

Abstract
Scanty information is available about the mechanisms underlying HLA class I Ag abnormalities in malignant cells exposed to strong T cell-mediated selective pressure. In this study, we have characterized the molecular defects underlying HLA class I Ag loss in five melanoma cell lines derived from recurrent metastases following initial clinical responses to T cell-based immuno-therapy. Point mutations in the translation initiation codon (ATG→ATA) and in codon 31 (TCA→TGA) of the β2-microglobulin (β2m) gene were identified in the melanoma cell lines 1074MEL and 1174MEL, respectively. A hot-spot CT dinucleotide deletion within codon 13–15 was found in the melanoma cell lines 1106MEL, 1180MEL, and 1259MEL. Reconstitution of β2m expression restored HLA class I Ag expression in the five melanoma cell lines; however, the HLA-A and HLA-B,-C gene products were differentially expressed by 1074MEL, 1106MEL, and 1259MEL cells. In addition, in 1259MEL cells, the Ag-processing machinery components calnexin, calreticulin, and low m.w. polypeptide 10 are down-regulated, and HLA-A2 Ags are selectively lost because of a single cytosine deletion in the HLA-A2 gene exon 4. Our results in conjunction with those in the literature suggest the emergence of a preferential β2m gene mutation in melanoma cells following strong T cell-mediated immune selection. Furthermore, the presence of multiple HLA class I Ag defects within a tumor cell population may reflect the accumulation of multiple escape mechanisms developed by melanoma cells to avoid distinct sequential T cell-mediated selective events.
In animal model systems, host's immune system has been convincingly shown to be able to mount an immune response against a naturally growing tumor (1). Recent evidence suggests that this immune response is not always beneficial because it may reshape a tumor cell population by favoring the outgrowth of tumor cells that have developed immunoresistance mechanisms. This phenomenon is referred to as immunoediting (2). In this regard, the current revival of the cancer immune surveillance theory has emphasized the role of immune selective pressure in the generation of tumor immune escape variants in the course of a malignant disease (2). Among the several immune effector mechanisms that may impose selective pressure on tumor cells, CTL are believed to be the major players because of their postulated crucial role in the control of tumor growth. Because the recognition of tumor cells by CTL is mediated by MHC class I Ags presenting tumor Ag (TA)3-derived peptides (1, 3), the immune selective pressure imposed by CTL on tumor cell populations may lead to the isolation and expansion of tumor cell subpopulations with impaired MHC class I Ag-TA-derived peptide complex expression. In humans, this possibility is supported, in part, by the frequent identification of malignant lesions with HLA class I Ag abnormalities (4) that are likely to have escaped recognition and destruction by HLA class I Ag-restricted, TA-specific CTL as a result of continuous CTL-mediated immune selective pressure. The negative impact of this phenomenon on the clinical course of the disease is implied by the correlation between HLA class I Ag abnormalities in tumor lesions and a poor clinical outcome in various types of malignant diseases (4).
Total HLA class I Ag loss by tumor cells results in their complete resistance to recognition and destruction by CTL (5). The mechanism underlying this abnormal phenotype has been frequently found to be loss of β2-microglobulin (β2m), which is required for the formation of the HLA class I H chain-β2m-peptide complex and its transport to the cell surface (6, 7). Two genetic events, namely, mutations of one copy of the β2m gene and loss of the other copy, i.e., loss of heterozygosity (LOH), have been found to underlie β2m loss in malignant cells, although the chronological sequence of the two events is not known (7). Several types of β2m gene mutations ranging from large deletions, which abolish transcription, to point mutations, which in most cases abolish translation, have been described (7). However, it is not clear whether a preferential β2m gene mutation would arise as a result of strong and continuous T cell selective pressure. To test this hypothesis, we set out to characterize the genetic defects underlying the β2m loss previously identified in the five melanoma cell lines, 1074MEL, 1106MEL, 1174MEL, 1180MEL, and 1259MEL (8). These melanoma cell lines were chosen because they were derived from recurrent metastases in patients who had initially experienced clinical responses to T cell-based immunotherapy (8). Therefore, they are likely to have been exposed to strong T cell selective pressure. In addition, we tested whether a host's immune system counteracts escape mechanisms developed by tumor cells by changing the target of its immune response. Lastly, we tested whether the change in the specificity of the selective pressure imposed on a tumor cell population results in the outgrowth of a tumor cell population that has accumulated multiple escape mechanisms to counteract changes in the specificity of the host's immune response. To this end, we determined whether β2m loss is associated with additional abnormalities in molecules involved in HLA class I-associated Ag processing and peptide presentation in the five melanoma cell lines.
Materials and Methods
Cell lines
Cultured human melanoma cell lines 1074MEL, 1106MEL, 1174MEL, 1180MEL, and 1259MEL were derived from recurrent metastases in five patients who had experienced clinical responses to T cell-based immunotherapy (8). These cell lines, the melanoma cell lines Colo38 and FO-1, and the lymphoblastoid cell line LG-2 were maintained in RPMI 1640 medium (Tissue Culture Media Facility, Roswell Park Cancer Institute) supplemented with 10% heat-inactivated FCS (Valeant Pharmaceuticals) or with serum plus (JRH Biosciences) in a 5% CO2 atmosphere at 37°C. The HLA class I phenotypes of patients 1074, 1106, 1174, 1180, and 1259, as determined by typing of PBMC, are A2/A3, B7/B62, Cw3/-; A11/A31, B38/-, −/−; A26/A33, B14/B37, Cw6/w7; A25/A25, B14/B18, −/−; and A1/A2, B8/B44, Cw5/w6, respectively.
mAbs and polyclonal Abs
The mouse mAbs W6/32, which recognize β2m-associated HLA-A, -B, -C, -E, and -G H chains (9, 10); mAb LGIII-147.4.1, which recognizes β2m-associated HLA-A H chains, excluding -A23, -A24, -A25, -A32 (11); mAb B1.23.1, which recognizes β2m-free and -associated HLA-B and -C H chains (12); anti-HLA-A2, -A24, -A28 mAb CR11–351 (13, 14); anti-HLA-A2, -A28 mAb KS-1 (14); anti-HLA-B7, -B27, -Bw42, -Bw54, -Bw55, -Bw56, -Bw67, -Bw73 mAb KS-4 (15); mAb HCA-2, which recognizes β2m-free HLA-A (excluding -A24), -B7301, and -G H chains (16, 17); mAb HC-10, which recognizes β2m-free HLA-A3, -A10, -A28, -A29, -A30, -A31, -A32, -A33, -B (excluding -B5702, -B5804, and -B73) H chain (16-18); anti-β2m mAb L368 (19); anti-tapasin mAb TO-3 (20); anti-calnexin mAb TO-5 (20); anti-ERp57 mAb TO-2 (20); anti-calreticulin mAb TO-11 (20); anti-TAP1 mAb NOB-1 (S. Ferrone, unpublished results); anti-20S proteasome constitutive β subunits δ, MB1, and Z mAb SY-5, SJJ-5, and NB-1, respectively (20) (S. Ferrone, unpublished results); anti-20S proteasome-inducible subunits low m.w. polypeptide (LMP)2, LMP7, and LMP10 mAb SY-1, HB-2, and TO-7, respectively (20); and anti-HLA-DP, -DQ, -DR mAb Q5/13 (21) were developed and characterized, as described. The anti-idiotypic mAb MK2–23, IgG1 (22) and F3C25, IgG2a (23) were used as isotype-matched irrelevant controls. R-PE-conjugated F(ab′)2 of goat anti-mouse Fc and HRP-conjugated goat anti-mouse Fc Abs were purchased from DakoCytomation and Jackson ImmunoResearch Laboratories, respectively.
HLA-A*0201-melanoma Ag (MA) recognized by T cells (MART)127–35 complex-specific single chain Ab fragment (scFv) tetramers
The monomeric HLA-A*0201-MART127–35-specific scFv 8.3 was isolated from the human synthetic VH + VL scFv library (Griffin.1 library) by panning with HLA-A*0201-MART127–35 peptide complexes (M. Campoli, unpublished results). To construct tetrameric scFv 8.3, monomeric scFv 8.3 were engineered with a C-terminal BirA biotinylation tag (scFv 8.3-BirA) and expressed from ampicillin-resistant bacterial colonies, as previously described (24-26). scFv 8.3-BirA was then purified with Ni-affinity chromatography (27), following the manufacturer's instructions. The purity of scFv 8.3-BirA was monitored by SDS-PAGE and silver staining. Tetrameric scFv 8.3 complexes were generated by linking monomeric scFv 8.3-BirA with streptavidin-PE, as described (28).
