

Abstract
The androgen receptor (AR) is a ligand-activated transcription factor which controls growth and survival of prostate cancer cells. In the present study, we investigated the regulation of AR activity by the receptor interacting protein RIP140. We first showed that RIP140 could be co-immunoprecipitated with the receptor when co-expressed in 293T cells. This interaction appeared physiologically relevant since ChIP assays revealed that under R1881 treatment, RIP140 could be recruited to the PSA encoding gene in LNCaP cells. In vitro GST pull-down assays evidenced that the carboxy-terminal domain of AR could interact with different regions of RIP140. By means of fluorescent proteins we demonstrated that ligand-activated AR was not only able to translocate to the nucleus but also to relocate RIP140 from very structured nuclear foci to a diffuse pattern. Overexpression of RIP140 strongly repressed AR-dependent transactivation by preferentially targeting the ligand binding domain-dependent activity. Moreover, disruption of RIP140 expression induced AR overactivation thus revealing RIP140 as a strong AR repressor. We analysed its mechanism of transrepression and first demonstrated that different regions of RIP140 could mediate AR-dependent repression. We then showed that the carboxy-terminal end of RIP140 could reverse transcriptional intermediary factor TIF2-dependent overactivation of AR. The use of mutants of RIP140 allowed us to suggest that CtBP played no role in RIP140-dependent inhibition of AR activity whereas HDACs partly regulated that transrepression. Finally, we provided evidence for a stimulation of RIP140 mRNA expression in LNCaP cells under androgen treatment, further emphasizing the role of RIP140 in androgen signalling.
Keywords: Adaptor Proteins, Signal Transducing; Alcohol Oxidoreductases; Animals; COS Cells; Cell Compartmentation; Cercopithecus aethiops; Cricetinae; DNA-Binding Proteins; metabolism; Gene Expression Regulation; Histone Deacetylases; metabolism; Humans; Metribolone; pharmacology; Nuclear Proteins; metabolism; Phosphoproteins; metabolism; Protein Binding; RNA, Messenger; metabolism; Receptors, Androgen; antagonists & inhibitors; Tumor Cells, Cultured; Up-Regulation; drug effects
Keywords: AR, RIP140, transcription repression, HDAC, LNCaP
INTRODUCTION
Defects in androgen signalling result in a large panel of clinical phenotypes ranging from perturbation in male sexual development to prostate cancer etiology (1, 2). Because androgen stimulation regulates prostate epithelial cell growth, treatment for advanced prostate cancer, the major malignancy in men in Western countries, can be achieved by eradication of androgen action through androgen withdrawal using chemical or surgical castration (3). However the disease invariably progresses to an androgen-independent state. Therefore, elucidation of mechanisms that regulate androgen actions is of critical importance.
The effects of androgens are mediated by the androgen receptor (AR), a transcription factor member of the nuclear receptor superfamily. Unliganded AR is a cytoplasmic protein associated in an inactive state with heat shock proteins (4). Under hormone binding, the receptor undergoes conformational changes which induce its translocation from the cytoplasm to the nucleus. In order to regulate transcription of target genes, AR binds to specific DNA sequences called androgen response elements (ARE) (1). The receptor harbors three main functional domains: the amino-terminal domain where the primary ligand-independent transactivation domain, AF1 (amino acids 142-337) supports the major transactivation function of the receptor (5), the central DNA-binding domain and the carboxy-terminal domain also called ligand-binding domain (LBD) (1). The AR LBD is highly conserved among the steroid receptor family of proteins and contains the weak transcriptional activation domain AF2 (6).
AR-mediated transactivation requires the concerted action of AF1 and AF2 (6). To date a great number of AR cofactors have been described to mediate androgens action (7). Gene activation by the AR is thought to require the general initiation factors that form preinitiation complexes on common core promoter element (8), and different general and specific coactivators that either modulate chromatin structure (9) or serve as direct adaptors between the receptor and general initiation factors (10). The interest in AR corepression rapidly developed in the recent years and subsequently the number of AR corepressors drastically increased (see (11) for a review). The mechanism of action for many corepressors remains to be discovered. However, recruitment of histone deacetylases (HDACs) is a common way to repress AR activity. In that category, are found different proteins (12–14) including the short heterodimer partner (SHP) (15), which can be all recruited by the agonist-activated AR.
RIP140 (receptor interacting protein of 140 kDa) is a protein of 1158 amino acids which is recruited by a large number of agonist-activated receptors, including ERα, TR, RAR and RXR (16), AR (17), VDR (18), PPARα/LXRα (19) or GR (20). It was also shown to interact with other nuclear receptors such as SF1 or DAX-1 (21) or other transcription factors including the aryl hydrocarbon receptor (22), 14-3-3 (23) or c-jun (24). Its mechanism of action not only involves a competition with coactivators such as those belonging to the p160 family (25) but it also implies active repression. We and others recently evidenced four repressive domains in the molecule involving complex mechanisms relying on multiple partners, including HDACs and C-terminal binding proteins (CtBPs) (26, 27).
Surprisingly, although widely depicted as a corepressor, a study by Ikonen et al. described RIP140 as a strong coactivator for AR (17). In order to decipher RIP140 mechanism of action we investigated further its role in the androgen signalling pathway. In the present paper, we first characterized the interaction between RIP140 and AR and provided evidence for a nuclear relocalization of RIP140 upon activation of the receptor. We showed that RIP140 is a strong AR repressor, and to shed light on the mechanism of RIP140-dependent inhibition, we investigated the role of CtBPs and HDAC as well as a competition with a p160 coactivator. Finally, we demonstrated that RIP140 mRNA expression in LNCaP cells was significantly increased by a treatment with R1881, further emphasizing the role of RIP140 in AR activity.
RESULTS
RIP140 interacts with AR
In order to determine whether AR could interact with the coregulator RIP140, we first performed immunoprecipitations between full-length proteins (Figure 1A). 293T cells were either non transfected (lanes 1 and 2) or transfected with pCMV-AR alone (lanes 3 and 4), pCMV-AR and pEF-c-mycRIP140 (lanes 5 and 6) or pEF-c-mycRIP140 alone (lanes 7 and 8), and treated with 10−8 M R1881. As shown in the Figure, when AR is expressed alone the use of an anti-AR antibody immunoprecipitated the receptor (lane 4, upper panel). When AR and c-mycRIP140 were co-expressed, the same antibody not only pulled-down AR (lane 6, upper panel) but also c-mycRIP140 (lane 6, lower panel). We noted the band corresponding to c-mycRIP140 was slightly retarded as compared to the input which could be due to differences in salt concentrations. It has to be noticed that, as a control for the immunoprecipitation, when pEF-c-mycRIP140 was transfected alone (lanes 7 and 8), the use of an anti-AR antibody could not pull-down c-mycRIP140, thus strengthening the specificity of the interaction.
Figure 1. RIP140 interacts with AR.
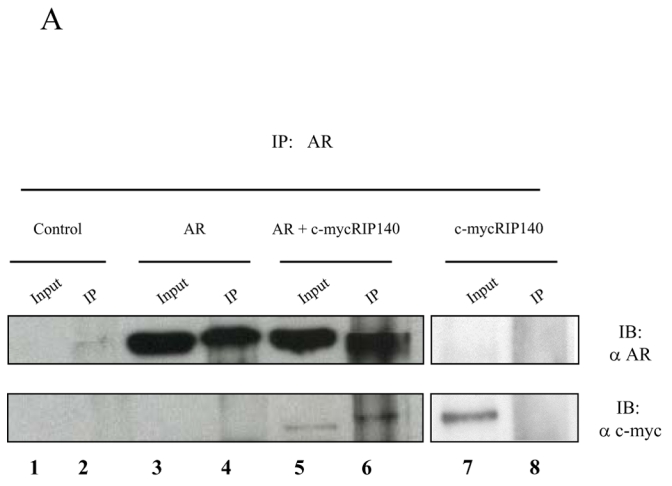
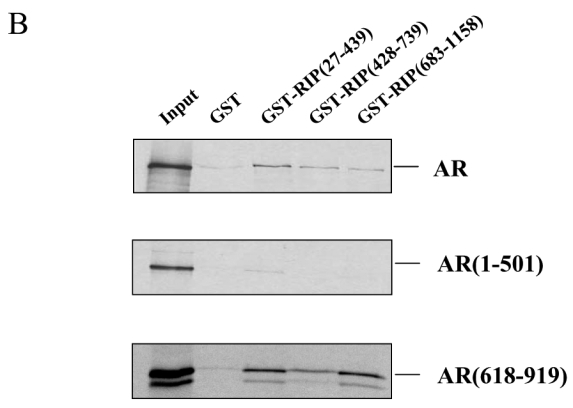
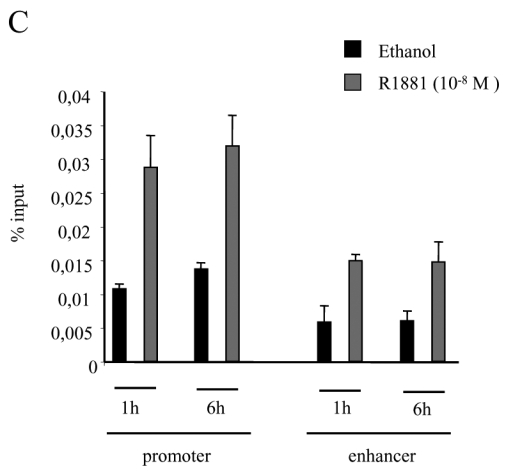
A. Immunoprecipitations. 293T cells were either non transfected (lanes 1 and 2), transfected with pCMV-AR alone (lanes 3 and 4), with pCMV-AR and pEF-c-mycRIP140 (lanes 5 and 6) or with pEF-c-mycRIP140 alone (lanes 7 and 8) and treated with R1881 (10−8 M). The immunoprecipitations were undertaken using an anti-AR monoclonal antibody and the western-blots were done with either an anti-AR polyclonal antibody (upper panel) or an anti-c-myc monoclonal antibody (lower panel). B. GST pull-downs assays. The assays were performed as described in the Materials and Methods using 35S labeled AR, AR(1-501) or AR(618-919), and purified GST, GST-RIP(27-439), GST-RIP(428-739) or GST-RIP(683-1158) in the presence of R1881 (10−6 M). GST alone was used as a control for the interaction. The inputs represent 10 % of the in vitro translated AR used in each assay. C. Interaction of RIP140 with PSA promoter and enhancer. LNCaP cells (2.106) were grown for 7 days in DMEM 3 % DCC before hormone treatment. They were then treated with R1881 10−8 M or vehicle (ethanol) for 1 or 6 hrs. ChIP assays were performed as described in Materials and Methods. Each experiment was repeated twice and quantitative PCR analyses were performed in duplicates (mean ± SD).
