

Abstract
Background & Aims
Specific mutations in the adenomatous polyposis coli (APC) gene can lead to an attenuated form of the Familial Adenomatous Polyposis (AFAP). Although AFAP mutation carriers have a 69% risk of colorectal cancer by age 80, clinical recognition remains a challenge in some cases, as they present with few colonic adenomas and are difficult to distinguish clinically from patients with sporadic polyps.
Methods
Family relationships were established using family history reports, the Utah Population Database, and the public records of the Mormon Church. Genetic analysis of representative family members was done using the Affymetrix 10K SNP array platform. Colonoscopy data were available on 120 individuals with the AFAP mutation.
Results
Two large AFAP kindreds with the identical APC disease causing mutation (c.426_427delAT) link to a founding couple who came to America from England around 1630. Genetic analysis demonstrates that the two families share a conserved haplotype of 7.17 Mbp surrounding the mutant APC allele. The data shows that 36.6% of the mutation-positive family members have fewer than 10 colonic adenomatous polyps, and 3 (6.8%) of these individuals were diagnosed with colorectal cancer.
Conclusions
In view of the apparent age of this mutation, a notable fraction of both multiple adenoma patients and perhaps even colon cancer cases in the U.S. could be related to this founder mutation. The colon cancer risk associated with the mutation makes genetic testing of considerable importance in patients with a personal or family history of either colonic polyps or cancer at a young age.
Familial adenomatous polyposis (APC [MIM 175100]) is a colon cancer syndrome characterized by the early age onset of hundreds to thousands of adenomatous polyps in the colon and nearly 100% risk of developing colon cancer at an mean age of 39 years if the colon is not removed 1, 2, 3. Mutation carriers may also present with polyps in the upper GI tract and have an increased risk of small bowel, thyroid, brain, and other malignancies. Mutations in the APC gene are the most common cause of this syndrome which is an autosomal-dominant condition with a prevalence estimated at 1:10,000 persons 4, 5. Extracolonic features can include osteomas, dental anomalies, cutaneous lesions, desmoids and congenital hypertrophy of the retinal pigment epithelia (CHRPE). Mutations in the proximal part of the APC gene (the first 5 exons), the distal end of the APC gene (3′ to codon 1596) and in exon 9 of the APC gene lead to an attenuated form of familial adenomatous polyposis (AFAP or AAPC) 3, 6–9. In contrast to FAP, AFAP is characterized by fewer colonic adenomas (clinically defined as less than 100 adenomatous polyps) which have a more proximal distribution (versus FAP, which has polyps covering the entire colon), and later age of onset 10–13. The mean age of colorectal cancer diagnosis in AFAP is reported at 51 to 58 years (15 to 20 years later than FAP) and the lifetime risk of colorectal cancer is decreased in AFAP compared to typical FAP 10–13. The molecular mechanism underlying the attenuated phenotype is not clear, but a variety of models have been proposed. In some cases, the classic FAP phenotypic may result from a dominant-negative truncating APC mutation that interferes with the normal APC protein 14. Deletions of the entire APC gene, however, can result in classic FAP 15 as well as AFAP 16 which argues against a dominant-negative model as a general mechanism. Somatic mutations and loss of heterozygosity have been observed in the attenuated APC allele (as well as the normal allele) in cancer suggesting that the attenuated APC allele retains some function that protects against the progression to cancer 17, 18. A putative alternative start site at codon 184 (exon 5) has been proposed to reinitiate protein synthesis downstream of a 5′ mutation 19. In addition, it has been suggested that AAPC mutations in exon 9 may produce normal APC protein by alternative splicing 20, 21. Recently, a recessive form of adenomatous polyposis due to mutations in the MYH gene was described and differentially named “MYH-associated polyposis” 22, 23.
Clinical characterization of large AFAP families has revealed a highly variable phenotype, even in individuals with the identical genetic mutation 10–13, 24. Polyp numbers from zero to many hundreds with both extremes observed even at older ages have been reported in mutation-carriers. Upper gastrointestinal polyps, both gastric and duodenal, are often, but not always, present and extracolonic features such as osteomas, epidermoid cysts, desmoids, and cutaneous lipomas are frequently reported. We recently published the detailed phenotype of two large AFAP families (Kindred 353 and Kindred 439) with the identical APC mutation (exon 4: c.426_427delAT) 13. This frameshift results in a downstream stop codon and a predicted truncated APC peptide of 145 amino acids. In this earlier report, initial colonoscopic evaluation of 120 individuals harboring this mutation revealed a median number of 25 colonic adenomas at a mean age of examination of 42 years. A total of 27 colorectal cancer cases at a mean age of 58 years were documented and a cumulative colorectal cancer risk of 69% by age 80 was estimated.
Founder mutations in hereditary colon cancer syndromes are commonly reported, in particular for the mismatch repair genes responsible for Lynch syndrome (MLH1, MSH2, MSH6 and PMS2). Of note is an “American Founder Mutation” in MSH2 that dates back to the 1700’s 25. Founder mutations in the APC gene, however, are rare. There is a founder FAP mutation in the Spanish Balearic islands 26, founder AFAP mutations in Newfoundland 24 and the family reported here, and an ancient I1307K variant of APC conferring a 2-fold risk of colon cancer 27.