IFN-γ and synthetic oligonucleotide primers
Human rIFN-γ was purchased from Roche. Oligonucleotide primers were purchased from Integrated DNA Technologies. They include β2m gene-specific primers 261M, 5′-CCTGAAGCTGACAGCATTCG-3′; 262M, 5′-ACCTCCATGATGCTGCTTACA-3′; 744M, 5′-CTCTAACCTGGCACTGCGTCGC-3′; 491M, 5′-CTGGCAATATTAATGTGTCTTTCC-3′; 468M, 5′-TTGAGAAGGAAGTCACGGAGCG-3′; and 462M, 5′-TCATACACAACTTTCAGCAGC-3′; HLA-A or HLA-A*0201gene-specific primers 5pE1A2, 5′-TCCTGCTACTCTCGGGGGCT-3′;5pE2A,5′-GACGCCGCGAGCCAGAGGAT-3′; AP2, 5′-TCACTTTCCGTGCTCCCC-3′; 3pE3A2, 5′-CTCCCACTTGTGCTTGGTGG-3′; and 3pE8A, 5′-AGTCACAAAGGGAAGGGCAGG-3′; A2E3–5′, 5′-GGAGCAGTGGAGAGC-3′; A2E4–3′, 5′-GGTGTATCTCTGCTCC-3′; and β-actin gene-specific primers sense 5′-GACTTCGAGCAAGAGATGGCCAC-3′ and antisense 5′-CAATGCCAGGGTACATGGTGGTG-3′.
Flow cytometry
Flow cytometric analysis of cells for HLA class I Ag expression was performed, as described (5). Briefly, cells (1 × 105) were incubated for 1 h at 4°C with primary mouse mAb at concentrations of 5–10 μg/ml before incubation for 30 min at 4°C with an optimal amount of R-PE-conjugated, mouse Fc-specific goat IgG F(ab′)2 (DakoCytomation). After washing off excess Abs, cells were fixed with 0.5% paraformaldehyde (Sigma-Aldrich) in PBS and analyzed on FACScan (BD Biosciences) with CellQuest software (BD Biosciences). Results are expressed as fold increase of mean fluorescence intensity (MFI) over the isotype control background (fold MFI). Intracellular Ag-processing machinery (APM) component, β2m, and HLA class I H chain expression were analyzed, as previously described (29). Briefly, cells (1 × 105) were fixed with 2% paraformaldehyde (Sigma-Aldrich) in PBS, heat denatured with microwave, and permeabilized with 0.1% saponin in 0.5% BSA/PBS before incubation with primary mAb. Following three washes, cells were stained with an optimal amount of R-PE-conjugated F(ab′)2 of goat anti-mouse Fc Abs (DakoCytomation) and analyzed on FACScan (BD Biosciences) with CellQuest software (BD Biosciences). Results are expressed as fold MFI. Melanoma cells were stained with the HLA-A*0201-MART127–35 complex-specific scFv 8.3 tetramer by incubating 1 × 106 cells (5 × 107 cells/ml) with the scFv 8.3 tetramer (10 μg) for 1 h at 4°C. After three washes with 0.5% BSA/PBS, cells were resuspended in 100 μl of PBS, fixed in 300 μl of 2% formaldehyde/PBS, and analyzed by flow cytometry. Results are expressed as fold MFI.
Western blotting
Western blot analysis of cell lysates for APM component, β2m, and HLA class I H chain expression was performed, as described (5). Briefly, cells (1 × 107) were solubilized in 1 ml of lysis buffer (1% Triton X-100, 50 mM Tris, pH 7.4, 150 mM NaCl, 5 mM EDTA) in the presence of a mixture of protease inhibitors (Calbiochem), and protein concentration was measured by Bradford assay (Bio-Rad). A total of 20 μg of total protein was separated by SDS-PAGE, semidry transferred (Bio-Rad) to polyvinylidene difluoride membranes (Millipore), and blocked with 5% dry milk/PBS before incubation with primary mAb (5∼50 μg/ml) overnight at 4°C. After washing off excess primary mAb, membranes were incubated with an optimal amount of HRP-conjugated goat anti-mouse Fc Abs for 30 min at room temperature. Ags were revealed by the ECL system (Amersham).
PCR and sequence analysis
Total RNA isolation, first-strand cDNA synthesis, and RT-PCR were conducted, as described elsewhere (5). Genomic DNA was extracted and gene fragments were amplified by PCR, as described elsewhere (5). PCR products were resolved by 1.5% agarose electrophoresis and purified with a QIAquick gel extraction kit (Qiagen). Direct sequencing of purified PCR products was performed using an ABI-PRISM model 377 sequencer (Applied Biosystems). Alternatively, PCR products were cloned with a pCR II-TOPO TA cloning kit (Invitrogen Life Technologies) and sequenced with T7 and Sp6 primers.
Transfection
Cells were transfected with a wild-type β2m gene or cDNA using lipofectamine-mediated gene transfer (Invitrogen Life Technologies), according to the manufacturer's instructions. Briefly, pb2m13 (30), pcDNA3-b2m (5), or the empty plasmid pcDNA3-neo (Invitrogen Life Technologies) was mixed with Lipofectamine 2000 before being added to melanoma cells grown in monolayers with a 90% confluence. Cells were selected 2 days after transfection in medium containing G418-sulfate (1 mg/ml) (Calbiochem). After 2∼3 wk of selection, G418-resistant clones were picked, expanded, and screened by flow cytometry for HLA class I Ag expression. Positive clones were then further expanded in complete medium supplemented with a maintenance dose (0.3 mg/ml) of G418.
Results
Identification of β2m mutations in the melanoma cell lines 1074MEL, 1106MEL, 1174MEL, 1180MEL, and 1259MEL
As shown in Fig. 1A, HLA class I Ags are not detectable on the surface of the melanoma cell lines 1074MEL, 1106MEL, 1174MEL, 1180MEL, and 1259MEL, because the five cell lines are not stained by anti-HLA class I mAb W6/32. Furthermore, β2m protein was not detected in the lysates of the five HLA class I− melanoma cell lines (Fig. 1B), although their β2m gene was transcribed (Fig. 1C). To test whether mutations in the coding region of the β2m gene exist, we purified and sequenced the RTPCR-amplified β2m cDNA fragments obtained from the five HLA class I− melanoma cell lines. Analysis of the nucleotide sequences of the β2m cDNA identified mutations in each of the five HLA class I− melanoma cell lines. Specifically, as shown in Fig. 2, a G was replaced with an A (ATG→ATA) at the initiation codon of the β2m gene in 1074MEL cells. This mutation is expected to abolish the initiation of translation of the β2m mRNA. In the cell line 1174MEL, a C was replaced with a G (TCA→TGA) at codon 31, resulting in the introduction of a premature TGA stop codon. In the cell lines 1106MEL, 1180MEL, and 1259MEL, one CT dinucleotide (delCT) was deleted within the 8-bp CT repeat region of codons 13–15 in exon 1. This mutation causes a reading frame shift, resulting in 40 missense codons, followed by the introduction of an early stop at codon 55.
FIGURE 1.