To investigate further the interaction and determine which domains of the proteins were involved, we performed in vitro GST pull-downs. In this series of experiments, three fragments of RIP140 spanning the whole protein were expressed as GST fusion proteins and either full-length or truncated domains of AR were in vitro translated. As indicated in Figure 1B, upper panel, in the presence of R1881, full-length AR interacted with the three regions of RIP140. However, the binding appeared stronger with GST-RIP140(27-439). As observed in Figure 1B, middle panel, only a very faint band corresponding to the binding between GST-RIP(27-439) and AR(1-501) could be detected whereas none was observed with either the central or the carboxy-terminal part of RIP140. In the lower panel was analysed the interaction with the carboxy-terminal part of the receptor in the presence of R1881. As observed, both GST-RIP(27-439) and GST-RIP(683-1118) appeared to have a strong affinity for AR(618-919) whereas GST-RIP(428-739) displayed a lower but still significant binding. Only a faint band was observed when GST was incubated with either full-length AR or AR(618-919) whereas none appeared with AR(1-501). It must be stated that the experiments with either full-length AR or AR(618-919) were also done in the absence of R1881 and gave the same degree of interaction (data not shown). Coomassie staining of the gels indicated that the amount of GST fusion proteins was kept constant in all experiments (data not shown).
To give further credit to the interaction we wondered whether RIP140 could be recruited to an androgen-dependent gene. To this end we performed chromatin immunoprecipitation (ChIP) assay with an anti-RIP140 antibody on LNCaP cells previously treated or not with 10−8 M R1881. Since a recent work (28) evidenced that transcription factors could differentially recruit the promoter and the enhancer of the PSA gene, these different regions of the gene were then amplified (Figure 1C). As observed on the figure either a 1-hour or 6-hour treatment with the AR agonist induced a clear amplification of both the PSA promoter and the enhancer as quantified by quantitative PCR demonstrating that an AR-responsive gene could be a target of RIP140.
We conclude from these experiments that RIP140 interacts with AR both in vitro and in intact cells. Furthermore the interaction is mediated on one hand by several regions covering the entire cofactor and on another hand by the ligand binding domain of AR.
AR relocalizes RIP140
Subcellular localization of transcription factors is tightly regulated. Therefore we questioned whether overexpression of one partner could affect the localization of the other. We first transfected COS7 cells with pYFP-RIP140 (see Figure 2A). As observed in the left panel, whatever the treatment of the cells YFP-RIP140 always formed foci in the nucleus, a structure already described (26). In Figure 2A, right panel, the cells were cotransfected with vectors expressing CFP-AR and YFP-RIP140. When the cells were incubated with ethanol, AR was localized to the cytoplasmic compartment, whereas RIP140 was nuclear and formed regular foci (upper panel). When treated with the agonist R1881, AR was entirely translocated to the nucleus (Figure 2A, middle panel). Remarkably, in the same cell, RIP140 presented a more evenly spread nuclear localization with only rare foci. Interestingly, when the cells were treated with the complete antagonist bicalutamide, AR was translocated to the nucleus as previously described (29) but there, RIP140 formed the same foci as observed in the presence of ethanol. Interestingly, when merged the two signals did not show a colocalization of the two proteins. From these observations we can conclude that translocation of the activated AR relocalized RIP140. Moreover the relocalization was specific to the activated receptor since the bicalutamide-liganded AR was not able to trigger it.
Figure 2. Intranuclear distribution of CFP-AR and YFP-RIP140.
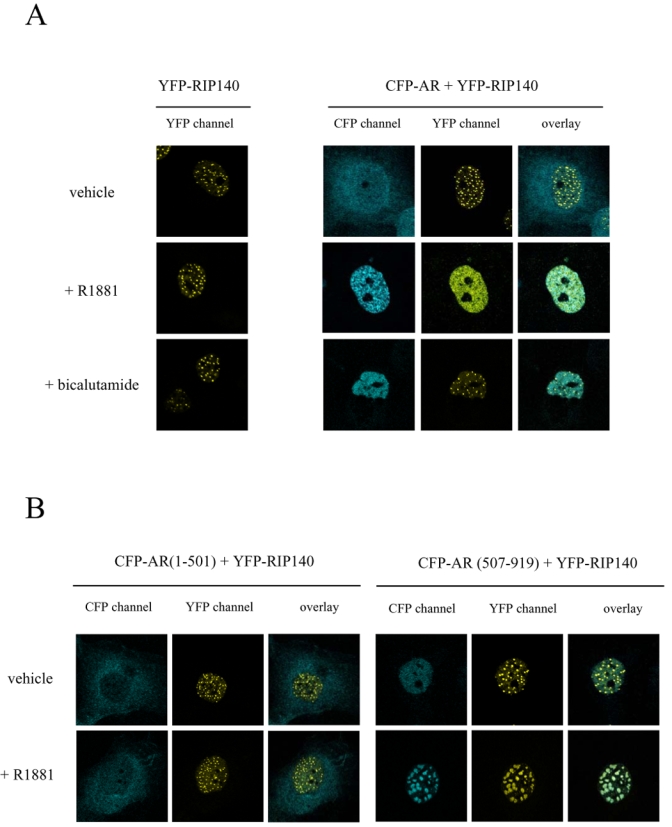
COS7 cells were transiently transfected with vectors expressing either the fusion protein YFP-RIP140 alone or together with CFP-AR (A), CFP-AR(1-501) (B, left panel) or CFP-AR(507-919) (B, right panel). Cells were incubated overnight in the presence of either vehicle or ligands (10−8 M R1881 or 10−6 M bicalutamide), fixed with 4 % paraformaldehyde and then observed using a Leica SP2 Confocal microscope. Pseudocolors were used (blue for CFP and yellow for YFP).
Then, we wondered whether AR amino- or carboxy-terminal domains would induce differential localizations of RIP140. Therefore, we cotransfected COS7 cells with YFP-RIP140 together with either CFP-AR(1-501) (Figure 2B, left panel) or CFP-AR(507-919) (Figure 2B, right panel). As observed in the left panel, whatever the treatment, CFP-AR(1-501) stayed in the cytoplasm whereas YFP-RIP140 was organized in nuclear foci. In Figure 2B, right panel, it is interesting to observe that CFP-AR(507-919) in the absence of agonist ligand was nuclear but evenly spread, whereas YFP-RIP140 still formed foci. Remarkably, under R1881 treatment CFP-AR(507-919) was organized in large foci. In the same conditions, YFP-RIP140 was organized in structures of the same size as CFP-AR(507-919). As observed on the overlay picture, the two proteins were perfectly colocalized. These data strengthen the evidence of a ligand-dependent intracellular interaction between RIP140 and AR mediated by the carboxy-terminal domain of the receptor.
RIP140 inhibits AR-dependent transactivation
We then investigated the role of RIP140 on AR-dependent transactivation. In a first series of experiments, CV1 cells were transfected with pCMV-AR and increasing amounts of pcRIP140 (Figure 3A). As shown in the figure, RIP140 dose-dependently inhibited AR-mediated transactivation with a maximal repression obtained with 2 μg of transfected pcRIP140.
Figure 3. RIP140 inhibits AR-dependent transactivation.
