

Abstract
Genetic and molecular studies indicate that dysbindin-1 plays a role in the pathophysiology of schizophrenia. We examined dysbindin-1 mRNA in the hippocampal formation of patients with schizophrenia and found reduced expression in dentate granule and polymorph cells and in hippocampal field CA3, but not in CA1. Furthermore, there were positive correlations between dysbindin-1 mRNA and expression of synaptic markers known to be reduced in schizophrenia. Our results indicate that previously reported dysbindin-1 protein reductions may be due in part to decreased dysbindin-1 mRNA and that reduced dysbindin-1 may contribute to hippocampal formation synaptic pathology in schizophrenia.
Keywords: schizophrenia, postmortem, hippocampus, spinophilin, synaptophysin, candidate gene, synapse, synaptic pathology
1. Introduction
Genetic variation in DTNBP1, which encodes dysbindin-1, is associated with schizophrenia in a number of populations (Riley and Kendler 2006; Schwab et al. 2003; Straub et al. 2002). One mechanism whereby these may alterations confer susceptibility to schizophrenia is via reduced dysbindin-1 expression, since risk SNPs and haplotypes appear to influence dysbindin-1 mRNA levels (Bray et al. 2003; Bray et al. 2005; Weickert et al. 2004). In patients with schizophrenia, reduced frontal cortex dysbindin-1 mRNA (Weickert et al. 2004) and lower hippocampal formation dysbindin-1 protein (Talbot et al. 2004) have been observed. We tested whether dysbindin-1 mRNA was reduced in the hippocampal formation of schizophrenia patients.
Abnormalities of glutamate neurotransmission, including reduction in AMPA receptors, have been consistently found in the hippocampal formation of patients with schizophrenia (Harrison and Eastwood 2001; Harrison et al. 2003). We and others have found significant reductions in synaptophysin (Eastwood et al. 1994; Webster et al. 2001) and spinophilin mRNA (Law et al. 2004) in the DGh (hilar or polymorph region of the dentate gyrus) and CA3 pyramidal cell regions. We hypothesized that synaptophysin and spinophilin mRNA, markers of the pre- and post-synaptic terminals, respectively, may be related to dysbindin-1 mRNA expression in specific hippocampal formation subregions.
2. Materials and Methods
2.1 Subjects
Mesial temporal lobes were cryostat-sectioned from postmortem brains (Law et al. 2004; Webster et al. 2001). Three sections per case (average) were anatomically matched at the genu and mid-body level. Demographic and diagnostic criteria were previously published [n=10 schizophrenics and n=10 normal controls (Law et al. 2004; Webster et al. 2001)]. Experimental groups were formed matching for group means in age, tissue pH and PMI [(controls= 55y, 6.5, 24h; schizophrenics =50y, 6.4, 34h) (all t<1.73, all p>0.10)]. Previous studies of cyclophilin, a control mRNA, showed no difference in expression between the schizophrenics and controls in this cohort (Law et al., 2004). Neuroleptic medication for each subject with schizophrenia was recorded after clinical chart review and doses were converted to milligram chlorpromazine (CPZ) equivalents. Neuroleptic exposure was measured as: 1) last recorded neuroleptic dose before death (last CPZ); 2) median daily neuroleptic dose for all medications prescribed during a subject’s life (daily CPZ); 3) lifetime neuroleptic exposure as median dose multiplied by treatment duration (lifetime CPZ).
2.2 In situ hybridization
Riboprobes to measure dysbindin-1 mRNA expression that include transcripts from GenBank Accession nos. BC011912, AF061734, and AL136637, were synthesized, validated, and applied to fresh frozen tissue sections as described (Weickert et al. 2004). Within the hippocampal formation, areas were sampled as regions of interest with the experimenter blind to diagnosis. Measurements were made on the dentate granule cell layer (DGg), the DGh, and the pyramidal cell layer of CA3 and CA1.
2.3 Statistical analyses
Diagnostic comparisons were made with ANOVA and Fisher’s Least Significant Difference Tests. Specific contrasts for each subregion were planned a priori and were investigated with one-tailed student t-tests. Pearson’s correlation among dysbindin-1 mRNA dysbindin-1 mRNA levels, synaptic mRNAs, demographic variables, spinophilin and synaptophysin mRNA levels were run (n=20) with and without a partial correlation for pH and PMI. Spearman correlations between the neuroleptic exposure in patients and dysbindin-1 mRNA were performed (n=10).