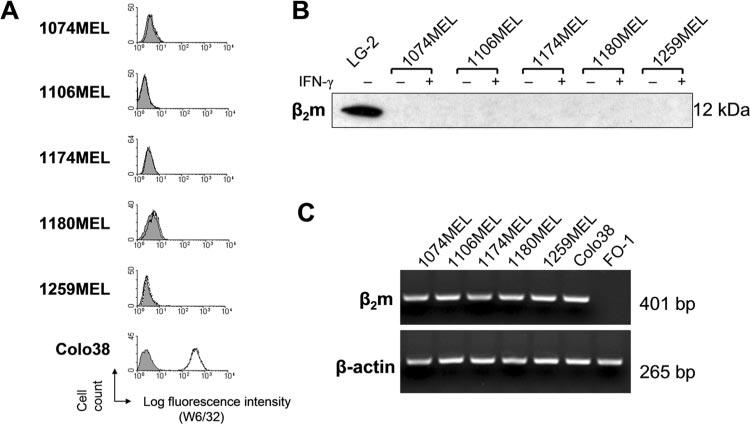
β2m loss by the melanoma cell lines 1074MEL, 1106MEL, 1174MEL, 1180MEL, and 1259MEL. A, Lack of HLA class I Ag expression by the melanoma cell lines 1074MEL, 1106MEL, 1174MEL, 1180MEL, and 1259MEL. Cells were stained with the mAb W6/32 (open profile) and analyzed by flow cytometry. The melanoma cell line Colo38 and the isotype-matched mAb F3C25 (filled profile) were used as controls. B, Lack of β2m protein expression in the melanoma cell lines 1074MEL, 1106MEL, 1174MEL, 1180MEL, and 1259MEL. Lysates prepared from control- and IFN-γ-treated (300 U/ml, 48 h at 37°C) melanoma cells were tested with the mAb L368 in Western blotting. A lymphoblastoid cell LG-2 lysate was used as a positive control. C, β2m mRNA expression by the melanoma cell lines 1074MEL, 1106MEL, 1174MEL, 1180MEL, and 1259MEL. Total RNA was isolated from cells, reversely transcribed to first-strand cDNA, and subjected to PCR analysis with β2m- and β-actin-specific primers. PCR products were run on an agarose gel and visualized by ethidium bromide staining. Colo38 and FO-1 (45) melanoma cells were used as a positive and negative control, respectively.
FIGURE 2.

Nucleotide and deduced amino acid sequence of β2m cDNA isolated from the melanoma cell lines 1074MEL, 1106MEL, 1174MEL, 1180MEL, and 1259MEL. Sequence conservation and nontranslated codons are indicated by dashes and dots, respectively. Point mutations and CT deletions are indicated by bold letters and small upward arrows (^), respectively. Exon-exon junctions are indicated by ▼. Consensus β2m cDNA sequence was obtained from the National Center for Biotechnology Information Gen-Bank (accession BC06491042).
The mutations found in the β2m cDNA fragments in the five melanoma cell lines analyzed were confirmed at the genomic DNA level, because identical mutations were detected in the β2m genes in these five cell lines (Fig. 3). These mutations are the only ones present in their β2m genes, because sequencing of PCR products corresponding to the 5′ flanking region, the entire coding region and the 3′ nontranslation region of the genes detected no additional mutations (data not shown). It is noteworthy that three of the five cell lines analyzed were found to have the same delCT mutation in the β2m gene, suggesting the presence of a mutational hot spot in that region of the gene.
FIGURE 3.
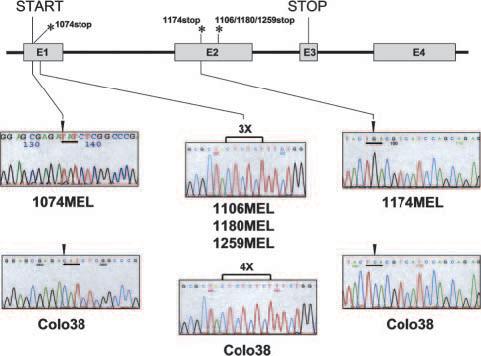
Identification of β2m gene mutations in genomic DNA extracted from melanoma cell lines 1074MEL, 1106MEL, 1174MEL, 1180MEL, and 1259MEL. A diagram of the β2m gene structure is shown on top with indications of the identified mutation sites. *, Indicate the premature stop codon in the β2m coding sequence from 1074MEL cells (1074stop), 1174MEL cells (1174stop), and 1106MEL, 1180MEL, and 1259MEL cells (1106/1180/1259stop). The sequencing histograms encompassing the β2m mutation sites are shown below where the histogram marked Colo38 represents the wild-type sequence. The mutated β2m gene sequence shown for 1074MEL cells (1074MEL) corresponds to the antisense strand, whereas the mutated β2m gene sequence shown for 1106MEL cells (1106MEL), 1174MEL cells (1174MEL), 1180MEL cells (1180MEL), and 1259MEL cells (1259MEL) corresponds to the sense strand.
Differential expression of HLA-A and HLA-B,-C gene products on the melanoma cell lines 1074MEL, 1106MEL, and 1259MEL transfected with a wild-type β2m gene or cDNA
Transient reconstitution of β2m expression has been previously shown to restore HLA class I Ag expression by the five melanoma cell lines (8). Furthermore, in preliminary experiments, we have found that the gene products of HLA-A, HLA-B, and HLA-C loci were differentially expressed on 1074MEL, 1106MEL, and 1259MEL cells, but not on 1174 MEL and 1180MEL cells (S. Ferrone, unpublished observations). To corroborate these findings, in the present study, we have established stable β2m transfectants for 1074MEL, 1106MEL, and 1259MEL melanoma cells and analyzed their HLA class I Ag expression. As shown in Fig. 4, HLA class I Ags were highly expressed on clones of β2m-transfected 1074MEL (1074MEL.β2.7), 1106MEL (1106MEL.β2), and 1259MEL (1259MEL.β2.18) cells, but were not detected on mock-transfected 1074MEL, 1106MEL, and 1259MEL cells. Upon stimulation with IFN-γ (300 U/ml) for 48 h at 37°C, the level of total HLA class I Ags was increased only on the β2m transfectants.
FIGURE 4.
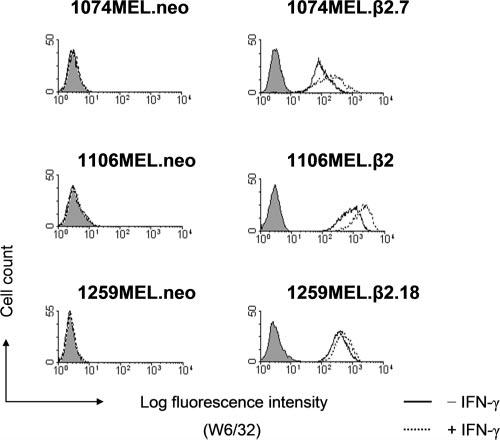
Restoration of HLA class I Ag expression on β2m-transfected melanoma cell lines 1074MEL, 1106MEL, and 1259MEL. Control-or IFN-γ-treated (300 U/ml, 48 h at 37°C) clones of neo- and β2m-transfected 1074MEL, 1106MEL, and 1259MEL cells were stained with the mAb W6/32 (open profile) and analyzed by flow cytometry. The isotype-matched mAb F3C25 (filled profile) was used as a control. Results of one of three representative experiments are shown.
As shown in Fig. 5A, the gene products of HLA-A, -B, and -C loci are differentially expressed by the stable β2m transfectants of 1074MEL, 1106MEL, and 1259MEL melanoma cells. Flow cytometric analysis of cells stained with HLA-A-specific mAb LGIII147.4.1 and HLA-B- and -C-specific mAb B1.23.1 detected a higher level of HLA-A Ags than of HLA-B,-C Ags on 1074MEL.β2.7 and 1259MEL.β2.18 cells, and a lower level of HLA-A Ags than of HLA-B,-C Ags on 1106MEL.β2 cells. Following incubation with IFN-γ (300 U/ml) for 48 h at 37°C, the down-regulated HLA-A Ags on 1106.MEL.β2 cells increased 3.5-fold, reaching a level comparable to the basal HLA-B and -C Ag level, while the down-regulated HLA-B and -C Ags on 1074MEL.β2.7 and 1259MEL.β2.18 cells increased 3.7- and 3.1-fold, respectively, remaining at a lower level than that of HLA-A Ags under basal conditions (Fig. 5A).
FIGURE 5.