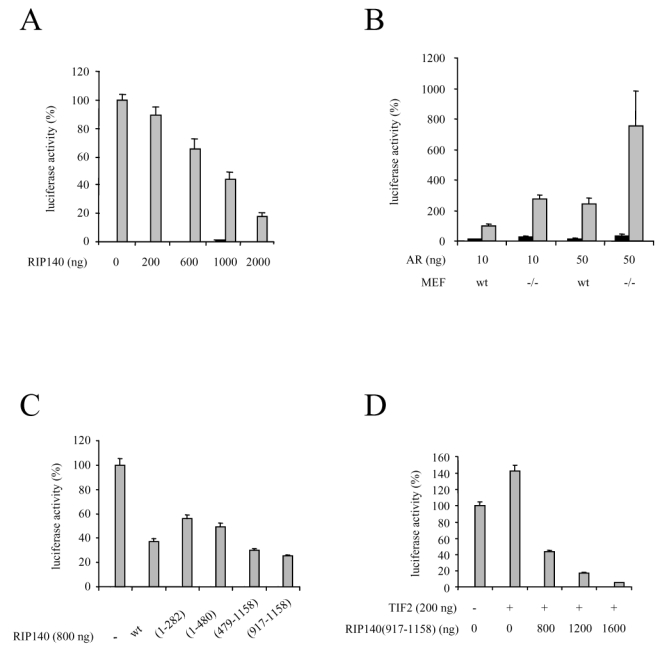
A. RIP140 represses AR-dependent transactivation. CV1 cells were transfected using the calcium phosphate method with 10 ng of pCMV-AR and increasing amounts of pcRIP140. The pcDNA empty vector was used to keep the amount of DNA transfected constant.
B. AR is overactivated in cells devoid of RIP140. Wild type (wt) or RIPKO (−/−) MEF cells were cotransfected with either 10 ng or 50 ng of pCMV-AR using the calcium phosphate method.
C. Different domains of RIP140 participate in AR-dependent transactivation. 10 ng of pCMV-AR as well as 800 ng of each mutant were transfected in CV1 cells using the calcium phosphate method.
D. RIP140 reverses TIF2-induced AR activity. CV1 cells were transfected with 10 ng of pCMV-AR, 200 ng of psg5-TIF2 and increasing amounts of pcRIP(917-1158).
The luciferase activity was expressed taking AR in the presence of R1881 as 100 % of the activity. The mean ± SD values from at least three independent experiments are shown.
■ = Ethanol;
 = R1881 (10−9 M).
= R1881 (10−9 M).
We then asked whether an extinction of RIP140 could affect AR transactivation. To this end we used mouse embryo fibroblasts lacking the RIP140 gene (termed RIPKO-1) (30) as well as the wild type counterpart cells. As observed in Figure 3B, whatever the dose of vector transfected, AR was repeatedly overactivated in RIPKO-1 cells treated with R1881 as compared to the wild-type. It has to be noticed that no difference in AR expression could be observed in either cell type (data not shown). This experiment allowed us to propose that endogenous RIP140 exert a significant repressive effect on AR-dependent transactivation further emphasizing results described above.
RIP140 possesses nine nuclear receptor boxes (31) and results from Figure 1 showed that several regions of RIP140 were able to mediate its interaction with AR. Therefore we investigated in CV1 cells the repressive potential of RIP140 constructs spanning different domains of the cofactor on AR-dependent transcription. As shown in Figure 3C, all the constructs tested displayed a high degree of repression. However, the two fragments encompassing the amino-terminal part of RIP140, i.e. RIP140(1-282) and RIP140(1-480) did not exhibit a repressive potential as strong as the wild type. By sharp contrast, the carboxy-terminal fragment of RIP140 and more precisely RIP140(917-1158) exhibited an even stronger repression than full-length RIP140. We concluded that different domains of RIP140 can mediate AR-dependent repression.
It has already been evidenced that RIP140 can compete away coactivators to bind nuclear receptors (25). Moreover, results presented above evidenced the carboxy-terminal domain of RIP140 as a strong inhibitor. Therefore we investigated whether RIP140(917-1158) could compete with an AR coactivator, TIF2, for repression of the receptor. As evidenced in Figure 3D, when CV1 cells were cotransfected with pSG5-TIF2, AR-dependent transcription was augmented. Remarkably, cotransfections with increasing amounts of pcRIP140(917-1158) not only reversed AR overactivation, but also completely down-regulated AR activity. Noticeably the same experiment was done with full-length RIP140 and the same results were obtained (data not shown). These data allowed us to propose that the carboxy-terminal part of RIP140 can act as a strong competitor for p160-mediated activation of AR.
AR exhibiting two transactivation domains, lying in the amino- and the carboxy-terminal parts of the receptor, we asked whether results from protein-protein interactions would be corroborated by transactivation assays. We first studied the effect of RIP140 on the constitutively active AR(1-660). CV1 cells were transfected with a constant dose of pCMV-AR(1-660) and increasing amounts of pcRIP140. As evidenced in Figure 4A, whatever the quantity of pcRIP140 transfected, AR(1-660)-dependent activity could not be modulated, indicating that the main activation domain of AR, when isolated, was not a target for RIP140. We then asked whether RIP140 could inhibit AR LBD-dependent transactivation. We thus used the deletion mutant AR(507-919) that lacks the amino-terminal domain and displays no transcriptional activity per se. In order to restore its transcriptional ability in a strictly ligand dependent manner, CHO cells were cotransfected with an AR coactivator, TIF2 (Figure 4B). In those conditions, as shown in the Figure, a dose as low as 50 ng pcRIP140 was sufficient to completely reverse TIF2-induced activity of AR(507-919). This series of data supports the conclusion that RIP140 represses AR-dependent activity by targeting its ligand binding domain.
Figure 4. RIP140 exerts its repression via the carboxy-terminal domain of AR.
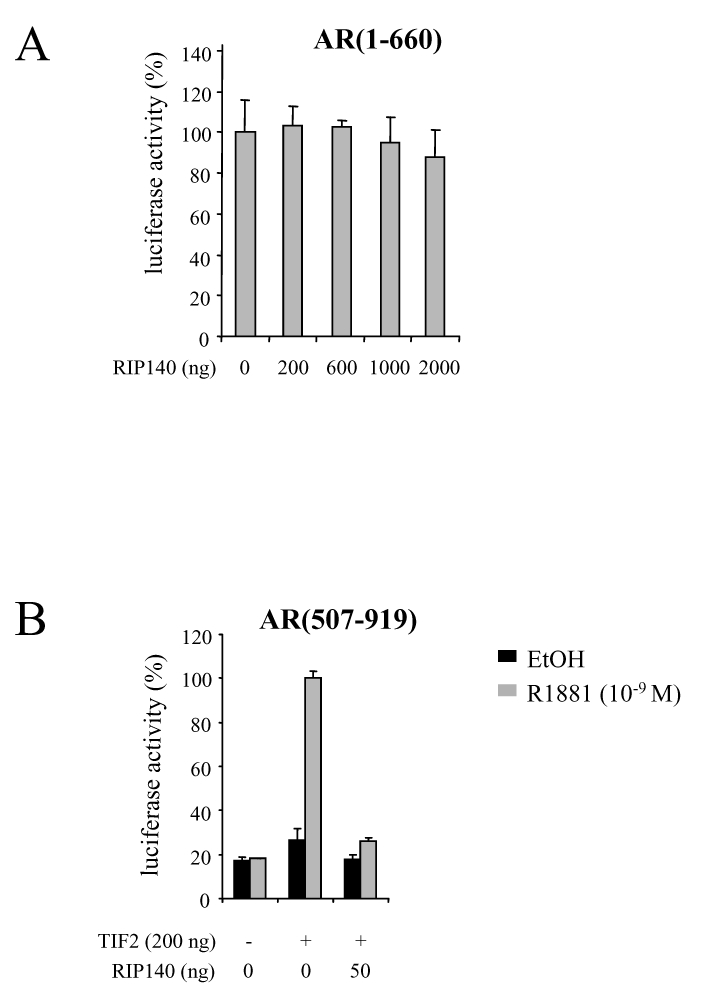
A. RIP140 has no effect on AR(1-660)-dependent transactivation. CV1 cells were transfected as above with 100 ng of pCMV-AR(1-660) and increasing amounts of pcRIP140. The luciferase value given by pCMV-AR(1-660) in the absence of pcRIP140 was determined as 100 %.
B. RIP140 inhibits AR(507-919)-dependent activity. CHO cells were transfected using the FUGENE-6 according to the manufacturer’s instructions with 100 ng of pCMV-AR(507-919), with or without psg5-TIF2 and pEF-RIP140. The luciferase activity was expressed taking AR(507-919) + TIF2 as 100 % of the activity.
The mean ± SD values from at least three independent experiments are shown.