3. Results
3.1 Dysbindin-1 expression
The antisense dysbindin-1 probe resulted in a robust signal in the neuronal fields of the hippocampus; whereas the dysbindin-1 sense strand control produced no discernable signal when hybridized to adjacent sections from the same case in the same experimental run with the same exposure time to film (Fig. 1A & B). Dysbindin-1 mRNA was most abundant in DGg, DGh and CA3 (Fig. 1 and 2). We found 20%–40% reductions in dysbindin-1 mRNA in patients with schizophrenia compared to controls (F = 4.69, df = 1, 18 p = 0.04, Fig. 2C) which were of a similar magnitude as dysbindin-1 protein reductions found previously by Talbot et al. (2004). Dysbindin-1 mRNA levels varied according to cell field (F = 19.78, df = 3, 54, p <0.0001). CA3 had significantly higher dysbindin-1 mRNA levels and CA1 significantly lower dysbindin-1 mRNA levels than all other cell fields studied (all p <0.01). An interaction effect between diagnosis and hippocampal region (F = 3.18, df = 3, 54, p = 0.03) was detected. Dysbindin-1 expression was significantly reduced in the dentate granule cells (DGg, t = −1.90, p = 0.04), dentate polymorph cells (DGh, t = −2.32, p = 0.02), and in CA3 (t = −1.99, p = 0.03), but not in CA1 (t = −1.33, p > 0.05) (Fig. 1C). In all subfields, dysbindin-1 mRNA correlated with pH (r = 0.50 to 0.75, all p < 0.05) and PMI (r range, −0.44 to −0.58, all p< or = 0.05). Dysbindin-1 mRNA did not correlate with age (all r < 0.30, all p > 0.20) or with last, daily or lifetime CPZ (average R = −0.22, all p > 0.29) confirming previous observations showing that dysbindin-1 mRNA was not altered by neuroleptics (Chiba et al. 2006; Weickert et al. 2004).
Figure 1.
Autoradiographic film images of the hybridization of dysbindin-1 riboprobe in human hippocampal formation with the adjacent section from the same brain are shown (panels A &B). Panel A is taken shows the image from the antisense strand and panel B shows the image from the sense strand control.
Figure 2.
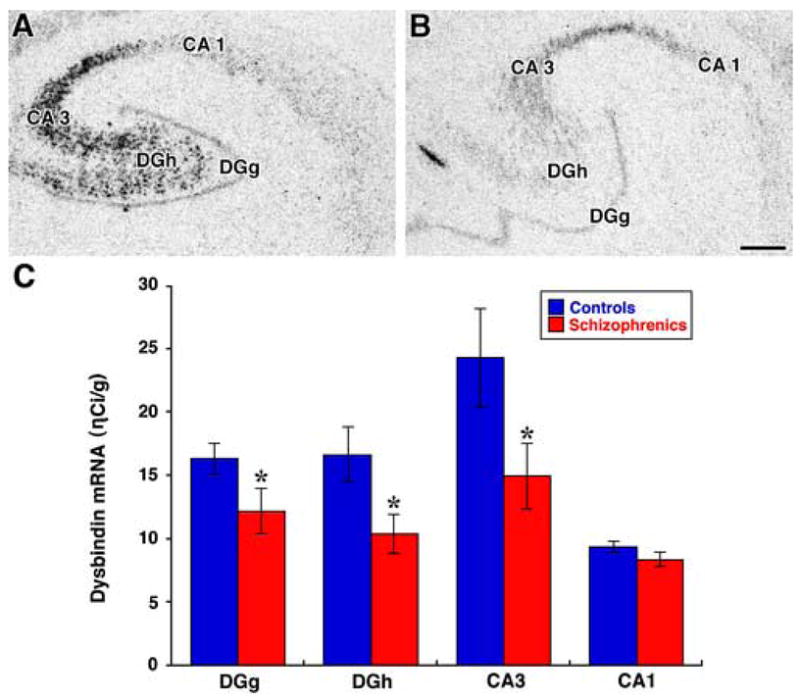
Autoradiographic film images of the hybridization of dysbindin-1 riboprobe in human hippocampal formation are shown (panels A &B). Panel A is taken from a normal control and panel B from a patient with schizophrenia. The subregions at the midbody level of the hippocampal formation demonstrate different expression levels of dysbindin-1 mRNA. Note the intense hybridization signal in the CA3 and DGh subfields. A distinct but fairly thin line of dysbindin-1 mRNA label is found overlying the DGg (panel A & B). The CA1 region has a modest dysbindin-1 hybridization signal. Scale bar = 1 mm. Panel C: Bar graph shows the mean OD reading from dysbindin-1 mRNA signal expressed as ηCi/g radioactivity. Significant differences between controls (blue bars) and schizophrenia patients (red bars) mRNA are found in the DG (mean (SEM) =16.3(1.2), 12.2 (1.8), respectively, p=0.04), DGh (mean (SEM) = 16.6(2.0), 10.4(1.6), respectively; p=0.02), and CA3 (mean (SEM) = 24.3(3.9), 14.9(2.6), respectively; p=0.03) subregions. There were no significant changes between the groups for CA1 mRNA (mean (SEM) = 9.4(0.43), 8.4(0.58), respectively; p>0.05).