We have long suspected that these two AFAP families originate from a common ancestor as this mutation has only been reported in families residing in North America 6. Kindred 353 is from Utah and more than 7000 descendents spanning nine generations are recorded in the Utah Population Database (UPDB). UPDB includes genealogies abstracted from the Utah Family History Library, maintained by the Church of Jesus Christ of Latter-day Saints (LDS or Mormon), linked to state-wide cancer records from the Utah Cancer Registry and the Cancer Data Registry of Idaho, Utah death certificates, and Utah birth certificates 28. The founding parents were born in Massachusetts and New York in the 1790’s and were part of the Mormon immigration to Utah in the 1850’s. Kindred 439 is from upstate New York and spans six generations. The founding parents were born in New York in the 1830’s. In this report, we provide genealogic and genetic evidence linking the two kindreds to a founding couple who were born in England in the 1590’s and came to America with their children around 1630. This extensive ancestry suggests that there may be many unidentified families harboring this founder mutation in North America, however, clinical recognition of this condition remains a challenge. We report that 36.6% of the mutation-positive family members have fewer than 10 colonic adenomatous polyps, and 13.3% have no family history of a first-degree relative with more than 10 adenomatous polyps. This often makes patients with AFAP difficult to distinguish clinically from patients with sporadic polyps.
Methods
Research Subjects and Genealogy
This study was approved by the University of Utah Institutional Review Board and by the University of Utah Resource for Genetic and Epidemiologic Research which oversees the appropriate use of the Utah Population Database. Informed consent was obtained from all research participants. Research participants included 490 members from two affected branches of Kindred 353 (145 of these are mutation-positive) and 99 members of Kindred 439 (36 of these are mutation-positive). Colonoscopy results with precise polyp number were available on 120 of the 181 mutation-positive individuals as previously reported 13. These results are extracted from the initial colonoscopy screening for the individual and in some cases, from a corresponding colectomy procedure.
Family relationships were established using family history reports, standard genealogy research methods, the Utah Population Database (K353), and the public records of the Mormon Church (familysearch.org web site). A list of common surnames from K353 and K439 ancestors was compared to facilitate identification of the founder.
SNP analysis
The Affymetrix GeneChip Human Mapping 10,000 SNP array (HMA10K) was used. Samples were processed according to the GeneChip Mapping Assay Manual (Affymetrix). Briefly, 250 ng of genomic DNA was digested with XbaI. The Adaptor Xba (Affymetrix) was then ligated to the digested DNA. Each ligation reaction was diluted and added to four separate 100-μl PCRs. Products were purified with QIAquick spin columns (Qiagen) then fragmented and end-labeled. A hybridization cocktail was added to the labeled DNA and then heat-denatured, cooled and incubated at 48°C for 2 min. Each sample was added to a HMA10K and hybridized at 48°C for 16 h in an Affymetrix GeneChip Hybridization Oven at 60 rpm. After 16 h, the probe arrays were washed and stained according to the GeneChip Mapping Assay Manual by use of the DNAARRAY_WS2 protocol on the Affymetrix Fluidics Station 400. Arrays were scanned once with the Affymetrix GeneChip® Scanner 3000 and analyzed with Affymetrix GeneChip DNA Analysis Software (GDAS) to generate genotype assignments for each of the SNP probes on the array.
The chromosome 5 haplotypes associated with the AFAP mutation were determined by analyzing DNA from the affected parent (and in one case both parents) and the indicated individual. NCBI build 35 from May 2004 was used to define physical map positions.
Four short tandem repeat (STR) genetic markers flanking the APC locus (D5S2501 at 111,037,094 Mbp, D5S2027 at 111,173,318 Mbp, D5S346 at 112,241,523 Mbp, and D5S421 at 112,882, 042 Mbp) were used to establish a shared haplotype for the APC locus. The corresponding heterozygosity for the markers are 65%, 73%, 85%, and 78% as reported by the CEPH genotype database browser V2.1 (http://www.cephb.fr/cephdb/php/eng/index.php). Genetic marker PCR products were resolved on Applied Biosystems 3130xl Genetic Analyzer with a 36cm capillary using POP-7 polymer.
Linkage analysis
Parametric linkage analysis under a dominant model was performed using the Variation List vs. Trait analysis of GeneSpring GT (Agilent; http://www.chem.agilent.com/). This program uses the Elston-Stuart (ES) algorithm 29 as described in section 9.2 by Ott in Analysis of Human Genetic Linkage, 30. The variation list was generated from the SNP data. Pedigree structure shown in Figure 1 was used and the 6 mutation-positive cases plus their mutation-positive parent were coded as having the trait (affected), the spousal parent was coded as unaffected, and upper generations were coded as unknown. The analysis also used the deCODE genetic map with Haldane mapping function and the Affymetrix Caucasian allele frequencies for the SNPs.
Figure 1. AFAP American Founder Pedigree.