HDACs but not CtBPs participate in RIP140-dependent repression
In a previous study, we precised the role of the two C-terminal binding proteins (CtBPs) on RIP140 activity (26). In that work, the use of RIP140 proteins harboring mutations preventing the interaction with CtBPs significantly affected RIP140 repressive potential. Therefore, in order to further enlighten the mechanism of action of RIP140-mediated inhibition of AR we questioned the role of CtBPs.
RIP140 possesses two CtBP interacting sites corresponding to the sequences PIDLS and PINLS (26). We used vectors coding for RIP140 with single or double mutations for the two interaction motifs.
As observed in Figure 5, it appeared that transfections of 293T cells with either RIP140-mutPIDLS, RIP140-mutPINLS or RIP140-mutPID/NLS did not result in any change in RIP140-mediated AR repression.
Figure 5. CtBPs do not participate in RIP140-dependent AR transactivation.
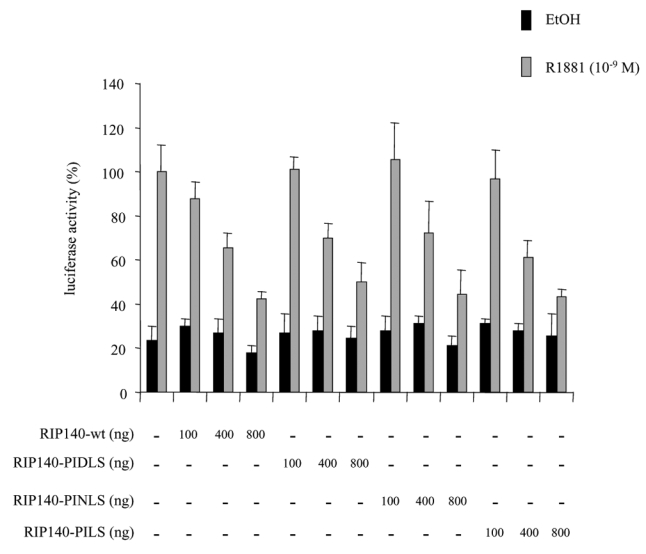
293T cells were transfected with 10 ng of pCMV-AR, increasing amounts of pEF-c-mycRIP140(wt), pEF-c-mycRIP140(PIDLS), pEF-c-mycRIP140(PINLS) or pEF-c-mycRIP140(PID/NLS). The luciferase activity was expressed taking AR in the presence of R1881 as 100 % of the activity. The mean ± SD values from at least three independent experiments are shown.
We concluded that in our experimental conditions, CtBP could not account for RIP140-dependent inhibition of AR.
In the same above mentioned study we showed that RIP140 could interact with class I and class II HDACs (26). Therefore, we asked whether such enzymes would participate in the repression of AR on two different promoters.
We first studied the routinely used mouse mammary tumour virus (MMTV) promoter (Figure 6A). When CV1 cells, first transfected with increasing amounts of RIP140 expressing vectors were then treated with 50 nM trichostatin A (TSA), a specific inhibitor of HDAC activity, we still observed a strong repressive effect of RIP140 on AR transactivation. However, as compared to RIP140-dependent activity without TSA, the same degree of repression was never reached. This first series of data indicated that HDAC activity could partly account for RIP140-dependent AR transcription.
Figure 6. TSA reverses the repressive effect of RIP140 on AR transactivation.
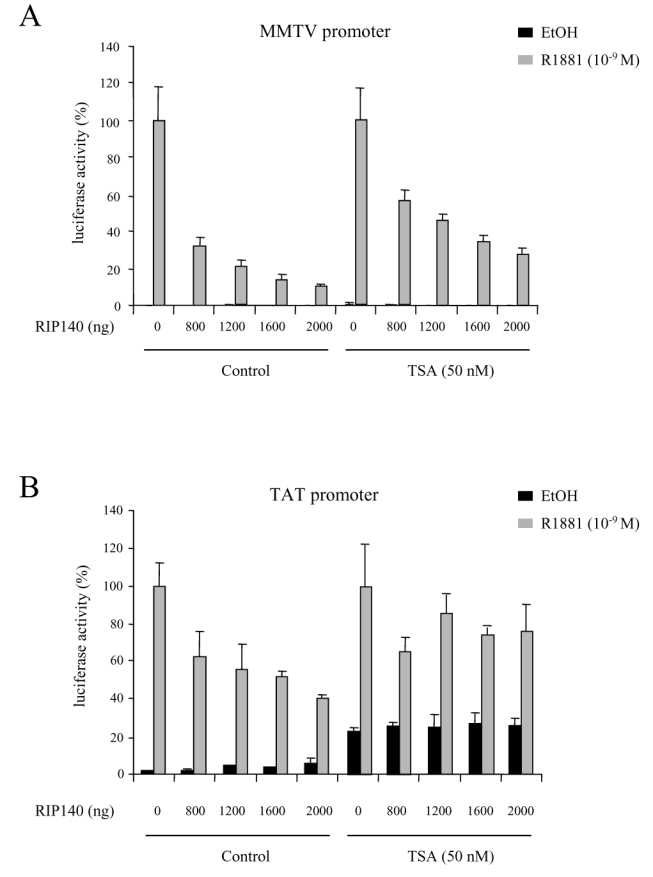
CV1 cells were transfected with 10 ng of pCMV-AR, increasing amounts of pEF-RIP140, and either 1 μg of MMTV (A) or 1 μg of TAT (B) promoters. The experiments were done both in the absence and in the presence of 50 nM TSA. The luciferase activity was expressed taking AR in the presence of R1881 as 100 % of the activity. The mean ± SD values from at least three independent experiments are shown.
To complete this investigation, we then studied the effect of the drug on another AR-responsive promoter, i.e the tyrosine aminotransferase (TAT) promoter. As shown in Figure 6B, RIP140 displayed a repressive effect although not as strong as observed on the MMTV promoter. Indeed, the maximal effect, obtained with 2 μg RIP140 transfected resulted in 60 % decrease of AR activity whereas a 90 % decrease was observed with the MMTV promoter. In the same conditions, under 50 nM TSA, the basal activity of AR was significantly augmented (Figure 6B, right panel). Nevertheless, as observed in the Figure, TSA had a striking effect on RIP140-dependent inhibition. Indeed, it appeared that under TSA treatment, RIP140 only induced between 20 and 30 % decrease of AR transactivation potential as compared to the 70 % decrease observed on a MMTV promoter.
We concluded from this study that TSA had an effect on RIP140-mediated repression of AR which is dependent on the nature of the promoter.
R1881 induces RIP140 mRNA expression in LNCaP cells
RIP140 mRNA expression was already shown to be under the control of different hormones/nuclear receptor ligands, including estradiol (32) and all-trans retinoic acid (33). We therefore asked whether androgens could as well stimulate RIP140 mRNA expression. To this end, we treated LNCaP cells with 10 nM R1881 and then performed a northern blot analysis (Figure 7). As shown in the figure non treated cells displayed barely detectable amounts of RIP140 mRNA. Remarkably, under stimulation with R1881, the amount of mRNA rapidly augmented after a 1-hour treatment to reach a peak at 6-hour induction. This experiment allowed us to propose that in prostate cells RIP140 expression is under the control of androgens thus revealing a potential feed-back loop.
Figure 7. R1881 stimulates RIP140 mRNA expression in LNCaP cells.
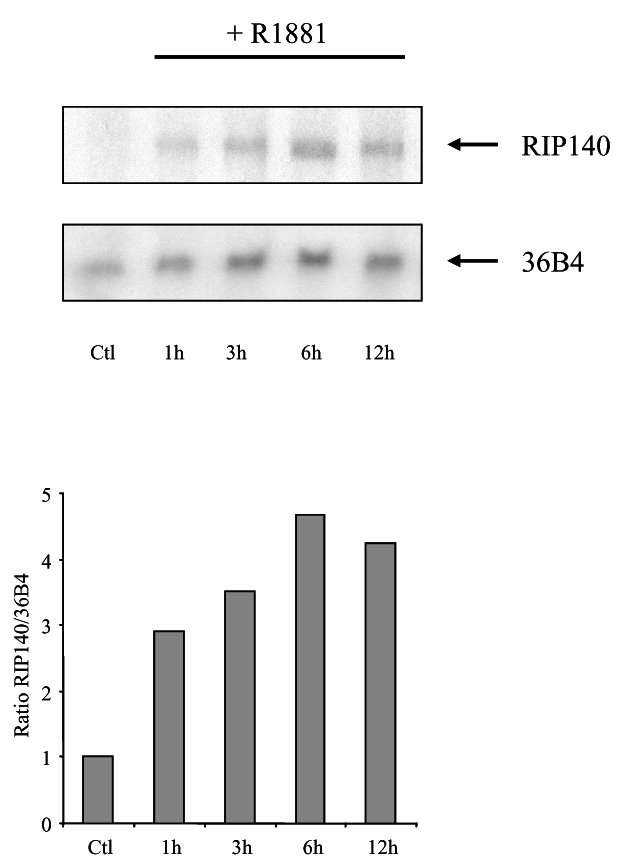
Northern blot analysis of RIP140 mRNA induction in LNCaP cells treated with 10 nM R1881. Upper panel: time course expression of RIP140 mRNA expression. The same membrane was also hybridized with a 36B4 probe to control for RNA loading and transfer. Lower panel: relative ratio of RIP140 to 36B4 signals as determined by phosphor imaging analysis. The figure shown is representative of three independent experiments. Ctl refers to control for which cells were incubated with vehicle for 12 h.