3.2 Dysbindin-1 expression and synaptic markers
In the same cohort of patients studied here, reductions in spinophilin and synaptophysin mRNA in the hippocampal formation of schizophrenics was found (Law et al. 2004; Webster et al. 2001). Correlations were tested among synaptic markers and dysbindin-1 as follows: 1) between mRNAs of the presynaptic proteins synaptophysin and dysbindin-1 within the same region; 2) between presynaptic and postsynaptic markers within target subfields; and 3) between dysbindin-1 and the postsynaptic marker (spinophilin) within the same neuronal field. This last analysis was done since recurrent collaterals where neurons synapse back on the originating projection field was identified as abnormal in schizophrenia by Talbot et al. (2004).
Synaptophysin and dysbindin-1 mRNA levels within the DGg (r = 0.68, p = 0.002), DGh (r = 0.82, p <0.0001) and CA3 (r = 0.58, p = 0.02) were positively correlated. The correlation between dysbindin-1 and synaptophysin mRNA levels within DGh remained significant after partial correlation for pH (r = 0.76, p < 0.001), while CA3 showed a trend (r = 0.46, p = 0.08), and the DGg was no longer significant.
Expression of dysbindin-1 in the DGg and of spinophilin in the CA3 subfield were correlated (r = 0.74, p = 0.0006) and showed a trend in that direction after partial correlation for pH (p = 0.06). Positive correlations were found for DGh dysbindin-1 and DGg spinophilin (r = 0.69, p = 0.002) and a trend was found after partial correlation for pH (r = 0.47, p = 0.07). CA3 dysbindin-1 and CA1 spinophilin mRNAs were not correlated (r = −0.31, p = 0.22).
Dysbindin-1 and spinophilin mRNA in CA3 were positively correlated (r = 0.60, p = 0.01), but the relationship was not significant when pH was taken into account. We found positive correlations between dysbindin-1 and spinophilin mRNA within the DGg (r = 0.76, p = 0.0003), which remained significant after partial correlation for pH (p < 0.01). The statistical significance of the correlations above were not affected by partial correlation for PMI.
4. Discussion
We detected a reduction of dysbindin-1 mRNA expression in multiple regions of the hippocampal formation in patients with schizophrenia. This observation extends our findings of reduced dysbindin-1 mRNA in the frontal cortex and midbrain of patients with schizophrenia to another brain region (Weickert et al. 2004). The fact that dysbindin-1 mRNA expression was not decreased in CA1 suggests that expression is not ubiquitously altered in schizophrenia. Talbot et al. (2004) likewise found that decreased dysbindin-1 protein in the hippocampal formation of schizophrenia cases was not accompanied by altered levels of dysbindin-1 in the anterior cingulate cortex of the same cases. It should be noted that Talbot et al (2004) did find significantly reduced dysbindin protein in neuropil areas of CA1, whereas we found no change in dysbindin-1 mRNA in cell bodies areas of CA1. Since the dysbindin-1 protein in the neuropil areas of CA1 could emanate from terminals from CA3 our lack of ability to find dysbindin mRNA reductions in CA1 are not necessarily inconsistent with the findings of Talbot et al. (2004).
Expression of dysbindin-1 in the human hippocampal formation is robust in DGg, DGh and CA3 subfields as compared to CA1. At the cellular level dysbindin-1 mRNA may be higher per neuron in CA3 as compared to CA1 (Talbot et al. 2004). The distinct neuroanatomical distribution of dysbindin-1 mRNA within the hippocampal formation suggests that it may relate to regional synaptic pathology previously reported within DGh and CA3, but not CA1, in schizophrenia patients (Harrison 2004).
Our data shows reduced dysbindin-1 mRNA in DGh, which is known to project to the inner molecular of the dentate gyrus, a region where dysbindin-1 protein is reduced in schizophrenia (Talbot et al. 2004). Thus, we suggest that the mechanism of the reduction in hippocampal dysbindin-1 protein involves reduced dysbindin-1 transcript levels, recognizing that increased degradation of the dysbindin-1 protein may also be involved. Reduction in CA3 dysbindin-1 mRNA may relate to protein reductions in CA3 and/or CA1. Our data supports the presumptive deficiency in recurrent collateral connectivity within the hippocampal subregions of patients with schizophrenia, as we find that dysbindin-1 and spinophilin correlate within hippocampal subregions. While we do not know if this is correlation represents a direct effect of dysbindin-1, recent detailed anatomical studies have localized dysbindin-1 in postsynaptic sites and along microtubules in the hippocampal formation suggesting a unique function for dysbindin in dendrites (Talbot et al. 2006).