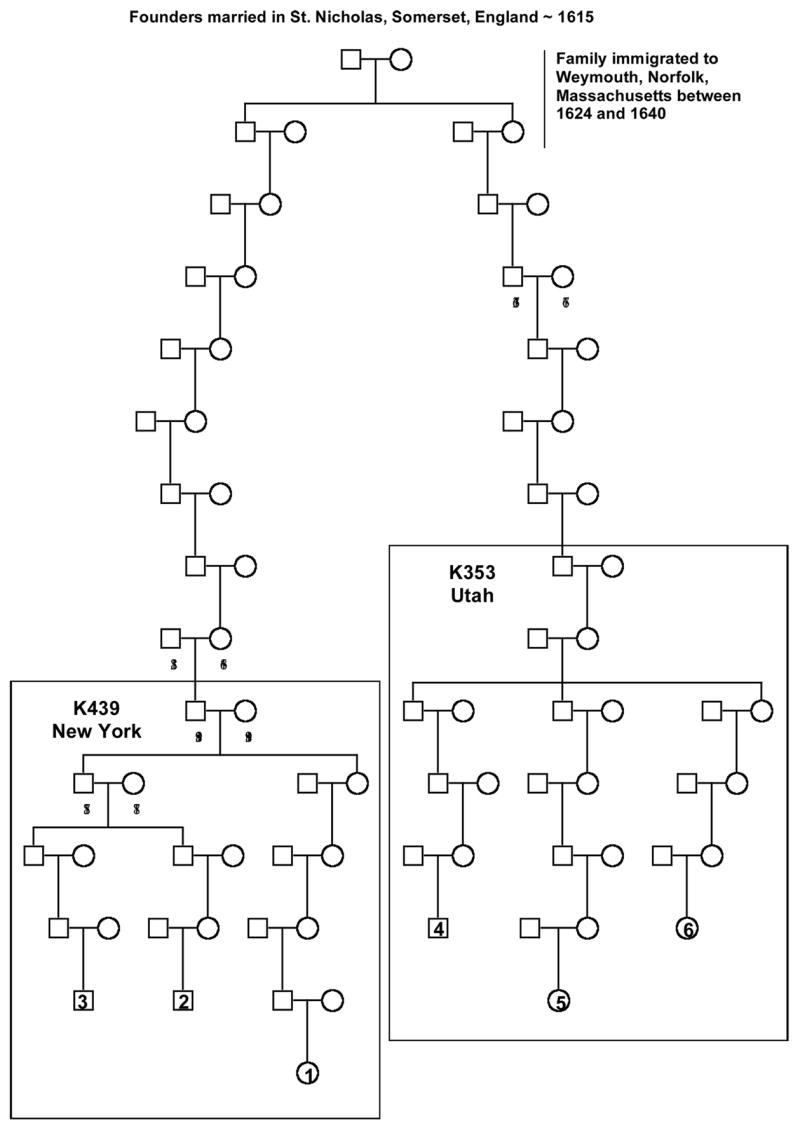
A linear pedigree is shown from the founding parents to the six AFAP mutation-positive individuals reported in the SNP haplotype analysis. The reference numbers for the six individuals are indicated on the pedigree (1 to 6) and correspond to the individual numbers in Figure 2. Birth years are also reported for each individual where the data is available. Birth decades are reported for the lower generations to protect the individuals’ identities. The separate 353 and 439 kindreds which have been extensively studied are shown within the pedigree.
Analysis of relationship between colorectal polyp number and cancer
The 120 mutation-positive subjects with a quantitative polyp number at their first exam were divided into three groups by polyp count (0–9, 10–50 and >50). The three categories were defined by clinically relevant criteria used by practicing endoscopists. A finding of less than ten polyps is often considered a sporadic polyp case. A finding of ten or more polyps is suggestive of a syndrome and indicates genetic testing. Cases with more than 50 polyps strongly indicate a syndrome and are often referred to surgery for colon resection. Nine of the 120 subjects were diagnosed with colorectal cancer at the time of their first exam as indicated in Table 1. Exact confidence intervals for the proportion of subjects with colon cancer in each group were calculated using R 2.1.1 statistical software (R Foundation for Statistical Computing). Logistical regression modeling was used to explore the effect of polyp count on the probability of cancer, while adjusting for age at exam and gender. No threshold effects of age or polyp count were evident from exploratory scatter plots. In the logistic regression models, age (in years) and polyp count were incorporated as linear predictors. Multivariate logistic regression models were also evaluated. Starting with a model with all three predictors and 2nd order interaction terms for gender with polyps and age with polyps, predictors that were not statistically significant were removed one at a time, (starting with the interaction terms), until the final model was obtained. None of the removed terms was statistically significant at the p = 0.10 significance level.
Table 1.
Prevalence estimate of cancer based on polyp number.
| Polyp number | # subjects without colorectal cancer | # subjects with colorectal cancer | Prevalence of cancer (95% CI) |
|---|---|---|---|
| 0–9 | 41 | 3 | 6.8 % (1.4% – 18.7%) |
| 10–50 | 31 | 3 | 8.8% (1.9% – 23.7%) |
| >50 | 39 | 3 | 7.1% (1.5% – 19.5%) |
| All groups | 111 | 9 | 7.5% (3.5% – 13.8%) |
Results
Identification of common founder
Using family history records and standard genealogy methods, Kindred 353 and Kindred 439 were traced to a common founding couple who were born in England in the 1590s (Figure 1). The couple was married in St. Nicholas, Somerset, England in 1615 and had four children born in England between 1615 and 1624. The couple, along with at least two of their children, arrived in America some time before 1640, when their daughter was married in Weymouth, Norfolk, Massachusetts. A son born in 1615 is the ancestor to Kindred 439 and a daughter, born in 1620, is the ancestor to Kindred 353. The birth year of each parent is indicated on the pedigree. To our knowledge, the APC mutation (c.426_427delAT) has not been found in England or Europe suggesting that the mutation originated with this couple (personal communications Ian Tomlinson, Anne Lyster Knudsen, Riccardo Fodde). The exception is one case from a German registry 31, however the reported individual is presumed to have originated from America and had family there (personal communication Waltraut Friedl).