DISCUSSION
This study, aimed at investigating the role of RIP140 on AR-dependent transactivation clearly evidences that RIP140 acts as a strong AR repressor. A controversy raised a few years ago when RIP140 was first described as a coactivator for AR (17). At that time, RIP140 was believed to have modulating effects which could vary with the amount of DNA transfected. Indeed, as first evidenced with ER, low doses of RIP140 were described to slightly increase ER-dependent transactivation whereas higher amounts had the opposite effect (34). In the study by Ikonen et al., the authors observed a paradoxical effect of RIP140: when cotransfected with AR it behaved as a strong coactivator whereas it strongly inhibited the interactions between amino and carboxy-terminal domains (17). Since then, the need for ligand-dependent intra-molecular interactions between the carboxy- and the amino-terminal interactions was extensively described for a full AR transactivation (35, 36). Therefore it appears puzzling to envisage that a protein which exerts a repression of the intramolecular interactions would act as a strong AR coactivator.
In the present study, we first provided clear evidence of the interaction between the two full-length proteins and further delineated the interaction between the ligand binding domain of AR and several domains of RIP140. It is noticeable that RIP140 displays nine so-called nuclear receptor boxes, i. e. the LXXLL motifs. These motifs are randomly distributed along the protein sequence which would explain why the three domains interacted with the carboxy-terminal domain of AR. Although it is not yet explained which motifs in RIP140 would be specifically recruited by AR, a previous study investigated the relative affinity of RIP140 LXXLL motifs for nuclear receptors, evidencing some selectivity (31). Along with this study, fluorescence anisotropy analysis showed that the RIP140 LXXLL motifs presented differential affinities for ERα and ERβ (our unpublished results).
RIP140 is a nuclear protein which was described to form small nuclear foci (37). Tazawa et al. described that when RIP140 was coexpressed with GR, stimulation of the cells with a glucocorticoid ligand induced a relocalization of the cofactor to a more diffuse pattern. In our experiments we described that RIP140 formed previously described nuclear foci. But, when coexpressed with AR previously liganded with R1881, RIP140 was completely relocalized to a diffuse pattern. This relocalization was very specific to the agonist since the antagonist-bound AR was not able to induce such a change. Our results concerning the intracellular localization of AR subdomains may appear contradictory of the study by Saitoh et al. (38) since they described AR-AF1-YFP to form foci whereas the AR amino-terminal domain had a diffuse pattern in our hands. It must be underlined that their construct include the constitutively active receptor which induce a nuclear localization of the fluorescent protein. Very interestingly, when CFP-AR(507-919) was coexpressed with YFP-RIP140 and treated with R1881, the cofactor was completely relocalized with AR LBD, further supporting the fact that AR carboxy-terminal domain of AR is the target for RIP140. It can be hypothesized that a relocalization of RIP140 concomitant to that of the interacting receptor could be a way to activate and render RIP140 available for its targets.
Both the in vitro interaction data and the cotransfection experiments showed that the ligand binding domain of AR was the target of RIP140 action. Many corepressors of AR were shown to target the carboxy-terminal part of the receptor, including AES (39), DAX-1 (40), hRAD9 (41), NCoR and SMRT (42), HBO1 (43), HDAC1 (44) and SHP (45). These corepressors are recruited by the agonist-activated receptor but their mechanisms of action, when elucidated are very different from one protein to another. Only a few can be recruited by the AR DNA-binding domain. Among them can be found ARR19 (13), DJBP (14) or SRY (39). By sharp contrast the corepressors able to be recruited by the amino-terminal domain of AR are rare: SHP (45), Daxx (46), Cyclin D1 (47) and Hey1 (48).
Based on our work it is tempting to postulate that RIP140 first interacts with AR ligand binding domain hence preventing the interaction between the amino- and carboxy-terminal ends of the receptor as evidenced by Ikonen et al. (17). In a previous study by Treuter et al. the authors demonstrated that RIP140 was capable of competing with SRC-1 for binding to nuclear receptors (25). Herein, we showed that RIP140 was able to reverse the overactivation of AR triggered by TIF2. In a similar way, we can propose that a second level of repression could be achieved by the competition between coactivators members of the p160 family of proteins and RIP140. Then, RIP140 could develop its repression activity by recruiting other proteins able to mediate AR inhibition.
Recently, CtBP was shown to be recruited by RIP140 and could partly account for the cofactor-dependent intrinsic repression (26, 49). However, in the present paper, the use of RIP140 mutants unable to bind to CtBPs allowed us to show that this negative modulator had no effect on RIP140-mediated AR activity.
The use of a specific inhibitor of HDAC activity, TSA, significantly affected RIP140 repression. As previously reported, the intrinsic repressive potential of RIP140 was shown to be sensitive to TSA (50). However, this evidence was then controversed by the report of Castet et al. where the effect of TSA was efficient on subdomains of RIP140 fused to a heterologous protein whereas the drug had no effect on the full-length protein (26). Still, in the same study, we described an interaction between a class II HDAC, precisely HDAC5, and RIP140. Along with this study, in our investigations, TSA had different effects according to the promoter context further supporting this hypothesis. We therefore hypothesize that depending on RIP140 targets and cellular context, HDAC proteins may be differentially recruited and therefore have very different effects on the nuclear receptor activity.
We and others (26, 27) evidenced that the carboxy-terminal domain of RIP140 displayed a strong intrinsic repression. Still, to date no protein was isolated and shown to mediate that repression. Moreover, results obtained with TSA suggest that non-HDAC proteins also participate in RIP140-dependent transrepression of AR. Therefore we are currently investigating that issue in order to decipher the complete mechanism of action of the cofactor. RIP140-dependent repression of AR is reminiscent of that found for other AR corepressors including SHP (15). Indeed, we described SHP as a cofactor able to exert its transcriptional repression through both a competition with AR coactivators and also by recruitment of HDAC activities. According to the cellular context, RIP140 could exert its repressive functions via various proteins. This large potential could well be a way to make sure the protein can exert a strong repressive action.
The present work also revealed that RIP140 mRNA was under the control of R1881 in LNCaP cells. The induction of mRNA expression was rapid and reached a maximum effect after 6h. At this point, it is impossible to conclude whether this induction requires de novo protein synthesis or whether it is directly mediated by AR-stimulation of the gene. However, isolation of the gene coding for RIP140 revealed that the promoter region contained at least three consensus androgen receptor elements (unpublished results) suggesting that the mRNA expression could be directly stimulated by the receptor. It must be underlined that such an induction is reminiscent of the previously described estradiol and retinoic acid-mediated RIP140 mRNA expression (33, 51).
Very interestingly, our data from ChIP assays clearly indicated that, under AR agonist treatment RIP140 was recruited to both the promoter and the enhancer regions of the PSA gene with a better recruitment to the promoter than to the enhancer. Recent studies were undertaken to investigate the dynamics of different transcription factors onto the PSA gene in response to androgens. Two different studies described AR as preferentially recruited to the enhancer but differed with description of either a receptor’s residence time being more transient on the enhancer (52) or a long term recruitment of the receptor to that region (28).
The two papers also differed in describing the recruitment of different receptor cofactors with either a loading of the p160 family members to both the enhancer and promoter of the PSA gene (52) or a preferential recruitment of the enhancer region of the gene (28). However very little is known about loading of AR corepressors to androgen-responsive genes. In that context, it would be of interest to undertake a kinetics study of RIP140 association to a specific AR-responsive gene such as PSA. That work would tell us when that recruitment to the promoter occurs with regards to other transcription factors (52) and would permit us to give substantial insight into the androgen-induced expression of RIP140 mRNA we evidenced. Overall, we believe that the potential physiological loop we observed in prostate cells is of importance since it would allow a precise control of the androgen activity in many physiopathological issues including cancer.
MATERIALS AND METHODS
Plasmids
Mammalian expression plasmids
pCMV-AR and pSG5-TIF2 were generous gifts from respectively Drs Terry Brown and Hinrich Gronemeyer.
pCMV-AR(1-660), pCMV-AR(507-919) (45) and pFC31-Luc (53) were already described. pEYFP-RIP140, pcRIP140, pcRIP(1-282), pcRIP(1-480), pcRIP(479-1158), pcRIP(917-1158), pcRIPmutPIDLS, pcRIPmutPINLS and pcRIPmutPID/NLS were described elsewhere (26).
pEF-c-mycRIP140 was created by inserting the PCR-amplified cDNA encoding c-myc fused to RIP140 amino-acids 1 to 479 into pEF-RIP140 previously digested with BclI and EcoRV.
pEF-RIPmutPIDLS was created by digestion of pcRIPmutPIDLS with BclI and EcoRV and insertion of the resulting fragment into pEF-RIP140. pEF-RIPmutPINLS was created by digestion of pcRIPmutPINLS with EcoRV and BlpI and insertion of the resulting fragment into pEF-RIP140. pEF-RIPmutPID/NLS was created by digestion of pcRIPmutPID/NLS with BclI and BlpI and insertion of the resulting fragment into pEF-RIP140.
pECFP-AR(507-919) was created by inserting the PCR-amplified cDNA encoding AR(507-919) into pECFP-C2 previously digested with BamHI. pGFP-AR(1-501) was digested with XhoI and XbaI and the resulting fragment was inserted into pECFP-AR previously digested with the same enzymes to create pECFP-AR(1-501). pECFP-AR was obtained by insertion of AR excised by NheI and BglII restriction sites from the previously described pGFP-AR (29) into pECFP-C1 (Clontech, Palo Alto, CA).
