.1, 37.5, 27.7, 27.4, 26.3, 26.0; CIHRMS: Calculated for [C19H27N3O7SNa+]: 464.1462, found: 464.1467.
4.1.15. (3aR,9S,9aR,9bS)-octahydro-2,2-dimethyl-[1,3]dioxolo[4,5-a]indolizin-9-ol (44)
To a solution of acetonide 43 (410 mg, 0.928 mmol) in dry EtOH/THF (4 mL, v/v = 1:1) was added Pd(OH)2/C (100 mg) and the mixture was stirred under an atmosphere of H2 at room temperature. After reacting for 1 day, the mixture was concentrated, simply passed a short pad of silica gel to remove palladium toxicity byproduct and concentrated again. The resulting residue was dissolved in EtOH/THF (4 mL, v/v = 1:1) and Pd(OH)2/C (200 mg) was added. The suspended mixture was stirred for another 6 days under an atmosphere of H2 at room temperature. Then the catalyst was filtered off through a short pad of Celite, concentrated under reduced pressure. The resulting crude product was purified using silica gel flash chromatography eluting with 30% MeOH/EtOAc to give protected (-)-8-epi-d-swainsonine 44 (164 mg, 0.769 mmol, 83%); a colorless needles, Rf = 0.77 (50% MeOH/EtOAc); (c = 1.08, CH2Cl2); mp: 57.4-59.3 °C; IR (neat, cm-1); 3513, 2982, 2937, 2786, 1464, 1373, 1262, 1209, 1135, 1018; 1H NMR (600 MHz, CDCl3) δ 4.67 (dd, J = 6.0, 4.2 Hz, 1H), 4.56 (dd, J = 6.0, 4.2 Hz, 1H), 3.79 (ddd, J = 10.9, 8.9, 4.4 Hz, 1H), 3.11 (d, J = 10.8 Hz, 1H), 2.96 (dt, J = 10.8, 3.0 Hz, 1H), 2.52 (br s, 1H), 2.09 (dd, J = 10.8, 4.8 Hz, 1H), 2.00-2.03 (m, 1H), 1.82 (ddd, J = 10.8, 10.8, 3.6 Hz, 1H), 1.61-1.65 (m, 1H), 1.59 (dd, J = 9.0, 4.8 Hz, 1H), 1.51 (s, 3H), 1.36-1.40 (m, 1H), 1.28 (s, 3H), 1.21-1.32 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 111.5, 82.0, 78.1, 68.7, 65.8, 60.1, 53.3, 31.4, 25.8, 24.2, 19.7; CIHRMS: Calculated for [C11H19NO3H+]: 214.1443, found: 214.1438.
4.1.16. (1S, 2R, 8R, 8aR)-octahydroindolizine-1, 2, 8-triol (Swainsonine 1):24
To a solution of diol 6 (4.0 g, 9.96 mmol) in dry EtOH (40 mL) was added Pd(OH)2/C (1.5 g) and the mixture was stirred under H2 at an 100 psi pressure for 3 days at room temperature. The catalyst was filtered off through a short pad of celite, concentrated under reduced pressure. The resulting crude product was applied to ion-exchange chromatography (Dowex 1C 8, 200 mesh, OH- form) eluting with water. Removal of water in vacuo to give colorless needles d-swainsonine 1 (1.48 g, 8.5 mmol, 86%): mp: 143-144 °C; (c =1.1, MeOH); [lit. (c = 0.98, MeOH);24 (c = 1.03, MeOH); 21b (c = 0.21, EtOH); 21c (c = 2.33, MeOH)].21d Rf = (25% MeOH/CHCl3, w 1% NH4OH) = 0.38; IR (thin film, cm-1) 3287, 2953, 2722, 1639, 1405, 1338, 1141, 1076; 1H NMR (600 MHz, CD3OD) 4.26 (ddd, J = 8.0, 6.0, 2.4, 1H), 4.24 (m, 1H), 3.83 (ddd, J =4.5, 9.3, 10.8 Hz, 1H), 2.95-2.97 (m, 2H), 2.45 (dd, J = 7.2, 10.2 Hz, 1H), 2.08 (m, 1H), 1.92 (ddd, J = 11.6, 11.4, 2.8 Hz, 1H), 1.75 (dd, J = 3.6, 9.6 Hz, 1H), 1.71-1.74 (m, 1H), 1.70 (m, 1H), 1.64 (qt, J =13.2, 4.2 Hz, 1H), 1.25 (qd, J =12.6, 4.8 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 75.4, 70.9, 70.0, 67.2, 63.3, 53.3, 34.3, 24.7.