Kindred 353 and Kindred 439 share an identical SNP haplotype around the APC gene
To rule out the possibility that the two kindreds share a common ancestor by coincidence and that the identical mutation in the APC gene arose independently, the SNP haplotype around the APC gene was examined. Six individuals and their affected parents (three from each kindred) were genotyped with SNPs to determine which SNPs were coinherited with the mutant APC allele and which SNPs came from the unaffected spouse (Figure 1). The six individuals clearly share a haplotype on the allele inherited from the affected parent, providing evidence that these individuals originate from a common founder with the APC mutation (Figure 2). The minimal non-recombinant region is 7.17 Mbp or approximately 5.43 cM based on the deCODE genetic map. Parametric linkage analysis under a dominant model gave a statistically significant maximum LOD score of 5.2 (p-value = 9.4 × 10−7) at SNP rs1822488, which is just distal to the APC gene (Figure 2). This indicates that the two branches have not coinherited this region by chance.
Figure 2. SNP analysis of 6 kindred members shows a minimal shared haplotype around the APC gene of 7.17 Mbp (5.43 cM).
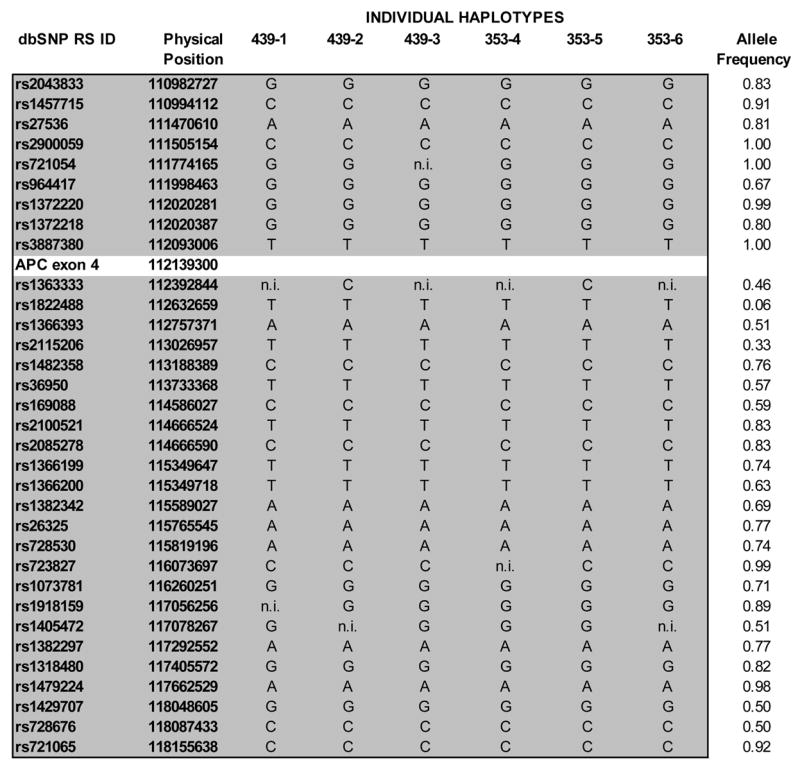
The individual’s numbers at the top of each column correspond to the number indicated on the pedigree (Figure 1). The minimal shared haplotype including the SNP identifiers and physical position (in Mbp) and specific allele are represented in the region shaded grey. The frequencies of the alleles as reported by Affymetrix for 118 unrelated Caucasian controls are listed. The APC position indicated represents exon 4 of the APC gene where the inherited mutation resides. “n.i.” indicates non-informative for that individual for that particular SNP.
Thirteen additional AFAP kindred share the founder mutation and an APC haplotype
The genealogic data is not sufficiently complete to estimate the number of descendents from the founding couple, but the number of generations suggests that there are likely to be many more North American families harboring the APC (c.426_427delAT) mutation. This founder mutation has been published in two additional American families ascertained by H. Lynch (K2764, Creighton University) and P. Lynch (K3101, M.D. Anderson) and all four kindreds (K353, K439, K2764, and K3101) are reported to share an identical disease haplotype 6. Furthermore, families harboring this mutation and not known to link to the Utah founder (K353) or the New York founder (K439) have been identified by clinicians in Michigan, New York, Vermont, Wisconsin, and Washington (personal communications Wendy Kohlmann, Wendy McKinnon, Stephen Gruber, Peter Levonian, and Sara Michelson). Four highly polymorphic STR genetic markers, D5S2501, D5S2027, D5S346, and D5S421, surrounding APC were used to genotype 15 previously unlinked families (Table 2). They all share an identical haplotype surrounding the APC locus including D5S2027, D5S346 and D5S421 (Table 2). The two Wisconsin families, have a different genotype at D5S2501, at the end of the linked region, demonstrating a likely recombination, narrowing the shared region to 6.69 Mbp, and ending near SNP, rs27536 (Figure 2).
Table 2.