Bacterial expression plasmids
pGEX-RIP(27-439), pGEX-RIP(428-739) and pGEX-RIP(683-1158) were described elsewhere (16).
Plasmids for in vitro expression
pGBK-AR, pGBK-AR(1-501) and pGBK-AR(627-919) were previously described (45).
Transient transfection
CV1, COS7 and 293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fœtal calf serum (FCS). For transient transfection experiments, cells were plated in 12-well dishes and transfected using the calcium phosphate method. CHO and MEF cells were cultured in DMEM-F12 supplemented with 10 % FCS and transfected using FuGENE 6 according to the manufacturer’s instructions (ROCHE) and calcium phosphate respectively. Whenever different amounts of an expressing vector were transfected, the quantity of DNA was kept constant by the use of the empty vector. Twelve hours after transfection, and 24–30 hours before lysis, R1881 was added to a final concentration of 10−9 M. When indicated, trichostatin A (TSA) was added 17 hours before lysis. Cells were then harvested in a lysis buffer (25 mM Tris-H3PO4 (pH 7.8), 2 mM DTT, 2 mM EDTA, 1 % TritonX-100 and 10 % glycerol) and the luciferase activity was measured by the reaction of lysate with the luciferin solution (270 μM coenzyme A, 470 μM luciferin, 530 μM ATP, 20 mM Tris-H3PO4, 1.05 mM MgCl2, 2.7 mM MgSO4, 0.1 mM EDTA and 33 mM DTT) on a luminometer. In all experiments pCMV-βgal was used to normalize the transfection efficiency.
Fluorescence microscopy and imaging analysis
COS7 cells were cultured on coverslips and then transfected with 1 μg of each plasmid using 3 μl of FuGENE 6 reagent (Roche) per dish. 24 hours after transfection, the culture medium was replaced with serum-free DMEM for overnight starvation. Cells were incubated with R1881 (10−8 M) or antihormones (10−6 M) for 8 hours, fixed with 4 % paraformaldehyde for 15 min, washed three times with PBS and mounted on slides with DAKO mounting medium. The cells were imaged using confocal laser scanning microscopy (Leica SP2 UV system, Leica Microsystems, Heidelberg, Germany). CFP was excited with a 457-nm argon laser line and CFP emission was sampled between 460 and 490 nm. The cells were imaged for yellow fluorescence by excitation with the 514-nm argon laser line and emission was sampled between 520 and 550 nm. The images were analyzed with LCS (Leica Confocal Software) and merged images were generated by Adobe Photoshop software.
In vitro transcription and translation
Expression plasmids pGBK-ARwt, pGBK-AR(1-501) and pGBK-AR(618-628) were transcribed and translated using the TNTT7-coupled reticulocyte lysate system (Promega, France) in the presence of 35S labeled methionin for 1h30 at 30 C according to the manufacturer’s instructions.
GST pull-down
GST, GST-RIP(27-439), GST-RIP(428-739) and GST-RIP(683-1158) were produced and purified as previously described (26). Each aliquot of 500 μl containing 30 μl of Glutathione Sepharose (Pharmacia) was mixed with either 35S labelled ARwt, AR (1-501) or AR (618-928). After incubation for 3 hours at 4° C, beads were washed four times with PDB (PBS containing 20 mM Hepes-KOH, pH 7.9, 10 % glycerol, 100 mM KCl, 5 mM MgCl2, 0.2 mM EDTA, 1 mM DTT, 0.2 mM PMSF), and boiled for 5 minutes in the presence of SDS buffer. Proteins were then separated on a 10 % SDS-PAGE. Gels were colored with coomassie blue, dried, and autoradiographies were performed with KODAK biomax films. The figures are representative of at least three independent experiments.
Immunoprecipitations
293T cells were transfected and treated with 10 nM R1881 as described above except that 100 mm dishes were used and 10 μg of pCMV-AR and pEF-c-mycRIP140 were transfected. The cells were resuspended in 500 μl of lysis buffer (50 mM Tris-HCl (pH 7.4), 100 mM NaCl, 5 mM CaCl2, 5 mM MgCl2, 1 % NP40, 1 % Triton) supplemented with protease inhibitors. 400 μl of each extract were first incubated with the anti-AR (AR-441, Santa Cruz) monoclonal antibody for 2 hours at 4 C, and then with Protein G Sepharose for an additional 16 hours at 4 C. Protein G-Sepharose containing the immune complex was then washed 3 times with the washing buffer (50 mM Tris-HCl (pH 7.4), 100 mM NaCl, 5 mM CaCl2, 5 mM MgCl2, 0.1 % NP40) and resuspended in SDS containing sample buffer. The proteins were resolved through a 6 % SDS-PAGE and immunoblotted with either an anti-c-myc monoclonal antibody or an anti-AR polyclonal antibody (N20, Santa Cruz). Signals were detected with the ECL method (Amersham Biosciences) using Kodak Biomax films.
ChIP analysis
ChIP assays were performed as described in Metivier et al. (54) with minor modifications. In brief, at the end of hormone treatment, medium was removed and replaced by PBS containing hormone or ethanol. Chromatin was crosslinked using 1 % formaldehyde at 25 C for 10 min followed by a 15 min incubation with 250 mM glycine. Cells were then rinsed twice with cold PBS and centrifuged. PBS was removed and cells were quickly frozen, using liquid nitrogen, until sonication process of all samples. Cells were then washed sequentially with buffer A (10 mM EDTA, 0.5 mM EGTA, 10 mM HEPES (pH 6.5) and 0.25 % Triton X-100) and buffer B (1 mM EDTA, 0.5 mM EGTA, 10 mM HEPES (pH 6.5) and 200 mM NaCl), containing antiproteases. They were then resuspended in Lysis buffer (10 mM EDTA, 50 mM Tris-HCl (pH 8.0), 1 % SDS, 0.5 % Empigen BB). Samples were sonicated 3 × 8 times for 4 s at 60 % settings (Bioblock Vibra cell, Model 7205) and centrifuged at 14000 rpm at 4 C. Immunoprecipitations (using 2 μg of H300 anti-RIP140 (Santa Cruz, France)) and washes were performed as described in (54), (except that samples were diluted 10 times with IP dilution buffer containing antiproteases). DNA was purified with QIAquick columns (Qiagen, France). Real time PCRs were performed using 3 μl of sample DNA, and 3 μl of diluted inputs. The primers were: PSA promoter, forward primer: TCTGCCTTTGTCCCCTAGAT; reverse primer: GGGAGGGAGAGCTAGCACTTG. PSA enhancer, forward primer: GCCTGGATCTGAGAGAGATATCATC, reverse primer: ACACCTTTTTTTTTCTGGATTGTTG.
RNA extraction and Northern blot analysis
LNCaP cells were cultured in RPMI1640 medium supplemented with 10 % charcoal-stripped FCS, 0.1 % glucose and puromycin, prior to incubation with or without 10 nM R1881 as indicated. Total RNA was isolated with TRI REAGENT (Molecular Research Center) as described by the manufacturer. RNA quantity was determined photometrically by absorption at 260 nm, stored in RNase-free H2O at −80 C until analysis. For Northern blot assays, 30 μg RNA were electrophoresed and then hybridized with [32P]ATP-labeled probes: RIP140 cDNA (51) and 36B4 cDNA (encoding the human acidic ribosomal phosphoprotein PO), used to correct variations in the amount of RNA loaded on each track (55). Hybridization was quantified by phosphorimager analysis using a Fujix-Bas 1000 phosphorimager (RAYTEST, Courbevoie, France).
Acknowledgments
We would like to thank Dr Jacky Marie for the kind gift of anti-c-myc antibodies. This paper is dedicated to the memory of Françoise Vignon.
S. C. received grants from “Association pour la Recherche sur les Tumeurs de la Prostate” and ”Ligue Nationale contre le Cancer”, J. G. and V. G. from “Association pour la Recherche contre le Cancer” and A. C. was a recipient of “Poste d’accueil INSERM”. This work was funded by the Institut National de la Santé et de la Recherche Médicale, Faculté de Médecine de Montpellier, “Association pour la Recherche contre le Cancer” (grant N° 3494), “Ligue Nationale contre le Cancer” (grant N° RAB05002FFA) and “Fondation Jérôme Lejeune”.