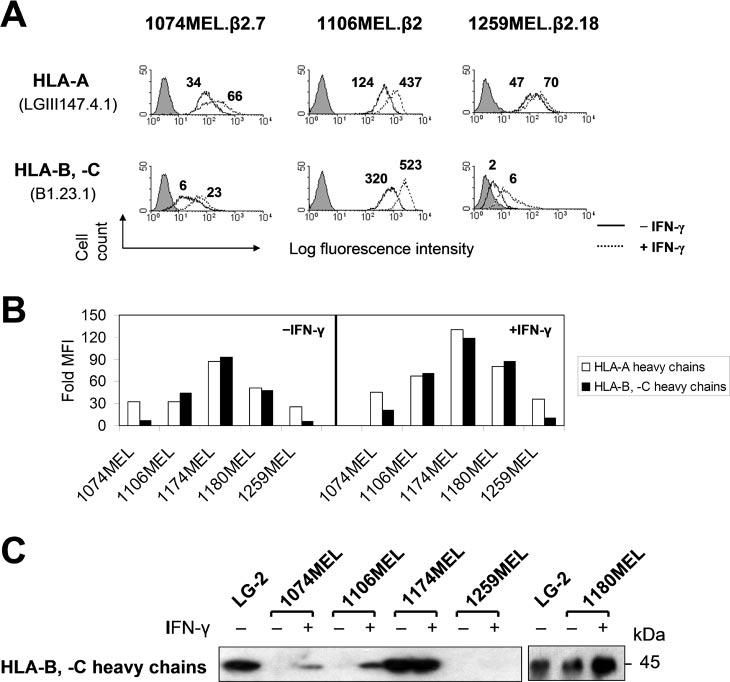
Differential HLA-A and HLA-B,-C gene product expression by the β2m-transfected melanoma cell lines 1074MEL.β2.7, 1106MEL.β2, and 1259MEL.β2.18. A, Higher levels of HLA-A than HLA-B,-C Ag expression by 1074MEL.β2.7 and 1259MEL.β2.18 cells and higher levels of HLA-B,-C than HLA-A Ag expression by 1106MEL.β2 cells. Cells were stained with the anti-HLA-A mAb LGIII147.41 and the anti-HLA-B,-C mAb B1.23.1 and analyzed by flow cytometry. The figure above each histogram indicates the fold increase of MFI over the isotype control background (fold MFI). Results of one of three representative experiments are shown. B and C, Higher levels of HLA-A than HLA-B,-C H chain expression by 1074MEL and 1259MEL cells and higher levels of HLA-B,-C than HLA-A H chain expression by 1106MEL cells. B, Control- and IFN-γ-treated (300 U/ml, 48 h at 37°C) cells were permeabilized, stained with the mAb HCA-2 and the mAb HC-10, and analyzed by flow cytometry. Results are expressed as fold MFI and shown in bars. C, Lysates prepared from control- and IFN-γ-treated (300 U/ml, 48 h at 37°C) cells were tested with the mAb HC-10 in Western blotting.
Differential HLA-A and HLA-B,-C H chain expression in the melanoma cell lines 1074MEL, 1106MEL, and 1259MEL
We next tested whether the differential HLA-A and -B,-C allospecificity expression on the cell membrane of β2m-transfected 1074MEL, 1106MEL, and 1259MEL cells is correlated with differential expression of the corresponding HLA class I H chains. As shown in Fig. 5B, using flow cytometry with intracellular staining, we found that HLA-B,-C H chains were expressed at a considerably lower level than HLA-A H chains in 1074MEL and 1259MEL cells, but at a higher level than HLA-A H chains in 1106MEL cells. In contrast, HLA-A, -B, and -C H chains were equally well expressed in 1174MEL and 1180MEL cells. The results obtained with flow cytometry were corroborated by Western blot analysis with HLA class I H chain-specific mAb of the lysates of the five melanoma cell lines (Fig. 5C and data not shown).
Selective HLA-A2 Ag loss caused by a single-base deletion in exon 4 of the HLA-A2 gene in 1259MEL cells
To test whether the differential expression of the gene products of both HLA-A and -B loci was detectable at the level of individual allospecificities, we analyzed HLA-A2 Ag expression on 1074MEL.β2.7 cells (patient's HLA-A phenotype: A2/A3) and 1259MEL.β2.18 cells (patient's HLA-A type: A1/A2), as well as HLA-B7 Ag expression on 1074MEL.β2.7 cells (patient's HLA-B phenotype: B7/B62), with HLA-A2-, -A24-, -A28-specific mAb CR11–351; HLA-A2-, -A28-specific mAb KS-1; and HLA-B7-cross-reacting group-specific mAb KS-4. While HLA-B7 Ags were expressed, although at a low level, on 1074MEL.β2.7 cells (data not shown), to our surprise, HLA-A2 Ags were not detectable on 1259MEL.β2.18 cells (Fig. 6). HLA-A2 Ags were not detected even following incubation with IFN-γ (300 U/ml) for 48 h at 37°C (Fig. 6). The lack of HLA-A2 Ag expression by IFN-γ-treated 1259MEL.β2.18 cells is not due to their unresponsiveness to the cytokine, because HLA-A, -B, and -C Ags (Fig. 5A) as well as HLA-DP, -DQ, and -DR Ags (Fig. 6) were up-regulated or induced upon IFN-γ stimulation. These results suggest that HLA-A2 Ags were selectively lost in 1259MEL cells. Additional experiments investigated HLA-A2 mRNA expression in 1259MEL cells. RT-PCR analysis detected bands with sizes similar to those found in the wild-type HLA-A2 mRNA isolated from 1074MEL cells (Fig. 7, A and B). Nucleotide sequence analysis of 1259MEL HLA-A2 cDNA and gene fragments identified a single cytosine deletion at nt 701 (delC701) in the exon 4 (Fig. 7C and data not shown). This mutation causes a reading frame shift, resulting in the introduction of five missense codons (234–238), followed by a premature stop codon TGA (Fig. 7D). The disrupted translation of HLA-A2 mRNA is expected to result in the loss of most of the HLA-A2 H chain α3 domain. The latter has been shown by the crystal structure of a HLA-A2-β2m-peptide complex (Fig. 8; reconstructed from Protein Data Bank ID 1AKJ) (31) to represent the major contact region for β2m.
FIGURE 6.
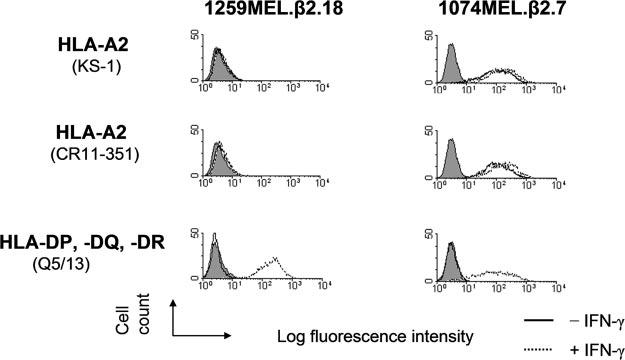
Selective HLA-A2 Ag loss by β2m-transfected 1259MEL melanoma cells. Control- and IFN-γ-treated (300 U/ml, 48 h at 37°C) β2m-transfected 1074MEL (1074MEL.β2.7) and β2m-transfected 1259MEL (1259MEL.β2.18) cells were stained with the mAb KS-1 and CR11–351 and analyzed by flow cytometry. The mAb Q5/13 was used to assess HLA class II Ag expression following treatment of these melanoma cells with IFN-γ. Results of one of three representative experiments are shown.
FIGURE 7.