APC haplotype of kindreds with AFAP founder mutation
| Kindred | Origin | Source of Kindred | Link to founder | # tested | APC Haplotype | CEPH allele frequencies |
|---|---|---|---|---|---|---|
| K353 | Utah | Spirio et al. 6 | Yes | 3 | 3:2:5:2 | 0.37: 0.31: 0.07: 0.15 |
| K439 | New York | Spirio et al. 6 | Yes | 1 | 3:2:5:2 | 0.37: 0.31: 0.07: 0.15 |
| K3101 | Texas | Spirio et al. 6 | UNK | 2 | 3:2:5:2 | 0.37: 0.31: 0.07: 0.15 |
| K2467 | Nebraska | Spirio et al. 6 | UNK | 2 | 3:2:5:2 | 0.37: 0.31: 0.07: 0.15 |
| K37326 | Washington | PC (S. Michelson) | UNK | 1 | 3:2:5:2 | 0.37: 0.31: 0.07: 0.15 |
| K63 | Michigan | PC (W Kohlmann, S Gruber) | UNK | 2 | 3:2:5:2 | 0.37: 0.31: 0.07: 0.15 |
| K206 | Michigan | PC (W Kohlmann, S Gruber) | UNK | 3 | 3:2:5:2 | 0.37: 0.31: 0.07: 0.15 |
| K631 | Michigan | PC (W Kohlmann, S Gruber) | UNK | 1 | 3:2:5:2 | 0.37: 0.31: 0.07: 0.15 |
| K2711 | Michigan | PC (W Kohlmann, S Gruber) | UNK | 1 | 3:2:5:2 | 0.37: 0.31: 0.07: 0.15 |
| K2739 | Michigan | PC (W Kohlmann, S Gruber) | UNK | 1 | 3:2:5:2 | 0.37: 0.31: 0.07: 0.15 |
| K5254 | Michigan | PC (W Kohlmann, S Gruber) | UNK | 1 | 3:2:5:2 | 0.37: 0.31: 0.07: 0.15 |
| K19342 | Wisconsin | PC (P Levonian) | UNK | 2 | 2:2:5:2 | 0.12: 0.31: 0.07: 0.15 |
| K20536 | Wisconsin | PC (P Levonian) | UNK | 2 | 2:2:5:2 | 0.12: 0.31: 0.07: 0.15 |
| K3506 | Vermont | PC (W McKinnon) | UNK | 1 | 3:2:5:2 | 0.37: 0.31: 0.07: 0.15 |
| K3031 | Vermont | PC (W McKinnon) | UNK | 3 | 3:2:5:2 | 0.37: 0.31: 0.07: 0.15 |
| K3877 | Vermont | PC (W McKinnon) | UNK | 0 | UNK |
PC indicates personal communication. UNK indicates unknown. The # tested represents the number of kindred members with the AFAP founder mutation tested for the APC haplotype. The APC haplotype alleles are in the following order: D5S2501:D5S2027: D5S346:D5S421. The reported allele numbers and corresponding CEPH allele frequencies are based on those reported by the CEPH genotype database browser V2.1 (http://www.cephb.fr/cephdb/php/eng/index.php). The CEPH allele frequencies are based on a minimum of 27 unrelated Caucasian individuals from 8 kindreds.
Clinical implications of a common AFAP mutation
One of the most striking aspects about an American founding mutation dating to the 1600’s is the potentially large number of mutation-positive descendents in North America. Since AFAP is an adult-onset disease and the mean age of colon cancer is ~58 years, there is little if any selective pressure against transmission of the mutant allele. The K353 Utah founding father who was born in 1794 and immigrated to Utah around 1850 has 9 descending generations with 7818 descendents (5247 still living) recorded in the Utah Population Database. The mutation is transmitted in two of five of the branches descending from this founder. Known mutation-positive individuals descending from this founder account for 0.15% of all colorectal cancers in Utah reported from 1966–1995. From 1996 to 2003, the mutation-positive family members accounted for 0.02% of colorectal cancers reported to Utah Cancer Registry which coincided with aggressive education and clinical intervention in the family.
Although AFAP is currently considered a rare condition, estimated to account for less than 1% of all colon cancers 1, it may be under-diagnosed. Many affected individuals are difficult to distinguish from individuals with sporadic adenomatous polyps or colon cancer without genetic testing 13. In our study of the two kindreds, 44 of 120 (36.6%) mutation-positive family members with colonoscopic data had zero to nine adenomatous polyps. Of these, 25 (20.9%) had zero to three adenomatous polyps. The median age of this zero to nine polyp group was 32.5 years (mean, 36.4; range 16 to 67). However, family history and age when polyps develop can help distinguish this hereditary condition. Of the zero to nine polyp group, 28 mutation-positive individuals (63.6 %) had a first-degree relative enrolled in the study with more than 10 polyps, nine (20.4%) had no first-degree relatives enrolled in the study with more than 10 polyps and seven (15.9%) did not have first-degree relatives with polyp data enrolled in the study. Taken together, 16 of 120 mutation-positive individuals (13.3%) have fewer than 10 polyps and no first-degree relatives with more than 10 polyps, thus appearing like a sporadic polyp case, even with reasonably skillful family history intake.
One surprising clinical detail is that the mutation-positive individuals with low polyp numbers do develop colorectal cancer. The total polyp numbers leading up to the cancer diagnosis were available on only 9 of 27 mutation-positive family members. Of the individuals with fewer than 10 polyps, three had colorectal cancer (6.8%; age 29, 57 and 53); of the individuals with 10 to 50 polyps, three had colorectal cancer (8.8%; age 35, 41, and 58); and of the individuals with more than 50 polyps, three had colorectal cancer (7.1%; age 36, 73, and 80) (Table 1). Logistical regression modeling was used to explore the effect of polyp count on the probability of cancer, while adjusting for age at exam and gender. Gender had no statistical significance, so polyp count and age were used as the only predictors in the final model. The estimated log-odds of colon cancer for each additional year of age was 0.053 (95% confidence interval 0.0055–0.10, p =0.03). The estimated log-odds associated with each additional polyp was −0.003 (95% confidence interval. −0.020 – 0.014, p = 0.72). The effects of age and polyp count were similar with and without the other predictor in the model. In each of the models, the estimated effect of polyp count on the risk of cancer was negative and not statistically significant.