Non standard abbreviations
- RIP140
receptor interacting protein of 140 kDa
- AR
androgen receptor
- ChIP
chromatin immunoprecipitation
- PSA
prostate specific antigen
- CtBP
C-terminal binding protein
- TIF2
transcriptional intermediary factor 2
- ARE
androgen receptor response element
- TSA
trichostatin A
- HDAC
histone deacetylase
- DBD
DNA binding domain
- LBD
ligand binding domain
- AF
activating function
Footnotes
Institute of Reproductive and Developmental Biology, Imperial College London, Du Cane Road, London W12 0NN, United Kingdom
This is an un-copyedited author manuscript copyrighted by The Endocrine Society. This may not be duplicated or reproduced, other than for personal use or within the rule of “Fair Use of Copyrighted Materials” (section 107, Title 17, U.S. Code) without permission of the copyright owner, The Endocrine Society. From the time of acceptance following peer review, the full text of this manuscript is made freely available by The Endocrine Society at http://www.endojournals.org/. The final copy edited article can be found at http://www.endojournals.org/. The Endocrine Society disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties.
present address: Institut Biologie Intégrative, 7, quai Saint-Bernard 75252 PARIS cedex 05, France
References
- 1.Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20:3001–15. doi: 10.1200/JCO.2002.10.018. [DOI] [PubMed] [Google Scholar]
- 2.McPhaul MJ. Molecular defects of the androgen receptor. J Steroid Biochem Mol Biol. 1999;69:315–22. doi: 10.1016/s0960-0760(99)00050-3. [DOI] [PubMed] [Google Scholar]
- 3.Culig Z, Klocker H, Bartsch G, Hobisch A. Androgen receptors in prostate cancer. Endocr Relat Cancer. 2002;9:155–70. doi: 10.1677/erc.0.0090155. [DOI] [PubMed] [Google Scholar]
- 4.Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18:306–60. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- 5.Jenster G, van der Korput HA, Trapman J, Brinkmann AO. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J Biol Chem. 1995;270:7341–6. doi: 10.1074/jbc.270.13.7341. [DOI] [PubMed] [Google Scholar]
- 6.Brinkmann AO, Blok LJ, de Ruiter PE, Doesburg P, Steketee K, Berrevoets CA, Trapman J. Mechanisms of androgen receptor activation and function. J Steroid Biochem Mol Biol. 1999;69:307–13. doi: 10.1016/s0960-0760(99)00049-7. [DOI] [PubMed] [Google Scholar]
- 7.Heinlein CA, Chang C. Androgen Receptor (AR) Coregulators: An Overview. Endocr Rev. 2002;23:175–200. doi: 10.1210/edrv.23.2.0460. [DOI] [PubMed] [Google Scholar]
- 8.Roeder RG. The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem Sci. 1996;21:327–35. [PubMed] [Google Scholar]
- 9.Neely KE, Workman JL. The complexity of chromatin remodeling and its links to cancer. Biochim Biophys Acta. 2002;1603:19–29. doi: 10.1016/s0304-419x(02)00067-7. [DOI] [PubMed] [Google Scholar]
- 10.Roeder RG. Role of general and gene-specific cofactors in the regulation of eukaryotic transcription. Cold Spring Harb Symp Quant Biol. 1998;63:201–18. doi: 10.1101/sqb.1998.63.201. [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Hsu CL, Chang C. Androgen receptor corepressors: An overview. Prostate. 2005;63:117–30. doi: 10.1002/pros.20170. [DOI] [PubMed] [Google Scholar]
- 12.Cheng S, Brzostek S, Lee SR, Hollenberg AN, Balk SP. Inhibition of the dihydrotestosterone-activated androgen receptor by nuclear receptor corepressor. Mol Endocrinol. 2002;16:1492–501. doi: 10.1210/mend.16.7.0870. [DOI] [PubMed] [Google Scholar]
- 13.Jeong BC, Hong CY, Chattopadhyay S, Park JH, Gong EY, Kim HJ, Chun SY, Lee K. Androgen Receptor Corepressor-19 kDa (ARR19), a Leucine-Rich Protein that Represses the Transcriptional Activity of Androgen Receptor through Recruitment of Histone Deacetylase. Mol Endocrinol. 2004;18:13–25. doi: 10.1210/me.2003-0065. [DOI] [PubMed] [Google Scholar]
- 14.Niki T, Takahashi-Niki K, Taira T, Iguchi-Ariga SM, Ariga H. DJBP: a novel DJ-1-binding protein, negatively regulates the androgen receptor by recruiting histone deacetylase complex, and DJ-1 antagonizes this inhibition by abrogation of this complex. Mol Cancer Res. 2003;1:247–61. [PubMed] [Google Scholar]
- 15.Gobinet J, Carascossa S, Cavaillès V, Vignon F, Nicolas JC, Jalaguier S. SHP Represses Transcriptional Activity via Recruitment of Histone Deacetylases. Biochemistry. 2005;44:6312–20. doi: 10.1021/bi047308d. [DOI] [PubMed] [Google Scholar]
- 16.L’Horset F, Dauvois S, Heery DM, Cavaillès V, Parker MG. RIP-140 interacts with multiple nuclear receptors by means of two distinct sites. Mol Cell Biol. 1996;16:6029–36. doi: 10.1128/mcb.16.11.6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ikonen T, Palvimo JJ, Jänne OA. Interaction between the amino- and carboxyl-terminal regions of the rat androgen receptor modulates transcriptional activity and is influenced by nuclear receptor coactivators. J Biol Chem. 1997;272:29821–29828. doi: 10.1074/jbc.272.47.29821. [DOI] [PubMed] [Google Scholar]
- 18.Masuyama H, Brownfield CM, St-Arnaud R, MacDonald PN. Evidence for ligand-dependent intramolecular folding of the AF-2 domain in vitamin D receptor-activated transcription and coactivator interaction. Mol Endocrinol. 1997;11:1507–17. doi: 10.1210/mend.11.10.9990. [DOI] [PubMed] [Google Scholar]
- 19.Miyata KS, McCaw SE, Meertens LM, Patel HV, Rachubinski RA, Capone JP. Receptor-interacting protein 140 interacts with and inhibits transactivation by, peroxisome proliferator-activated receptor alpha and liver-X-receptor alpha. Mol Cell Endocrinol. 1998;146:69–76. doi: 10.1016/s0303-7207(98)00196-8. [DOI] [PubMed] [Google Scholar]
- 20.Subramaniam N, Treuter E, Okret S. Receptor interacting protein RIP140 inhibits both positive and negative gene regulation by glucocorticoids. J Biol Chem. 1999;274:18121–7. doi: 10.1074/jbc.274.25.18121. [DOI] [PubMed] [Google Scholar]
- 21.Sugawara T, Abe S, Sakuragi N, Fujimoto Y, Nomura E, Fujieda K, Saito M, Fujimoto S. RIP 140 modulates transcription of the steroidogenic acute regulatory protein gene through interactions with both SF-1 and DAX-1. Endocrinology. 2001;142:3570–7. doi: 10.1210/endo.142.8.8309. [DOI] [PubMed] [Google Scholar]
- 22.Kumar MB, Tarpey RW, Perdew GH. Differential recruitment of coactivator RIP140 by Ah and estrogen receptors. Absence of a role for LXXLL motifs. J Biol Chem. 1999;274:22155–64. doi: 10.1074/jbc.274.32.22155. [DOI] [PubMed] [Google Scholar]
- 23.Zilliacus J, Holter E, Wakui H, Tazawa H, Treuter E, Gustafsson JÅ. Regulation of glucocorticoid receptor activity by 14-3-3-dependent intracellular relocalization of the corepressor rip140. Mol Endocrinol. 2001;15:501–11. doi: 10.1210/mend.15.4.0624. [DOI] [PubMed] [Google Scholar]
- 24.Teyssier C, Belguise K, Galtier F, Cavaillès V, Chalbos D. Receptor-Interacting Protein 140 Binds c-Jun and Inhibits Estradiol-Induced Activator Protein-1 Activity by Reversing Glucocorticoid Receptor-Interacting Protein 1 Effect. Mol Endocrinol. 2003;17:287–99. doi: 10.1210/me.2002-0324. [DOI] [PubMed] [Google Scholar]
- 25.Treuter E, Albrektsen T, Johansson L, Leers J, Gustafsson JÅ. A regulatory role for RIP140 in nuclear receptor activation. Mol Endocrinol. 1998;12:864–81. doi: 10.1210/mend.12.6.0123. [DOI] [PubMed] [Google Scholar]
- 26.Castet A, Boulahtouf A, Versini G, Bonnet S, Augereau P, Vignon F, Khochbin S, Jalaguier S, Cavaillès V. Multiple domains of the Receptor-Interacting Protein 140 contribute to transcription inhibition. Nucleic Acids Res. 2004;32:1957–66. doi: 10.1093/nar/gkh524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christian M, Tullet JM, Parker MG. Characterization of four autonomous repression domains in the corepressor receptor interacting protein 140. J Biol Chem. 2004;279:15645–51. doi: 10.1074/jbc.M313906200. [DOI] [PubMed] [Google Scholar]