We suggest, as others have (Talbot et al. 2004), that alterations in dysbindin-1 may relate to the observed abnormalities in synaptic connectivity found in schizophrenia. Indeed, in cultured cells, antisense dysbindin-1 RNA impacts synaptic proteins and impairs glutamate release (Numakawa et al. 2004). In our study, the correlation between DGh dysbindin-1 mRNA and DGh synaptophysin mRNA is particularly strong. However, this finding is somewhat surprising as an earlier study of dysbindin-1 and synaptic marker proteins, did not find a reduction in synaptophysin protein in terminal fields of the DGh neurons (Talbot et al. 2004). Since the earlier study measured protein, it is possible that the increased synaptophysin protein in the inner molecular layer in the dentate gyrus originates from terminals other than the DGh. However, since non-DGh inputs to the inner molecular layer (DGiml) are minor in normals, we speculate that a significant deviation from the “normal” anatomical connectivity could be present in schizophrenia patients to account for this.
Another possible explanation for the apparent difference between the postmortem studies is that synaptophysin mRNA and protein changes are not detectable in all patient groups due to clinical heterogeneity or limits of quantitative techniques. But unlike the somewhat inconsistent findings on synaptic change reported in postmortem studies of patients with schizophrenia, controlled studies on sandy mice show that absence of DGh dysbindin-1 protein could cause reductions in DGiml synaptophysin (Talbot et al 2005) Furthermore, the loss of dysbindin-1 in homozygous sandy mice is associated with reductions in synaptophysin detected via western blotting of whole brain (Talbot et al. 2005). These results would favor the interpretation that the reductions in dysbindin-1 mRNA may more directly cause reductions in synaptophysin mRNA in the same neuron. In contrast, in patients with schizophrenia, who do not entirely lack dysbindin-1 protein as do the sandy mice, it is possible that the dysbindin mRNA reductions are downstream of a more generalized synaptic pathology.
In summary, we report reductions of dysbindin-1 mRNA in specific regions of the schizophrenic hippocampal formation, particularly those implicated in the synaptic pathology. Our expression results are concordant with reported decreased dysbindin-1 protein levels (Talbot et al. 2004). We provide further evidence that the control of dysbindin-1 gene transcription and/or dysbindin-1 transcript stability is altered in brains of patients with schizophrenia. Moreover, reduced dysbindin-1 may contribute to the changes in synaptic connectivity and glutamate signaling found in schizophrenia. Altered expression of schizophrenia susceptibility genes in patients might be argued to be predominantly upstream, or primary in nature (Bray et al. 2003; Bray et al. 2005; Weickert et al. 2004). To test this hypothesis quantitatively for dysbindin-1, a better understanding of how risk haplotypes influence transcription, splicing, and translation is required that includes a comparison of controls to patients with schizophrenia, bipolar disorder, and other psychiatric conditions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bray NJ, Buckland PR, Owen MJ, O’Donovan MC. Cis-acting variation in the expression of a high proportion of gene in human brain. Human Genet. 2003;113:149–153. doi: 10.1007/s00439-003-0956-y. [DOI] [PubMed] [Google Scholar]
- Bray NJ, Preece A, Williams NM, Moskvina V, Buckland PR, Owen MJ, O’Donovan MC. Haplotypes at the dystrobrevin binding protein 1 (DTNBP1) gene locus mediate risk for schizophrenia through reduced DTNBP1 expression. Hum Mol Genet. 2005;14:1947–54. doi: 10.1093/hmg/ddi199. [DOI] [PubMed] [Google Scholar]
- Chiba S, Hashimoto R, Hattori S, Yohda M, Lipska B, Weinberger DR, Kunugi H. Effect of antipsychotic drugs on DISC1 and dysbindin expression in mouse frontal cortex and hippocampus. J Neural Transm. 2006;113:1337–46. doi: 10.1007/s00702-005-0414-1. [DOI] [PubMed] [Google Scholar]