Comment
We have previously described the clinical phenotype of two American kindreds with the identical APC mutation, and have long suspected common ancestry. Here we present clear evidence linking both pedigrees to a founding couple who came to America from England in the early 1600s. Additionally, K353 and K439 members share an identical genetic haplotype of 7.17 Mbp around the APC locus. Thirteen additional American kindreds with the identical mutation share the APC haplotype. Taken together, these results strongly indicate that the ancestral pair identified in this study represent the founding AFAP couple of the mutation under consideration. Since families harboring this mutation have not been reported in England, it is not known if the mutation originated from the husband or wife.
Nevertheless, clinical recognition of AFAP remains a challenge. Consistent with reports of other AFAP families, clinical evaluation of these two families demonstrates a highly variable phenotype and a substantial percentage of affected individuals whose clinical presentation would be difficult to distinguish from sporadic polyps in the absence of genetic testing 10, 11, 24. The variable phenotype may be attributable to genetic and/or environmental modifiers. The estimated cumulative risk of colorectal cancer by age 80 is 69%, with a corresponding standardized incidence ratio (SIR) of 17.9 (95% CI: 8.4–24.7) and, as described here, early colorectal cancers occur even in those with low polyp numbers. By comparison, a case-control study of individuals with two or more adenomas on initial sigmoidoscopy examination and followed over an average of 14 years, demonstrated a SIR of colon cancer of 4.8 (95%CI: 2.4–8.7) 32. Currently, the American Gastroenterological Association recommends genetic testing for AFAP in those with ≥ 20 adenomas but no known family history 33. However, the AGA technical review of this subject details that “no clear standard exists concerning the number of colorectal adenomas needed to suspect this diagnosis in the index case” 34. Based on our observation of mutation carriers developing colon cancer, regardless of polyp number, this guideline may be insensitive. More recently, we and others have suggested that genetic testing should be considered when 10 or more colonic adenomatous polyps have been found in a patient at one examination or over time 1, 35. Even this suggestion is an over simplification, as 36.6% of our mutation-positive family members who were examined by colonoscopy had fewer than 10 adenomas. Other large AFAP studies have observed fewer than 10 polyps in mutation-positive individuals, suggesting that this is not specific to this family and mutation 10, 11, 24. A family history of colonic polyps or cancer in combination with any number of adenomatous polyps at a young age may be the key to recognizing potential AFAP patients from the vast number of individuals with sporadic adenomas. We suggest that genetic testing should be considered in any individual with a family history of 10 or more colonic adenomas based on the observation that 86.7% of mutation-positive individuals in this family would be captured with this criterion
One can speculate that this specific mutation may constitute a notable and possibly definable fraction of the multiple adenoma patients and even the colorectal cancer burden in the U.S. Mutations in the APC gene have been reported to occur in at least 10% of individuals with multiple, but less than 100, adenomas (range=3–96; median=6.5; mean=14) 36. Determination of the actual prevalence of this American founder mutation in colonic adenoma and cancer patients will help define the optimal clinical situation in which to offer genetic testing.
Understanding that both K353 and K439 are from the same ancestor is important, as we analyze the clinical similarities and differences between branches. Additionally, identification and inclusion of new families harboring the AFAP founder mutation will expand our current knowledge regarding the similar and variable components of this genetic disorder. Further investigation of environmental and genetic factors that modify all aspects of the AFAP phenotype will be the key to unraveling the complexities of clinical risks as well as the basic biology of colon cancer progression.
Acknowledgments
Grant support
This research is supported by NCI grants P01-CA073992 (RWB) R01-CA040641(RWB), the Utah Cancer Registry, which is funded by contract # NCI-CN-67000, the Utah Department of Health, the University of Utah, and Huntsman Cancer Foundation. Partial support of UPDB is provided by University of Utah and Huntsman Cancer Institute.
Thank you to over 1000 family members who have participated in this research over the years. Thank you to our collaborators who shared their AFAP families and DNA samples, Stephen Gruber, Wendy Kohlmann, Peter Levonian, Henry Lynch, Patrick Lynch, Wendy McKinnon and Sara Michelson. We also acknowledge Jared Cox for generating pedigrees and Therese Tuohy for her critical reading of this manuscript.