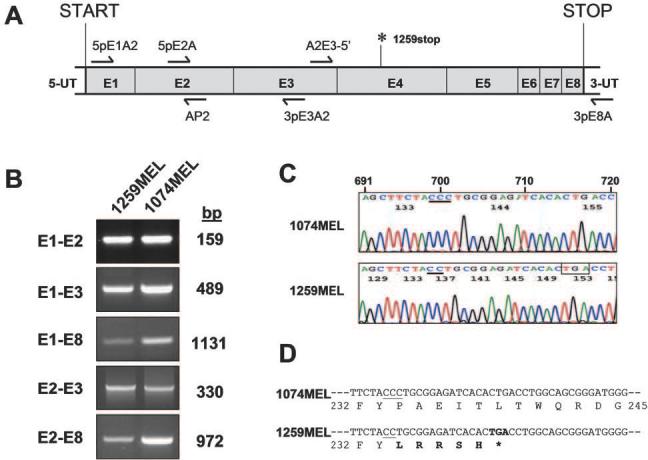
Selective HLA-A2 Ag loss caused by a single cytosine deletion in the HLA-A2 gene exon 4 in 1259MEL melanoma cells. A, Diagram of HLA-A*0201 mRNA structure marked with the location of the primers (arrows) and of the premature stop codon (1259stop) identified in 1259MEL cells. B, HLA-A*0201 mRNA expression by 1259MEL cells. Total RNA was isolated from cells, reversely transcribed to first-strand cDNA, and subjected to PCR analysis with different combinations of HLA-A- or HLA*0201 gene-specific primers. PCR products were run on an agarose gel and visualized by ethidium bromide staining. C, Sequencing histogram of the single cytosine deletion found in the HLA-A*0201 cDNA isolated from 1259MEL cells. RT-PCR-amplified HLA-A2 exon1-exon8 (E1-E8) cDNA fragments from 1259MEL and 1074MEL cells were gel purified and subjected to nucleotide sequencing analysis with sense primers 5pE1A2, 5pE2A, and A2E3–5′ or antisense primers AP2, 3pE3A2, A2E4–3′, and 3pE8A. The histograms obtained with primer A2E3–5′ are shown. Nucleotides are numbered according to Arnett and Parham (46) and indicated above the 1074MEL histogram. The cytosine stretch starting from nt 699 is underlined. The premature stop codon TGA generated as a result of a shifted reading frame is boxed. D, Nucleotide and deduced amino acid sequence of the partial HLA-A2 exon 4 in 1074MEL and 1259MEL cells. The regions corresponding to codon 232–245 are shown. The cytosine stretches are underlined. The missense codon-deduced amino acids are indicated in bold letters.
FIGURE 8.
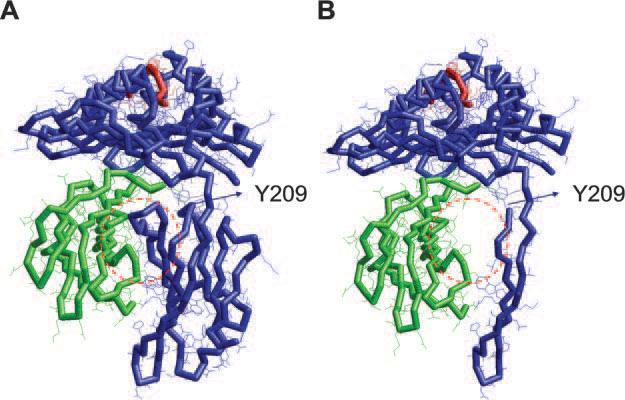
Three-dimensional model of a HLA-A2-β2m-peptide complex with a C terminus-truncated HLA-A2 H chain. Backbone diagram of the HLA-A2-β2m-HIVpol peptide complex (A) and the H chain-truncated HLA-A2-β2m-HIVpol peptide complex (B) is shown. Blue, green, and red backbones indicate the HLA-A2 H chain, β2m, and the HIVpol peptide, respectively. The models are generated by using the RasTop 1.3.1 molecular graphics program (P. Valadon) based on the coordinates retrieved from PDB ID 1AKJ (31). The coordinates corresponding to HLA-A2 H chain position 1∼209 (codon +24∼233) were used to generate the HLA-A2-β2m-HIVpol peptide complex model with a truncated HLA-A2 H chain. Y209 indicates a tyrosine at position 209 (codon 233), which is the last residue before the missense codon-deduced residue leucine at position 210 (codon 234) in the 1259MEL HLA-A2 H chain. The red-dashed circles indicate the HLA-A2 α3 domain-β2m contact area, which is postulated to be missing in the truncated version of HLA-A2-β2m-HIVpol peptide complex.
Heterogeneous APM component expression in the melanoma cell lines 1074MEL, 1106MEL, 1174MEL, 1180MEL, and 1259MEL
APM components play a critical role in the generation of peptides to be loaded onto HLA class I Ags in cells (32) and have been found to be frequently down-regulated in a variety of tumor types (33). Therefore, we analyzed the five melanoma cell lines for possible defects in the APM components, which include the endoplasmic reticulum chaperones calnexin, calreticulin, ERp57, and tapasin; the peptide transporter TAP1; the constitutive proteasome subunits δ, MB1, and Z; and the IFN-γ-inducible proteasome subunits LMP2, LMP7, and LMP10. As shown in Fig. 9A, flow cytometric analysis of melanoma cells intracellularly stained with APM component-specific mAb detected all of these components at comparable levels in the five melanoma cell lines tested, except for a marked calnexin, calreticulin, and LMP10 down-regulation in 1259MEL cells. Of the latter three molecules, only LMP10 expression can be slightly restored by the administration of IFN-γ. The results obtained with flow cytometry were corroborated by Western blot analysis of the five cell line lysates with APM component-specific mAb (Fig. 9B).
FIGURE 9.
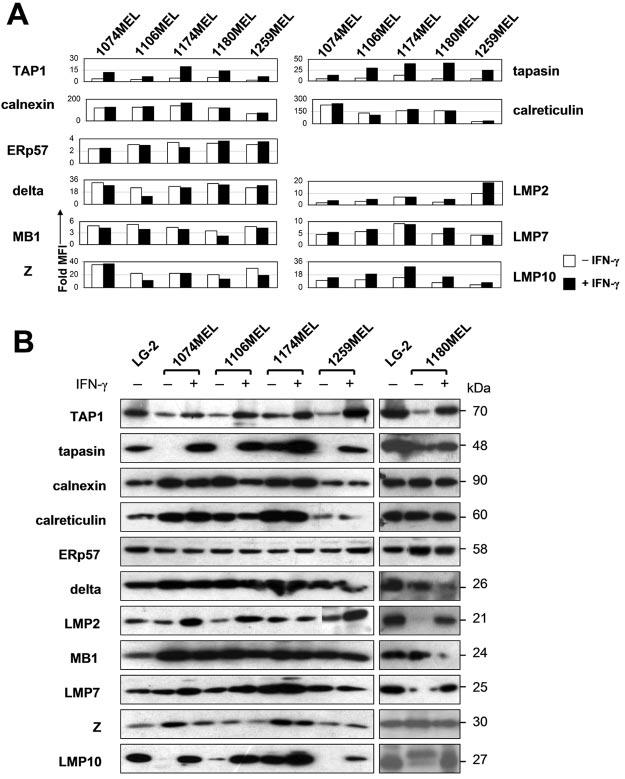
Heterogeneous APM component expression in the melanoma cell lines 1074MEL, 1106MEL, 1174MEL, 1180MEL, and 1259MEL. A, Control- and IFN-γ-treated (300 U/ml, 48 h at 37°C) cells were permeabilized; stained with the mAb TO-2, TO-5, TO-11, TO-3, NOB-1, SY-5, SY-1, SJJ-3, HB-2, NB-1, and TO-7; and analyzed by flow cytometry. Results are expressed as fold MFI and shown in bars. B, Lysates prepared from control- and IFN-γ-treated (300 U/ml, 48 h at 37°C) cells were tested with APM-specific mAb in Western blotting.
HLA-A*0201-MART127–35 complex expression on β2m-transfected 1074MEL cells
To determine the functionality of APM in melanoma cells analyzed in this study, we assessed HLA-A*0201-MART127–35 peptide complex expression by 1074MEL cells. As shown in Fig. 10, β2m-transfected 1074MEL cells were stained by the HLAA*0201-MART127–35 peptide complex-specific scFv8.3 tetramer. The staining is specific because HLA-A*0201-MART127–35 peptide complexes were detected on β2m-transfected 1074MEL cells, but not on mock-transfected 1074MEL cells, β2m-transfected 1106MEL cells (HLA-A11/A31), mock- and β2m-transfected 1259MEL cells (HLA-A1/A2delC701), and LG2 cells (HLA-A2+). The expression of HLA-A*0201-MART127–35 peptide complexes on β2m-transfected 1074MEL cells is compatible with their fully functional APM capable of generating HLA class I Ag-restricted, MA-derived peptides.