- 28.Wang Q, Carroll JS, Brown M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol Cell. 2005;19:631–42. doi: 10.1016/j.molcel.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 29.Georget V, Terouanne B, Nicolas JC, Sultan C. Mechanism of antiandrogen action: key role of hsp90 in conformational change and transcriptional activity of the androgen receptor. Biochemistry. 2002;41:11824–31. doi: 10.1021/bi0259150. [DOI] [PubMed] [Google Scholar]
- 30.Christian M, Kiskinis E, Debevec D, Leonardsson G, White R, Parker MG. RIP140-targeted repression of gene expression in adipocytes. Mol Cell Biol. 2005;25:9383–91. doi: 10.1128/MCB.25.21.9383-9391.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heery DM, Hoare S, Hussain S, Parker MG, Sheppard H. Core LXXLL Motif Sequences in CREB-binding Protein, SRC1, and RIP140 Define Affinity and Selectivity for Steroid and Retinoid Receptors. J Biol Chem. 2001;276:6695–6702. doi: 10.1074/jbc.M009404200. [DOI] [PubMed] [Google Scholar]
- 32.Thénot S, Bonnet S, Boulahtouf A, Margeat E, Royer CA, Borgna JL, Cavaillès V. Effect of ligand and DNA binding on the interaction between human transcription intermediary factor 1alpha and estrogen receptors. Mol Endocrinol. 1999;13:2137–50. doi: 10.1210/mend.13.12.0387. [DOI] [PubMed] [Google Scholar]
- 33.Kerley JS, Olsen SL, Freemantle SJ, Spinella MJ. Transcriptional activation of the nuclear receptor corepressor RIP140 by retinoic acid: a potential negative-feedback regulatory mechanism. Biochem Biophys Res Commun. 2001;285:969–75. doi: 10.1006/bbrc.2001.5274. [DOI] [PubMed] [Google Scholar]
- 34.Cavaillès V, Dauvois S, L’Horset F, Lopez G, Hoare S, Kushner PJ, Parker MG. Nuclear factor RIP140 modulates transcriptional activation by the estrogen receptor. Embo J. 1995;14:3741–51. doi: 10.1002/j.1460-2075.1995.tb00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He B, Kemppainen JA, Voegel JJ, Gronemeyer H, Wilson EM. Activation Function 2 in the Human Androgen Receptor Ligand Binding Domain Mediates Interdomain Communication with the NH(2)-terminal Domain. J Biol Chem. 1999;274:37219–37225. doi: 10.1074/jbc.274.52.37219. [DOI] [PubMed] [Google Scholar]
- 36.He B, Kemppainen JA, Wilson EM. FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J Biol Chem. 2000;275:22986–94. doi: 10.1074/jbc.M002807200. [DOI] [PubMed] [Google Scholar]
- 37.Tazawa H, Osman W, Shoji Y, Treuter E, Gustafsson JÅ, Zilliacus J. Regulation of subnuclear localization is associated with a mechanism for nuclear receptor corepression by RIP140. Mol Cell Biol. 2003;23:4187–98. doi: 10.1128/MCB.23.12.4187-4198.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saitoh M, Takayanagi R, Goto K, Fukamizu A, Tomura A, Yanase T, Nawata H. The Presence of Both the Amino- and Carboxyl-Terminal Domains in the AR Is Essential for the Completion of a Transcriptionally Active Form with Coactivators and Intranuclear Compartmentalization Common to the Steroid Hormone Receptors: A Three-Dimensional Imaging Study. Mol Endocrinol. 2002;16:694–706. doi: 10.1210/mend.16.4.0812. [DOI] [PubMed] [Google Scholar]
- 39.Yu X, Li P, Roeder RG, Wang Z. Inhibition of Androgen Receptor-Mediated Transcription by Amino-Terminal Enhancer of split. Mol Cell Biol. 2001;21:4614–25. doi: 10.1128/MCB.21.14.4614-4625.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holter E, Kotaja N, Mäkela S, Strauss L, Kietz S, Janne OA, Gustafsson JÅ, Palvimo JJ, Treuter E. Inhibition of Androgen Receptor (AR) Function by the Reproductive Orphan Nuclear Receptor DAX-1. Mol Endocrinol. 2002;16:515–28. doi: 10.1210/mend.16.3.0804. [DOI] [PubMed] [Google Scholar]
- 41.Wang L, Hsu CL, Ni J, Wang PH, Yeh S, Keng P, Chang C. Human checkpoint protein hRad9 functions as a negative coregulator to repress androgen receptor transactivation in prostate cancer cells. Mol Cell Biol. 2004;24:2202–13. doi: 10.1128/MCB.24.5.2202-2213.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jackson TA, Richer JK, Bain DL, Takimoto GS, Tung L, Horwitz KB. The partial agonist activity of antagonist-occupied steroid receptors is controlled by a novel hinge domain-binding coactivator L7/SPA and the corepressors N-CoR or SMRT. Mol Endocrinol. 1997;11:693–705. doi: 10.1210/mend.11.6.0004. [DOI] [PubMed] [Google Scholar]
- 43.Sharma M, Zarnegar M, Li X, Lim B, Sun Z. Androgen receptor interacts with a novel MYST protein, HBO1 [In Process Citation] J Biol Chem. 2000;275:35200–8. doi: 10.1074/jbc.M004838200. [DOI] [PubMed] [Google Scholar]
- 44.Gaughan L, Logan IR, Cook S, Neal DE, Robson CN. Tip60 and Histone Deacetylase 1 Regulate Androgen Receptor Activity through Changes to the Acetylation Status of the Receptor. J Biol Chem. 2002;277:25904–13. doi: 10.1074/jbc.M203423200. [DOI] [PubMed] [Google Scholar]
- 45.Gobinet J, Auzou G, Nicolas JC, Sultan C, Jalaguier S. Characterization of the interaction between androgen receptor and a new transcriptional inhibitor, SHP. Biochemistry. 2001;40:15369–77. doi: 10.1021/bi011384o. [DOI] [PubMed] [Google Scholar]
- 46.Lin DY, Fang HI, Ma AH, Huang YS, Pu YS, Jenster G, Kung HJ, Shih HM. Negative modulation of androgen receptor transcriptional activity by Daxx. Mol Cell Biol. 2004;24:10529–41. doi: 10.1128/MCB.24.24.10529-10541.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burd CJ, Petre CE, Moghadam H, Wilson EM, Knudsen KE. Cyclin D1 binding to the androgen receptor (AR) NH2-terminal domain inhibits activation function 2 association and reveals dual roles for AR corepression. Mol Endocrinol. 2005;19:607–20. doi: 10.1210/me.2004-0266. [DOI] [PubMed] [Google Scholar]
- 48.Belandia B, Powell SM, Garcia-Pedrero JM, Walker MM, Bevan CL, Parker MG. Hey1, a mediator of notch signaling, is an androgen receptor corepressor. Mol Cell Biol. 2005;25:1425–36. doi: 10.1128/MCB.25.4.1425-1436.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vo N, Fjeld C, Goodman RH. Acetylation of nuclear hormone receptor-interacting protein RIP140 regulates binding of the transcriptional corepressor CtBP. Mol Cell Biol. 2001;21:6181–8. doi: 10.1128/MCB.21.18.6181-6188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei LN, Hu X, Chandra D, Seto E, Farooqui M. Receptor-interacting Protein 140 Directly Recruits Histone Deacetylases for Gene Silencing. J Biol Chem. 2000;275:40782–40787. doi: 10.1074/jbc.M004821200. [DOI] [PubMed] [Google Scholar]
- 51.Thénot S, Charpin M, Bonnet S, Cavaillès V. Estrogen receptor cofactors expression in breast and endometrial human cancer cells. Mol Cell Endocrinol. 1999;156:85–93. doi: 10.1016/s0303-7207(99)00139-2. [DOI] [PubMed] [Google Scholar]
- 52.Kang Z, Janne OA, Palvimo JJ. Coregulator recruitment and histone modifications in transcriptional regulation by the androgen receptor. Mol Endocrinol. 2004;18:2633–48. doi: 10.1210/me.2004-0245. [DOI] [PubMed] [Google Scholar]
- 53.Gouilleux F, Sola B, Couette B, Richard-Foy H. Cooperation between structural elements in hormono-regulated transcription from the mouse mammary tumor virus promoter. Nucleic Acids Res. 1991;19:1563–9. doi: 10.1093/nar/19.7.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–63. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 55.Cavaillès V, Augereau P, Garcia M, Rochefort H. Estrogens and growth factors induce the mRNA of the 52K-pro-cathepsin-D secreted by breast cancer cells. Nucleic Acids Res. 1988;16:1903–19. doi: 10.1093/nar/16.5.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]