To a solution of protected swainsonine (ent)-31 (100 mg, 0.47 mmol) in THF was added 6 N HCl (0.47 mL) at room temperature over night. The resulting mixture was concentrated under reduced pressure and then eluted with water through an ion exchange column (Dowex 1X8 200 OH, 1 g). Removal of water in vacuo gives colorless needles swainsonine 2 (77.2 mg, 0.45 mmol, 95%). l-swainsonine 2: 1H, 13C NMR and IR are same as d-swainsonine 1, mp: 142-143 °C; (c = 0.92, MeOH); [lit. (c = 1.02, H2O); (c = 0.5, MeOH)].
4.1.17. (8R, 8aS)-octahydroindolizin-8-ol (4)
To a solution of mesylate 33 (42 mg, 0.1148 mmol) in dry EtOH/THF (0.5 mL, v/v = 1:1) was added Pd(OH)2/C (10 mg) and the mixture was stirred under an atmosphere of H2 at room temperature. After reacting for 1 day, the mixture was concentrated, simply passed a short celite pad to remove byproduct and concentrated again. The resulting residue was dissolved in EtOH/THF (0.5 mL, v/v = 1:1) and Pd(OH)2/C (10 mg) was added. The suspended mixture was stirred under H2 at a100 psi pressure for another 2 days at room temperature. Then the catalyst was filtered off through a short pad of celite, concentrated under reduced pressure. The resulting crude product was applied to ion-exchange chromatography (Dowex 1× 8, 200 mesh, OH- form) eluting with water. Removal of water in vacuo to give 8-hydroxyindolizidine 4 (11 mg, 0.078 mmol, 68%); Rf = 0.25 (MeOH); (c = 0.4, CH3OH); IR (thin film, cm-1) 3365, 2936, 2805, 1639, 1328, 1069; 1H NMR (600 MHz, CD3OD) δ 3.32 (ddd, J = 10.2, 9.0, 4.8 Hz, 1H), 3.06 (ddd, J = 9.0, 9.0, 3.0 Hz, 1H), 2.99 (m, 1H), 2.21 (q, J = 9.0 Hz, 1H), 2.11 (dddd, J = 12.0, 10.2, 6.6, 3.6 Hz, 1H), 1.96-2.01 (m, 2H), 1.70-1.83 (m, 4H), 1.59-1.67 (m, 1H), 1.48-1.57 (m, 1H), 1.22 (dddd, J = 15.0, 12.6, 10.2, 4.2 Hz, 1H),; 13C NMR (150 MHz, CD3OD) δ 73.4, 71.6, 55.1, 52.9, 34.5, 29.2, 25.1, 21.4; CIHRMS: Calculated for [C8H15NOH+]: 142.1232, found: 142.1228.
4.1.18. (1S, 2R, 8S, 8aR)-octahydroindolizine-1, 2, 8-triol ((-)-8-epi-d-swainsonine) (3):22
To a solution of protected (-)-8-epi-d-swainsonine 44 (120 mg, 0.56 mmol) in THF (1.2 mL) was added 6 N HCl (0.59 mL) at room temperature over night. The resulting mixture was concentrated under reduced pressure and then eluted with water through an ion exchange column (Dowex 1X8 200 OH-, 1 g). Removal of water in vacuo gave white powder (-)-8-epi-d-swainsonine 3 (92 mg, 0.53 mmol, 94%): Rf= (50 % MeOH/EtOAc) = 0.31; mp: 89-91 °C [lit. mp: 92-93 °C];22a (c = 0.75, MeOH); [lit. (c = 0.3, MeOH);22b (c = 0.67, MeOH)];22c IR (thin film, cm-1) 3360, 2937, 2789, 1645, 1329, 1138, 1013; 1H NMR (600 MHz, CD3OD) δ 4.32 (m, 2H), 4.20 (ddd, J = 7.2, 6.6, 1.8 Hz, 1H), 3.11 (m, 1H), 2.99 (dd, J = 10.2, 1.8 Hz, 1H), 2.34 (dd, J = 10.2, 7.2 Hz, 1H), 1.98-2.10 (m, 3H), 1.85-1.88 (m, 1H), 1.44-1.53 (m, 2H); 13C NMR (150 MHz, CD3OD) δ 74.3, 70.0, 69.5, 67.6, 63.1, 54.4, 32.2, 20.8; 22c,d CIHRMS: Calculated for [C8H15NO3H+]: 174.1130, found: 174.1125.
Supplementary Material
Complete experimental procedures and spectral data for all new compounds can be found in the Supporting Information. This material is available free of charge via the Internet.