FIGURE 10.
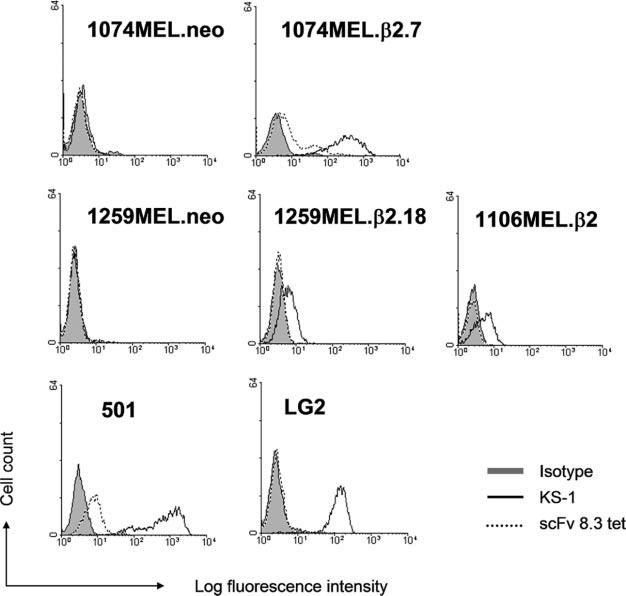
HLA-A2-MART-127–35 complex expression on 1074MEL.β2.7 melanoma cells. The neo (1074MEL.neo)- and β2m-transfected (1074MEL.β2.7) 1074MEL cells, the neo (1259MEL.neo)- and β2m-transfected (1259MEL.β2.18) 1259MEL cells, and the β2m-transfected 1106MEL cells (1106MEL.β2) were stained with the HLA-A2-MART-127–35 complex-specific scFv 8.3 tetramer (scFv 8.3 tet) and analyzed by flow cytometry. The HLA-A2, -A28-specific mAb KS-1, the HLA-A2+MART-1+ melanoma cells 501, and the HLA-A2+MART-1− lymphoblastoid cells LG-2 were used as controls.
Discussion
Taking advantage of a panel of β2m-, HLA class I H chain-, and APM component-specific mAb, we have performed a comprehensive analysis of all the molecules known to be involved in functional HLA class I Ag expression in five melanoma cell lines exposed to strong T cell selective pressure. Multiple molecular defects that compromise the presentation of MA-derived peptides to HLA class I Ag-restricted CTL do exist in at least three of the five melanoma cell lines tested. These findings thus provide for the first time in the human system the molecular mechanisms underlying the described immunoediting phenomenon (2).
Three types of β2m gene mutations were identified in the five melanoma cell lines, all of which do not affect the transcription, but result in lack of translation of the β2m protein. The mutation harbored by the cell line 1074MEL (ATG to ATA) is novel, because it is different from two other initiation codon mutations identified in the Burkitt lymphoma cell line Daudi (ATG to ATC) (34) and in the melanoma cell line LB1622-MEL (ATG to AAG) (35). The nonsense mutation identified in the cell line 1174MEL (TCA to TGA), which introduces a premature stop in codon 31 of exon 2 of the β2m gene (31Ser→Stop), is identical with the recently described mutation in the melanoma cell line BB74-MEL (35). It is noteworthy that both LB1622-MEL and BB74-MEL cell lines were derived from lesions in patients who had undergone T cell-based immunotherapy, suggesting that these cells have been exposed to strong T cell selective pressure. In contrast, the 31Ser→Stop mutation did not underlie the HLA class I Ag loss by other five melanoma cell lines originated from patients who had not been treated with T cell-based immunotherapy. Interestingly, the delCT mutation identified in the cell lines 1106MEL, 1180MEL, and 1259MEL is present in three of five melanoma cell lines originated from patients receiving T cell-based immunotherapy, but has only been described in one (Me1386) (5) of five melanoma cell lines established from patients not treated with T cell-based immunotherapy. Taken together, these findings clearly suggest an association between the preferential selection of a β2m gene mutational hot spot (especially delCT) and T cell-based immunotherapy in melanoma cells. If confirmed by the analysis of a large number of melanoma cell lines, this association may suggest a relationship between the outgrowth of melanoma cells harboring certain types of genomic instability and the type/extent of immune selective pressure introduced by T cell-based immunotherapy. Whether the same rule applies to other types of tumors remains to be determined. In this regard, the delCT mutation in the β2m gene has been found only in two colorectal carcinoma cell lines with HLA class I Ag loss (36, 37). As far as we know, the two patients from whom the two cell lines had been originated had not been treated with T cell-based immunotherapy. Whether the two patients had developed a strong T cell immunity against their own tumors is not known.
In melanoma, like in many other tumor types, LOH was found to associate with β2m gene mutation (7). This phenomenon also applies to the melanoma cell lines that we have analyzed in this study, because no wild-type β2m gene sequence was found to superimpose the mutated ones. The cause of LOH of β2m gene is not known, but is probably an interstitial deletion because of a mitotic recombination event (38).
Reconstitution of β2m expression following transfection with a wild-type β2m gene or cDNA was sufficient to restore total HLA class I Ag expression on the five melanoma cell lines. These results confirm the pivotal role of β2m in HLA class I Ag expression and show that β2m defects represent the only mechanism underlying HLA class I Ag loss by the five melanoma cell lines that we have analyzed. However, differential expression of the gene products of HLA-A and HLA-B,-C loci was found in the β2m-transfected cell lines 1074MEL, 1106MEL, and 1259MEL. This phenotype, which is correlated with the differential HLA-A and HLA-B,-C H chain down-regulation, appears to be caused by abnormalities in regulatory mechanisms, because transcription of the corresponding HLA class I alleles is down-regulated, but can be restored by the administration of IFN-γ. This possibility is supported by the lack of locus-specific trans-acting factors, which results in selective HLA class I locus down-regulation, as has been reported in melanoma cell lines as well as in colorectal carcinoma cell lines (7, 39).
The frame shift mutation identified in the HLA-A2 gene exon 4 in 1259MEL cells is novel because it is different from those described in the literature. The latter include a large deletion from the 5′-flanking region to exon 4 and loss of one copy of chromosome 6 in the melanoma cell lines 970604 and 950822, respectively (40), as well as a 5′-flanking region deletion of the gene, chromosome loss, and exon skipping in the melanoma cell lines SK-MEL-29.1.22, SK-MEL-29.1.29, and 624MEL28, respectively (41, 42). In the case of 1259MEL cells, it remains to be determined whether the truncated HLA-A2 H chain can still be synthesized and fold properly to associate with β2m and peptides and even secreted by 1259MEL cells. Our findings highlight the importance to analyze tumor cells for polymorphic HLA class I allospecificity expression and not to rely on the use of mAb-recognizing monomorphic and locus-specific determinants. In the latter cases, abnormalities in a given HLA class I locus or an individual allele will not be found, because they are masked by the positive staining of the gene products encoded by any of the remaining loci.
Only in 1259MEL cells the APM components calnexin, calreticulin, and LMP10 were found to be markedly down-regulated. The reduced APM component expression did not appear to have functional implications, because β2m-restored 1259MEL cells were still sensitive to lysis by H-2 Kb-restricted, vaccinia virus-specific murine CTL upon transient transduction of H-2 Kb molecules, as were the other four melanoma cell lines (8). These results are consistent with those reported in the literature that do not support a critical role for calnexin, calreticulin, and LMP10 in the presentation of MHC class I Ag-restricted peptides. However, the data obtained with the viral system may not be applicable to the presentation of MA-derived peptides and their role in eliciting CTL responses because: 1) the amount of MA synthesized in cells is not likely to be comparable to that of viral Ags produced in cells, and 2) the T cell repertoire for viral Ags is not likely to be the same as that for MA. It remains to be determined whether β2m-transfected 1259MEL cells are sensitive to lysis by autologous, MA-specific CTL. In contrast, the detection of HLA*0201-MART127–35 peptide complexes by the β2m-transfected HLA-A2+ 1074MEL cells (1074MEL.β2.7) suggests that 1074MEL cells carry a functional APM capable of generating HLA class I Ag-restricted, MA-derived peptides. Furthermore, this result parallels our previous finding that β2m-transduced 1074MEL cells were susceptible to lysis by HLA-A2 Ag-restricted, MA-specific CTL (8).