- Eastwood SL, Burnet PW, McDonald B, Clinton J, Harrison PJ. Synaptophysin gene expression in human brain: a quantitative in situ hybridization and immunocytochemical study. Neuroscience. 1994;59:881–92. doi: 10.1016/0306-4522(94)90292-5. [DOI] [PubMed] [Google Scholar]
- Harrison PJ. The hippocampus in schizophrenia: a review of the neuropathological evidence and its pathophysiological implications. Psychopharmacology (Berl) 2004;174:151–62. doi: 10.1007/s00213-003-1761-y. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Eastwood SL. Neuropathological studies of synaptic connectivity in the hippocampal formation in schizophrenia. Hippocampus. 2001;11:508–19. doi: 10.1002/hipo.1067. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Law AJ, Eastwood SL. Glutamate receptors and transporters in the hippocampus in schizophrenia. Ann N Y Acad Sci. 2003;1003:94–101. doi: 10.1196/annals.1300.006. [DOI] [PubMed] [Google Scholar]
- Law AJ, Weickert CS, Hyde TM, Kleinman JE, Harrison PJ. Reduced spinophilin but not microtubule-associated protein 2 expression in the hippocampal formation in schizophrenia and mood disorders: molecular evidence for a pathology of dendritic spines. Am J Psychiatry. 2004;161:1848–55. doi: 10.1176/ajp.161.10.1848. [DOI] [PubMed] [Google Scholar]
- Numakawa T, Yagasaki Y, Ishimoto T, Okada T, Suzuki T, Iwata N, Ozaki N, Taguchi T, Tatsumi M, Kamijima K, Straub RE, Weinberger DR, Kunugi H, Hashimoto R. Evidence of novel neuronal functions of dysbindin, a susceptibility gene for schizophrenia. Hum Mol Genet. 2004;13:2699–708. doi: 10.1093/hmg/ddh280. [DOI] [PubMed] [Google Scholar]
- Riley B, Kendler KS. Molecular genetic studies of schizophrenia. Eur J Hum Genet. 2006;14:669–80. doi: 10.1038/sj.ejhg.5201571. [DOI] [PubMed] [Google Scholar]
- Schwab SG, Knapp M, Mondabon S, Hallmayer J, Borrmann-Hassenbach M, Albus M, Lerer B, Rietschel M, Trixler M, Maier W, Wildenauer DB. Support for association of schizophrenia with genetic variation in the 6p22.3 gene, dysbindin, in sib-pair families with linkage and in an additional sample of triad families. Am J Hum Genet. 2003;72:185–90. doi: 10.1086/345463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub RE, Jiang Y, MacLean CJ, Ma Y, Webb BT, Myakishev MV, Harris-Kerr C, Wormley B, Sadek H, Kadambi B, Cesare AJ, Gibberman A, Wang X, O’Neill FA, Walsh D, Kendler KS. Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet. 2002;71:337–48. doi: 10.1086/341750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot K, Cho DS, Ong WY, Benson MA, Han LY, Kazi HA, Kamins J, Hahn CG, Blake DJ, Arnold SE. Dysbindin-1 is a synaptic and microtubular protein that binds brain snapin. Hum Mol Genet. 2006;15:3041–54. doi: 10.1093/hmg/ddl246. [DOI] [PubMed] [Google Scholar]
- Talbot K, Cho D, Ong W, Kazi HA, Siegel SJ, Blake DJ, Novak EK, Swank RT, Arnold SE. Sandy (Sdy) mice display both a loss of dysbindin and elevated vesicular glutamate transporter-1 (VGlut-1) in the same synaptic fields of the hippocampal formation 2005 [Google Scholar]
- Talbot K, Eidem WL, Tinsley CL, Benson MA, Thompson EW, Smith RJ, Hahn CG, Siegel SJ, Trojanowski JQ, Gur RE, Blake DJ, Arnold SE. Dysbindin-1 is reduced in intrinsic, glutamatergic terminals of the hippocampal formation in schizophrenia. J Clin Invest. 2004;113:1353–63. doi: 10.1172/JCI20425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MJ, Shannon Weickert C, Herman MM, Hyde TM, Kleinman JE. Synaptophysin and GAP-43 mRNA levels in the hippocampus of subjects with schizophrenia. Schizophr Res. 2001;49:89–98. doi: 10.1016/s0920-9964(00)00052-9. [DOI] [PubMed] [Google Scholar]
- Weickert CS, Straub RE, McClintock BW, Matsumoto M, Hashimoto R, Hyde TM, Herman MM, Weinberger DR, Kleinman JE. Human dysbindin (DTNBP1) gene expression in normal brain and in schizophrenic prefrontal cortex and midbrain. Arch Gen Psychiatry. 2004;61:544–55. doi: 10.1001/archpsyc.61.6.544. [DOI] [PubMed] [Google Scholar]