Acknowledgments
We are grateful to NIH (GM63150) and NSF (CHE-0415469) for the support of our research program and NSF-EPSCoR (0314742) for a 600 MHz NMR at WVU.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.(a) Michael JP. Nat Rep. 2004;19:625–649. doi: 10.1039/b310689f. [DOI] [PubMed] [Google Scholar]; (b) Asano N, Nash RJ, Molyneux RJ, Fleet GWJ. Tetrahedron: Asymmetry. 2000;11:1645–1680. [Google Scholar]
- 2.(a) Guengerich FP, DiMari SJ, Broquist HP. J Am Chem Soc. 1973;95:2055–2056. [Google Scholar]; (b) Schneider MJ, Ungemach FS, Broquist HP, Harris TM. Tetrahedron. 1983;39:29–32. [Google Scholar]
- 3.(a) Hino M, Nakayama O, Tsurumi Y, Adachi K, Shibata T, Terano H, Kohsaka M, Aoki H, Imanaka H. J Antibiot. 1985;38:926–935. doi: 10.7164/antibiotics.38.926. [DOI] [PubMed] [Google Scholar]; (b) Patrick M, Adlard MW, Keshavarz T. Biotechnol Lett. 1993;15:997–1000. [Google Scholar]
- 4.(a) Colegate SM, Dorling PR, Huxtable CR. Aust J Chem. 1979;32:2257–2264. [Google Scholar]; (b) Colegate SM, Dorling PR, Huxtable CR. Plant Toxicol. 1985:249–254. [Google Scholar]
- 5.(a) Molyneux RJ, James LF. Science. 1982;216:190–191. doi: 10.1126/science.6801763. [DOI] [PubMed] [Google Scholar]; (b) Davis D, Schwarz P, Hernandez T, Mitchell M, Warnock B, Elbein AD. Plant Physiol. 1984;76:972–775. doi: 10.1104/pp.76.4.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liao YF, Lal A, Moremen KW. J Biol Chem. 1996;271:28348–28358. doi: 10.1074/jbc.271.45.28348. [DOI] [PubMed] [Google Scholar]
- 7.(a) Elbein AD, Solf R, Dorling PR, Vosbeck K. Proc Natl Acad Sci U S A. 1981;78:7393–7397. doi: 10.1073/pnas.78.12.7393. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kaushal GP, Szumilo T, Pastuszak I, Elbein AD. Biochemistry. 1990;29:2168–2176. doi: 10.1021/bi00460a030. [DOI] [PubMed] [Google Scholar]; (c) Pastuszak I, Kaushal GP, Wall KA, Pan YT, Sturm A, Elbein AD. Glycobiology. 1990;1:71–82. doi: 10.1093/glycob/1.1.71. [DOI] [PubMed] [Google Scholar]
- 8.(a) Goss PE, Baker MA, Carver JP, Dennis JW. Clin Cancer Res. 1995;1:935–944. [PubMed] [Google Scholar]; (b) Das PC, Robert JD, White SL, Olden K. Oncol Res. 1995;7:425–433. [PubMed] [Google Scholar]; (c) Goss PE, Reid CL, Bailey D, Dennis JW. Clin Cancer Res. 1997;3:1077–1086. [PubMed] [Google Scholar]
- 9.Davis B, Bell AA, Nash RJ, Watson AA, Griffiths RC, Jones MG, Smith C, Fleet GWJ. Tetrahedron Lett. 1996;37:8565–8568. [Google Scholar]
- 10.For a review of swainsonine syntheses, see: Nemr AE. Tetrahedron. 2000;56:8579–8629.. For more recent syntheses, see: Martin R, Murruzzu C, Pericas MA, Riera A. J Org Chem. 2005;70:2325–2328. doi: 10.1021/jo048172s.Heimgaertner G, Raatz D, Reiser O. Tetrahedron. 2005;61:643–655.Song L, Duesler EN, Mariano PS. J Org Chem. 2004;69:7284–7293. doi: 10.1021/jo040226a.Lindsay KB, Pyne SG. Aus J Chem. 2004;57:669–672.Pearson WH, Ren Y, Powers JD. Heterocycles. 2002;58:421–430.Lindsay KB, Pyne SG. J Org Chem. 2002;67:7774–7780. doi: 10.1021/jo025977w.Buschmann N, Rueckert A, Blechert S. J Org Chem. 2002;67:4325–4329. doi: 10.1021/jo025589u.Zhao H, Hans S, Cheng X, Mootoo DR. J Org Chem. 2001;66:1761–1767. doi: 10.1021/jo001447t.Ceccon J, Greene AE, Poisson JF. Org Lett. 2006;8:4739–4712. doi: 10.1021/ol0617751.Au CWG, Pyne SG. J Org Chem. 2006;71:7097–7099. doi: 10.1021/jo0610661.Dechamps I, Pardo D, Cossy J. ARKIVOC. 2007;5:38–45.Guo H, O’Doherty GA. Org Lett. 2006;8:1609–1612. doi: 10.1021/ol0602811.. For the first syntheses, see: Mezher HA, Hough L, Richardson AC. J Chem Soc Chem Commun. 1984:447–448.Fleet GWJ, Gough MJ, Smith PW. Tetrahedron Lett. 1984;25:1853–1856.