β2m gene and HLA class I H chain abnormalities were both present in 1259MEL cells, raising the question of their chronological sequence of appearance. In light of immune system's ability to reshape tumor cell populations over time (2), we favor the possibility that the multiple HLA class I Ag defects found in 1259MEL cells may have developed sequentially in the order of HLA-A2 Ag loss, HLA-B and -C Ag down-regulation, and β2m loss, although the first two can also occur in the reverse order. The β2m gene mutation is postulated to be a late event. In this situation, HLA class I H chain abnormalities would provide 1259MEL cells with a mechanism to escape lysis by CTL that recognize immunodominant epitopes before a complete HLA class I-loss phenotype is obtained. In contrast, if total HLA class I Ag loss caused by β2m loss occurs early, abnormalities in H chains would not be advantageous to β2m-deficient 1259MEL cells because they are not expected to be recognized by CTL. If our interpretation is correct, β2m loss is also likely to be a late event in 1074MEL and 1106MEL cell lines, because both cell lines have HLA class I H chain down-regulation in addition to β2m gene mutations.
Disease progression in patients with melanoma associated with HLA class I Ag loss in metastases raises the question as to why tumor growth is not controlled by NK cells in vivo, given the increased in vitro and in vivo sensitivity to NK cell-mediated lysis of target cells that have lost MHC class I Ag expression (43). One can postulate at least two possible underlying mechanisms: either NK cells have an impaired migratory/effector function in vivo or the melanoma cells lack expression of molecules that activate NK cells. The latter possibility is supported by the markedly reduced sensitivity in vitro to NK cell-mediated lysis of 1074MEL and 1106MEL cells that express low levels of NK cell-activating ligands MHC class I chain-related A and UL16-binding proteins (44). These findings thus provide an explanation for the lack of control of tumor cells with HLA class I Ag loss by NK cells in vivo. In contrast, the melanoma cell lines 1174MEL and 1259MEL are highly sensitive to NK cell-mediated lysis, which is correlated with a high level of MHC class I chain-related A expression on the cell surface (44). Therefore, NK cells may have failed to migrate to the lesions from which the cell lines 1174MEL and 1259MEL were derived.
Acknowledgments
We are grateful to Bitao Liang for initial contribution to the project, and Michelle M. Detwiler for technical assistance.
Footnotes
This work was supported by Public Health Service Grants RO1 CA67108, P30 CA16056, and T32 CA85183 awarded by the National Cancer Institute, Department of Health and Human Services (to S.F.), by a Susan G. Komen Breast Cancer Foundation predoctoral fellowship (to C.-C.C.), and by a Department of Defense predoctoral fellowship (to M.C.).
Abbreviations used in this paper: TA, tumor Ag; APM, Ag-processing machinery; β2m, β2-microglobulin; LMP, low m.w. polypeptide; LOH, loss of heterozygosity; MA, melanoma Ag; MART, MA recognized by T cells; MFI, mean fluorescence intensity; scFv, single chain Ab fragment.
References
- 1.Schreiber H. Tumor immunology. In: Paul ED, editor. Fundamental Immunology. Lippincott Williams & Wilkins; Philadelphia: 2003. pp. 1557–1592. [Google Scholar]
- 2.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 2002;3:991. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 3.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv. Immunol. 2000;74:181. doi: 10.1016/s0065-2776(08)60911-6. [DOI] [PubMed] [Google Scholar]
- 5.Hicklin DJ, Wang Z, Arienti F, Rivoltini L, Parmiani G, Ferrone S. β2-Microglobulin mutations, HLA class I antigen loss, and tumor progression in melanoma. J. Clin. Invest. 1998;101:2720. doi: 10.1172/JCI498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arce-Gomez B, Jones EA, Barnstable CJ, Solomon E, Bodmer WF. The genetic control of HLA-A and B antigens in somatic cell hybrids: requirement for β2 microglobulin. Tissue Antigens. 1978;11:96. doi: 10.1111/j.1399-0039.1978.tb01233.x. [DOI] [PubMed] [Google Scholar]
- 7.Ferrone S, editor. Semin. Cancer Biol. Vol. 12. 2002. Tumor immune escape; p. 1. [DOI] [PubMed] [Google Scholar]
- 8.Restifo NP, Marincola FM, Kawakami Y, Taubenberger J, Yannelli JR, Rosenberg SA. Loss of functional β2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J. Natl. Cancer Inst. 1996;88:100. doi: 10.1093/jnci/88.2.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barnstable CJ, Bodmer WF, Brown G, Galfre G, Milstein C, Williams AF, Ziegler A. Production of monoclonal antibodies to group A erythrocytes, HLA and other human cell surface antigens: new tools for genetic analysis. Cell. 1978;14:9. doi: 10.1016/0092-8674(78)90296-9. [DOI] [PubMed] [Google Scholar]
- 10.Kovats S, Main EK, Librach C, Stubblebine M, Fisher SJ, DeMars R. A class I antigen, HLA-G, expressed in human trophoblasts. Science. 1990;248:220. doi: 10.1126/science.2326636. [DOI] [PubMed] [Google Scholar]
- 11.Wang X, Liang B, Rebmann V, Lu J, Celis E, Kageshita T, Grosse-Wilde H, Ferrone S. Specificity and functional characteristics of anti-HLA-A mAbs LGIII-147.4.1 and LGIII-220.6.2. Tissue Antigens. 2003;62:139. doi: 10.1034/j.1399-0039.2003.00087.x. [DOI] [PubMed] [Google Scholar]
- 12.Rebai N, Malissen B. Structural and genetic analyses of HLA class I molecules using monoclonal xenoantibodies. Tissue Antigens. 1983;22:107. doi: 10.1111/j.1399-0039.1983.tb01176.x. [DOI] [PubMed] [Google Scholar]
- 13.Russo C, Ng AK, Pellegrino MA, Ferrone S. The monoclonal antibody CR11–351 discriminates HLA-A2 variants identified by T cells. Immunogenetics. 1983;18:23. doi: 10.1007/BF00401353. [DOI] [PubMed] [Google Scholar]
- 14.Tsujisaki M, Sakaguchi K, Igarashi M, Richiardi P, Perosa F, Ferrone S. Fine specificity and idiotype diversity of the murine anti-HLA-A2, A28 monoclonal antibodies CR11–351 and KS1. Transplantation. 1988;45:632. doi: 10.1097/00007890-198803000-00026. [DOI] [PubMed] [Google Scholar]
- 15.Sakaguchi K, Ono R, Tsujisaki M, Richiardi P, Carbonara A, Park MS, Tonai R, Terasaki PI, Ferrone S. Anti-HLA-B7, B27, Bw42, Bw54, Bw55, Bw56, Bw67, Bw73 monoclonal antibodies: specificity, idiotypes, and application for a double determinant immunoassay. Hum. Immunol. 1988;21:193. doi: 10.1016/0198-8859(88)90071-7. [DOI] [PubMed] [Google Scholar]
- 16.Stam NJ, Spits H, Ploegh HL. Monoclonal antibodies raised against denatured HLA-B locus heavy chains permit biochemical characterization of certain HLA-C locus products. J. Immunol. 1986;137:2299. [PubMed] [Google Scholar]
- 17.Sernee MF, Ploegh HL, Schust DJ. Why certain antibodies cross-react with HLA-A and HLA-G: epitope mapping of two common MHC class I reagents. Mol. Immunol. 1998;35:177. doi: 10.1016/s0161-5890(98)00026-1. [DOI] [PubMed] [Google Scholar]
- 18.Perosa F, Luccarelli G, Prete M, Favoino E, Ferrone S, Dammacco F. β2-microglobulin-free HLA class I heavy chain epitope mimicry by monoclonal antibody HC-10-specific peptide. J. Immunol. 2003;171:1918. doi: 10.4049/jimmunol.171.4.1918. [DOI] [PubMed] [Google Scholar]