Abbreviations
- FAP
familial adenomatous polyposis
- AFAP
attenuated familial adenomatous polyposis
- SNP
single nucleotide polymorphism
- CHRPE
congenital hypertrophy of the retinal pigment epithelia
- UPDB
Utah Population Database
- STR
short tandem repeat
Footnotes
No conflicts of interest exist
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Burt R, Neklason DW. Genetic testing for inherited colon cancer. Gastroenterology. 2005;128:1696–716. doi: 10.1053/j.gastro.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 2.Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919–32. doi: 10.1056/NEJMra012242. [DOI] [PubMed] [Google Scholar]
- 3.Burt RW, Jacoby R. Polyposis syndromes. In: Yamada T, editor. Textbook of Gastroenterology. 4. Philadelphia: Lippincott Raven; 2003. pp. 1914–1939. [Google Scholar]
- 4.Burn J, Chapman P, Delhanty J, Wood C, Lalloo F, Cachon-Gonzalez MB, Tsioupra K, Church W, Rhodes M, Gunn A. The UK Northern region genetic register for familial adenomatous polyposis coli: use of age of onset, congenital hypertrophy of the retinal pigment epithelium, and DNA markers in risk calculations. J Med Genet. 1991;28:289–96. doi: 10.1136/jmg.28.5.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bisgaard ML, Fenger K, Bulow S, Niebuhr E, Mohr J. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat. 1994;3:121–5. doi: 10.1002/humu.1380030206. [DOI] [PubMed] [Google Scholar]
- 6.Spirio L, Olschwang S, Groden J, Robertson M, Samowitz W, Joslyn G, Gelbert L, Thliveris A, Carlson M, Otterud B, Lynch H, Watson P, Lynch P, Laurent-Puig P, Burt R, Hughes JP, Thomas G, Leppert M, White R. Alleles of the APC gene: an attenuated form of familial polyposis. Cell. 1993;75:951–957. doi: 10.1016/0092-8674(93)90538-2. [DOI] [PubMed] [Google Scholar]
- 7.Leppert M, Burt R, Hughes JP, Samowitz W, Nakamura Y, Woodward S, Gardner E, Lalouel JM, White R. Genetic analysis of an inherited predisposition to colon cancer in a family with a variable number of adenomatous polyps. N Engl J Med. 1990;322:904–8. doi: 10.1056/NEJM199003293221306. [DOI] [PubMed] [Google Scholar]
- 8.Knudsen AL, Bisgaard ML, Bulow S. Attenuated familial adenomatous polyposis (AFAP). A review of the literature. Fam Cancer. 2003;2:43–55. doi: 10.1023/a:1023286520725. [DOI] [PubMed] [Google Scholar]
- 9.Hernegger GS, Moore HG, Guillem JG. Attenuated familial adenomatous polyposis: an evolving and poorly understood entity. Dis Colon Rectum. 2002;45:127–34. doi: 10.1007/s10350-004-6127-y. discussion 134–6. [DOI] [PubMed] [Google Scholar]
- 10.Giardiello FM, Brensinger JD, Luce MC, Petersen GM, Cayouette MC, Krush AJ, Bacon JA, Booker SV, Bufill JA, Hamilton SR. Phenotypic expression of disease in families that have mutations in the 5′ region of the adenomatous polyposis coli gene. Ann Intern Med. 1997;126:514–519. doi: 10.7326/0003-4819-126-7-199704010-00003. [DOI] [PubMed] [Google Scholar]
- 11.Soravia C, Berk T, Madlensky L, Mitri A, Cheng H, Gallinger S, Cohen Z, Bapat B. Genotype-phenotype correlations in attenuated adenomatous polyposis coli. Am J Hum Genet. 1998;62:1290–301. doi: 10.1086/301883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lynch HT, Smyrk T, McGinn T, Lanspa S, Cavalieri J, Lynch J, Slominski-Castor S, Cayouette MC, Priluck I, Luce MC. Attenuated familial adenomatous polyposis (AFAP). A phenotypically and genotypically distinctive variant of FAP. Cancer. 1995;76:2427–2433. doi: 10.1002/1097-0142(19951215)76:12<2427::aid-cncr2820761205>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 13.Burt RW, Leppert MF, Slattery ML, Samowitz WS, Spirio LN, Kerber RA, Kuwada SK, Neklason DW, Disario JA, Lyon E, Hughes JP, Chey WY, White RL. Genetic testing and phenotype in a large kindred with attenuated familial adenomatous polyposis. Gastroenterology. 2004;127:444–51. doi: 10.1053/j.gastro.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 14.Dihlmann S, Gebert J, Siermann A, Herfarth C, von Knebel Doeberitz M. Dominant Negative Effect of the APC1309 Mutation: A Possible Explanation for Genotype-Phenotype Correlations in Familial Adenomatous Polyposis. Cancer Res. 1999;59:1857–1860. [PubMed] [Google Scholar]
- 15.Sieber OM, Lamlum H, Crabtree MD, Rowan AJ, Barclay E, Lipton L, Hodgson S, Thomas HJ, Neale K, Phillips RK, Farrington SM, Dunlop MG, Mueller HJ, Bisgaard ML, Bulow S, Fidalgo P, Albuquerque C, Scarano MI, Bodmer W, Tomlinson IP, Heinimann K. Whole-gene APC deletions cause classical familial adenomatous polyposis, but not attenuated polyposis or “multiple” colorectal adenomas. Proc Natl Acad Sci U S A. 2002;99:2954–8. doi: 10.1073/pnas.042699199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pilarski RT, Brothman AR, Benn P, Shulman Rosengren S. Attenuated familial adenomatous polyposis in a man with an interstitial deletion of chromosome arm 5q. Am J Med Genet. 1999;86:321–4. doi: 10.1002/(sici)1096-8628(19991008)86:4<321::aid-ajmg4>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 17.Spirio LN, Samowitz W, Robertson J, Robertson M, Burt RW, Leppert M, White R. Alleles of APC modulate the frequency and classes of mutations that lead to colon polyps. Nat Genet. 1998;20:385–8. doi: 10.1038/3865. [DOI] [PubMed] [Google Scholar]