- 11.Our initial effort towards the synthesis of swainsonine was previously disclosed, see ref. 10m.
- 12.Both d- and l-swainsonine have been prepared in our group, for simplicity herein we only show the approach to d-swainsonine.
- 13.(a) Fujii A, Hashiguchi S, Uematsu N, Ikariya T, Noyori R. J Am Chem Soc. 1996;118:2521–2522. [Google Scholar]; (b) Wang F, Liu H, Cun L, Zhu J, Deng J, Jiang Y. J Org Chem. 2005;70:9424–9429. doi: 10.1021/jo0514826. [DOI] [PubMed] [Google Scholar]
- 14.Achmatowicz O, Bielski R. Carbohydr Res. 1977;55:165–176. doi: 10.1016/s0008-6215(00)84452-3. For its recent use in carbohydrate synthesis, see: Guo H, O’Doherty GA. Org Lett. 2005;7:3921–3924. doi: 10.1021/ol051383e.
- 15.(a) Zhou M, O’Doherty GA. Org Lett. 2006;8:4342. [Google Scholar]; (b) Guppi SR, Zhou M, O’Doherty GA. J Org Chem. 2007;72:4966–4969. doi: 10.1021/jo070326r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Babu RS, O’Doherty GA. J Am Chem Soc. 2003;125:12406–12407. doi: 10.1021/ja037097k. For its application in the de novo syntheses see: Babu RS, Zhou M, O’Doherty GA. J Am Chem Soc. 2004;126:3428–3429. doi: 10.1021/ja039400n.Guo H, O’Doherty GA. Angew Chem Int Ed. 2007;46:5206–5208. doi: 10.1002/anie.200701354.
- 17.While the use of CeCl3 was not required for the stereoselectivity, the Luche conditions provided faster reactions, see: Luche JL. J Am Chem Soc. 1978;100:2226–2227.Haukaas MH, O’Doherty GA. Org Lett. 2001;3:401–404. doi: 10.1021/ol006907j.
- 18.(a) VanRheenen V, Kelly RC, Cha DY. Tetrahedron Lett. 1976;17:1973–1976. [Google Scholar]; (b) Shan M, O’Doherty GA. Org Lett. 2006;8:5149–5152. doi: 10.1021/ol062076r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Oliveira RN, Cottier L, Sinou D, Srivastava RM. Tetrahedron. 2005;61:8271–8281. [Google Scholar]
- 20.(a) Hoffmann RW, Brückner D. New J Chem. 2001;25:369–373. [Google Scholar]; (b) Martín-López MJ, Rodriquez R, Bermejo F. Tetrahedron. 1998;54:11623–11636. [Google Scholar]; (c) Magnus P, Padilla AI. Org Lett. 2006;8:2569–2571. doi: 10.1021/ol061391a. [DOI] [PubMed] [Google Scholar]; (d) Hoffmann RW, Brückner D, Gerusz VJ. Heterocycles. 2000;52:121–124. [Google Scholar]
- 21.(a) Oishi T, Iwakuma T, Hirama M, Ito S. Synlett. 1995:404–406. [Google Scholar]; (b) Naruse M, Aoyagi S, Kibayashi C. J Org Chem. 1994;59:1358–1364. [Google Scholar]; (c) Adams CE, Walker FJ, Sharpless KB. J Org Chem. 1985;50:420–422. [Google Scholar]; (d) Bennett RB, Choi JR, Montgomery WD, Cha JK. J Am Chem Soc. 1989;111:2580–2582. [Google Scholar]
- 22.(a) Ikota N, Hanaki A. Chem Pharm Bull. 1990;38:2712–18. [Google Scholar]; (b) Kim YG, Cha JK. Tetrahedron Lett. 1989;30:5721–5724. [Google Scholar]; (c) Austin GN, Baird PD, Fleet GWJ, Peach JM, Smith PW, Watkin DJ. Tetrahedron. 1987;43:3095–3108. [Google Scholar]; (d) Tadano K, Limura Y, Hotta Y, Fukabori C, Suami T. Bull Chem Soc Jpn. 1986;59:3885–3892. [Google Scholar]
- 23.Presented in this Experimental Section are the experimental procedures and spectral data for all new compounds. Complete experimental procedures and spectral data for all compounds are presented in Supporting Information.
- 24.Pearson WH, Hembre EJ. J Org Chem. 1996;61:7217–7221. doi: 10.1021/jo961101b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete experimental procedures and spectral data for all new compounds can be found in the Supporting Information. This material is available free of charge via the Internet.