- 19.Lampson LA, Fisher CA, Whelan JP. Striking paucity of HLA-A, B, C and β2-microglobulin on human neuroblastoma cell lines. J. Immunol. 1983;30:2471. [PubMed] [Google Scholar]
- 20.Ogino T, Wang X, Kato S, Miyokawa N, Harabuchi Y, Ferrone S. Endoplasmic reticulum chaperone-specific monoclonal antibodies for flow cytometry and immunohistochemical staining. Tissue Antigens. 2003;62:382. doi: 10.1034/j.1399-0039.2003.00114.x. [DOI] [PubMed] [Google Scholar]
- 21.Quaranta V, Walker LE, Pellegrino MA, Ferrone S. Purification of immunologically functional subsets of human Ia-like antigens on a monoclonal antibody (Q5/13) immunoadsorbent. J. Immunol. 1980;125:1421. [PubMed] [Google Scholar]
- 22.Chen ZJ, Yang H, Ferrone S. Human high molecular weight melanoma-associated antigen mimicry by mouse antiidiotypic monoclonal antibody MK2–23: characterization of the immunogenicity in syngeneic hosts. J. Immunol. 1991;147:1082. [PubMed] [Google Scholar]
- 23.Perosa F, Ferrone S. Syngeneic antiidiotypic monoclonal antibodies to the murine anti-HLA-DR, DP monoclonal antibody CR11–462. Hum. Immunol. 1988;23:255. doi: 10.1016/0198-8859(88)90061-4. [DOI] [PubMed] [Google Scholar]
- 24.Skerra A, Pluckthun A. Assembly of a functional immunoglobulin Fv fragment in Escherichia coli. Science. 1988;240:1038. doi: 10.1126/science.3285470. [DOI] [PubMed] [Google Scholar]
- 25.Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization: human antibodies from V-gene libraries displayed on phage. J. Mol. Biol. 1991;222:581. doi: 10.1016/0022-2836(91)90498-u. [DOI] [PubMed] [Google Scholar]
- 26.Schier R, Bye J, Apell G, McCall A, Adams GP, Malmqvist M, Weiner LM, Marks JD. Isolation of high-affinity monomeric human anti-c-erbB-2 single chain Fv using affinity-driven selection. J. Mol. Biol. 1996;255:28. doi: 10.1006/jmbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- 27.Canaan-Haden L, Ayala M, Fernandez-de-Cossio ME, Pedroso I, Rodes L, Gavilondo JV. Purification and application of a single-chain Fv antibody fragment specific to hepatitis B virus surface antigen. BioTechniques. 1995;19:606. [PubMed] [Google Scholar]
- 28.Altman JD, Moss PA, Goulder PJ, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94. [PubMed] [Google Scholar]
- 29.Ogino T, Wang X, Ferrone S. Modified flow cytometry and cell-ELISA methodology to detect HLA class I antigen processing machinery components in cytoplasm and endoplasmic reticulum. J. Immunol. Methods. 2003;278:33. doi: 10.1016/s0022-1759(03)00224-2. [DOI] [PubMed] [Google Scholar]
- 30.Wang Z, Cao Y, Albino AP, Zeff RA, Houghton A, Ferrone S. Lack of HLA class I antigen expression by melanoma cells SK-MEL-33 caused by a reading frameshift in β2-microglobulin messenger RNA. J. Clin. Invest. 1993;91:684. doi: 10.1172/JCI116249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao GF, Tormo J, Gerth UC, Wyer JR, McMichael AJ, Stuart DI, Bell JI, Jones EY, Jakobsen BK. Crystal structure of the complex between human CD8α and HLA-A2. Nature. 1997;387:630. doi: 10.1038/42523. [DOI] [PubMed] [Google Scholar]
- 32.Pamer E, Cresswell P. Mechanisms of MHC class I-restricted antigen processing. Annu. Rev. Immunol. 1998;16:323. doi: 10.1146/annurev.immunol.16.1.323. [DOI] [PubMed] [Google Scholar]
- 33.Seliger B, Maeurer MJ, Ferrone S. Antigen-processing machinery breakdown and tumor growth. Immunol. Today. 2000;21:455. doi: 10.1016/s0167-5699(00)01692-3. [DOI] [PubMed] [Google Scholar]
- 34.Rosa F, Berissi H, Weissenbach J, Maroteaux L, Fellous M, Revel M. The β2-microglobulin mRNA in human Daudi cells has a mutated initiation codon but is still inducible by interferon. EMBO J. 1983;2:239. doi: 10.1002/j.1460-2075.1983.tb01412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benitez R, Godelaine D, Lopez-Nevot MA, Brasseur F, Jimenez P, Marchand M, Oliva MR, van Baren N, Cabrera T, Andry G, et al. Mutations of the β2-microglobulin gene result in a lack of HLA class I molecules on melanoma cells of two patients immunized with MAGE peptides. Tissue Antigens. 1998;52:520. doi: 10.1111/j.1399-0039.1998.tb03082.x. [DOI] [PubMed] [Google Scholar]
- 36.Gattoni-Celli S, Kirsch K, Timpane R, Isselbacher KJ. β2-microglobulin gene is mutated in a human colon cancer cell line (HCT) deficient in the expression of HLA class I antigens on the cell surface. Cancer Res. 1992;52:1201. [PubMed] [Google Scholar]
- 37.Bicknell DC, Rowan A, Bodmer WF. β2-microglobulin gene mutations: a study of established colorectal cell lines and fresh tumors. Proc. Natl. Acad. Sci. USA. 1994;91:4751. doi: 10.1073/pnas.91.11.4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Natrajan R, Louhelainen J, Williams S, Laye J, Knowles MA. High-resolution deletion mapping of 15q13.2-q21.1 in transitional cell carcinoma of the bladder. Cancer Res. 2003;63:7657. [PubMed] [Google Scholar]
- 39.Griffioen M, Ouwerkerk IJ, Harten V, Schrier PI. HLA-B locus-specific down-regulation in human melanoma requires enhancer A as well as a sequence element located downstream of the transcription initiation site. Immunogenetics. 2000;52:121. doi: 10.1007/s002510000262. [DOI] [PubMed] [Google Scholar]
- 40.Geertsen R, Boni R, Blasczyk R, Romero P, Betts D, Rimoldi D, Hong X, Laine E, Willers J, Dummer R. Loss of single HLA class I allospecificities in melanoma cells due to selective genomic abbreviations. Int. J. Cancer. 2002;99:82. doi: 10.1002/ijc.10284. [DOI] [PubMed] [Google Scholar]
- 41.Wang Z, Seliger B, Mike N, Momburg F, Knuth A, Ferrone S. Molecular analysis of the HLA-A2 antigen loss by melanoma cells SK-MEL-29.1.22 and SK-MEL-29.1.29. Cancer Res. 1998;58:2149. [PubMed] [Google Scholar]
- 42.Wang Z, Marincola FM, Rivoltini L, Parmiani G, Ferrone S. Selective histocompatibility leukocyte antigen (HLA)-A2 loss caused by aberrant pre-mRNA splicing in 624MEL28 melanoma cells. J. Exp. Med. 1999;190:205. doi: 10.1084/jem.190.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ljunggren H-G, Karre K. In search of the “missing self”: MHC molecules and NK cell recognition. Immunol. Today. 1990;11:237. doi: 10.1016/0167-5699(90)90097-s. [DOI] [PubMed] [Google Scholar]
- 44.Pende D, Rivera P, Marcenaro S, Chang C-C, Biassoni R, Conte R, Kubin M, Cosman D, Ferrone S, Moretta L, Moretta A. Major histocompatibility complex class I-related chain A and UL16-binding protein expression on tumor cell lines of different histotypes: analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res. 2002;62:6178. [PubMed] [Google Scholar]
- 45.D'Urso CM, Wang Z, Cao Y, Tatake R, Zeff RA, Ferrone S. Lack of HLA class I antigen expression by cultured melanoma cells FO-1 due to a defect in β2m gene expression. J. Clin. Invest. 1991;87:284. doi: 10.1172/JCI114984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arnett KL, Parham P. HLA class I nucleotide sequences, 1995. Tissue Antigens. 1995;46:217. doi: 10.1111/j.1399-0039.1995.tb03124.x. [DOI] [PubMed] [Google Scholar]