- 18.Su LK, Barnes CJ, Yao W, Qi Y, Lynch PM, Steinbach G. Inactivation of germline mutant APC alleles by attenuated somatic mutations: a molecular genetic mechanism for attenuated familial adenomatous polyposis. Am J Hum Genet. 2000;67:582–90. doi: 10.1086/303058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heppner Goss K, Trzepacz C, Tuohy TMF, Groden J. Attenuated APC alleles produce functional protein from internal translation initiation. PNAS. 2002;99:8161–8166. doi: 10.1073/pnas.112072199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van der Luijt RB, Vasen HF, Tops CM, Breukel C, Fodde R, Meera Khan P. APC mutation in the alternatively spliced region of exon 9 associated with late onset familial adenomatous polyposis. Hum Genet. 1995;96:705–10. doi: 10.1007/BF00210303. [DOI] [PubMed] [Google Scholar]
- 21.Varesco L, Gismondi V, Presciuttini S, Groden J, Spirio L, Sala P, Rossetti C, De Benedetti L, Bafico A, Heouaine A, et al. Mutation in a splice-donor site of the APC gene in a family with polyposis and late age of colonic cancer death. Hum Genet. 1994;93:281–6. doi: 10.1007/BF00212023. [DOI] [PubMed] [Google Scholar]
- 22.Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS, Sampson JR, Cheadle JP. Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nat Genet. 2002;30:227–32. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- 23.Jones S, Emmerson P, Maynard J, Best JM, Jordan S, Williams GT, Sampson JR, Cheadle JP. Biallelic germline mutations in MYH predispose to multiple colorectal adenoma and somatic G:C-->T:A mutations. Hum Mol Genet. 2002;11:2961–7. doi: 10.1093/hmg/11.23.2961. [DOI] [PubMed] [Google Scholar]
- 24.Spirio L, Green J, Robertson J, Robertson M, Otterud B, Sheldon J, Howse E, Green R, Groden J, White R, Leppert M. The identical 5′ splice-site acceptor mutation in five attenuated APC families from Newfoundland demonstrates a founder effect. Hum Genet. 1999;105:388–98. doi: 10.1007/s004390051121. [DOI] [PubMed] [Google Scholar]
- 25.Lynch HT, Coronel SM, Okimoto R, Hampel H, Sweet K, Lynch JF, Barrows A, Wijnen J, van der Klift H, Franken P, Wagner A, Fodde R, de la Chapelle A. A founder mutation of the MSH2 gene and hereditary nonpolyposis colorectal cancer in the United States. Jama. 2004;291:718–24. doi: 10.1001/jama.291.6.718. [DOI] [PubMed] [Google Scholar]
- 26.Gonzalez S, Blanco I, Campos O, Julia M, Reyes J, Llompart A, Cabeza E, Germa JR, Obrador A, Capella G. Founder mutation in familial adenomatous polyposis (FAP) in the Balearic Islands. Cancer Genet Cytogenet. 2005;158:70–4. doi: 10.1016/j.cancergencyto.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 27.Niell BL, Long JC, Rennert G, Gruber SB. Genetic anthropology of the colorectal cancer-susceptibility allele APC I1307K: evidence of genetic drift within the Ashkenazim. Am J Hum Genet. 2003;73:1250–60. doi: 10.1086/379926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wylie JE, Mineau GP. Biomedical databases: protecting privacy and promoting research. Trends Biotechnol. 2003;21:113–6. doi: 10.1016/S0167-7799(02)00039-2. [DOI] [PubMed] [Google Scholar]
- 29.Elston RC, Stewart J. A general model for the genetic analysis of pedigree data. Hum Hered. 1971;21:523–42. doi: 10.1159/000152448. [DOI] [PubMed] [Google Scholar]
- 30.Ott J. Analysis of Human Genetic Linkage. The Johns Hopkins University Press; 1999. [Google Scholar]
- 31.Friedl W, Aretz S. Familial Adenomatous Polyposis: Experience from a Study of 1164 Unrelated German Polyposis Patients. Hereditary Cancer in Clinical Practice. 2005;3:95–114. doi: 10.1186/1897-4287-3-3-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Atkin WS, Morson BC, Cuzick J. Long-term risk of colorectal cancer after excision of rectosigmoid adenomas. N Engl J Med. 1992;326:658–62. doi: 10.1056/NEJM199203053261002. [DOI] [PubMed] [Google Scholar]
- 33.American Gastroenterological Association medical position statement: hereditary colorectal cancer and genetic testing. Gastroenterology. 2001;121:195–7. doi: 10.1053/gast.2001.25580. [DOI] [PubMed] [Google Scholar]
- 34.Giardiello FM, Brensinger JD, Petersen GM. AGA technical review on hereditary colorectal cancer and genetic testing. Gastroenterology. 2001;121:198–213. doi: 10.1053/gast.2001.25581. [DOI] [PubMed] [Google Scholar]
- 35.Winawer SJ, Zauber AG, Fletcher RH, Stillman JS, O’Brien MJ, Levin B, Smith RA, Lieberman DA, Burt RW, Levin TR, Bond JH, Brooks D, Byers T, Hyman N, Kirk L, Thorson A, Simmang C, Johnson D, Rex DK. Guidelines for colonoscopy surveillance after polypectomy: a consensus update by the US Multi-Society Task Force on Colorectal Cancer and the American Cancer Society. Gastroenterology. 2006;130:1872–85. doi: 10.1053/j.gastro.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 36.Lamlum H, Al Tassan N, Jaeger E, Frayling I, Sieber O, Reza FB, Eckert M, Rowan A, Barclay E, Atkin W, Williams C, Gilbert J, Cheadle J, Bell J, Houlston R, Bodmer W, Sampson J, Tomlinson I. Germline APC variants in patients with multiple colorectal adenomas, with evidence for the particular importance of E1317Q. Hum Mol Genet. 2000;9:2215–21. doi: 10.1093/oxfordjournals.hmg.a018912. [DOI] [PubMed] [Google Scholar]