

Summary
In order to quantitatively distinguish between highly similar RNA sequences, specific primers or probes must be designed. Unfortunately, consistent and reliable results are not always obtained with conventional techniques. This study uses reverse transcription-PCR coupled with direct terminator sequencing to economically and efficiently distinguish between sequence types in pooled samples while providing accurate relative quantification. As an example, the method is applied to measure template concentration of two Barley yellow dwarf virus (BYDV; family Luteoviridae) species in doubly infected wheat plants. A PERL script (polySNP) was developed that uses PHRED to automatically extract relative peak areas and heights from sequencing chromatograms at polymorphic sites. Peak measurements from experimental samples were compared to a standard curve generated by mixing in vitro transcribed RNA from BYDV-PAV and PAS templates in several ratios (ranging from 1:9 to 9:1 PAV:PAS) prior to RT-PCR amplification and sequencing. The relative amount of RNA template added to a sample was regressed onto the proportion of the chromatogram peak height or area corresponding to one virus species. The function of the best fit line was used to calculate template frequency in the experimental samples.
Keywords: single nucleotide polymorphisms, quantitative nucleotide sequencing, virus competition, Barley yellow dwarf virus
1. Introduction
The virus population in a host community (Kong et al., 2000, Kurath et al., 1993, Tsompana et al., 2005) and within an individual host (Schneider & Roossinck, 2000) can exhibit a high degree of genetic diversity. Consequently, for a given genotype other genetic variants will be part of the selective environment within a host. Theoretical models have demonstrated that competitive interactions between pathogen genotypes have important consequences for within-host pathogen population dynamics, pathogen transmission between hosts, and the evolution of pathogen virulence (Levin & Pimentel, 1981, Nowak & May, 1994). Despite their evolutionary importance, empirical studies of virus genetic variants in mixed infections are still quite rare due, in part, to the lack of an effective method for quantifying the population size of closely related virus genotypes in a single host. This study utilizes a new method called quantitative sequencing to measure the population size of two species of Barley yellow dwarf virus (BYDV; family Luteoviridae) in mixed infections. BYDV has a positive sense ssRNA genome of approximately 5.6 kb (Miller et al., 2002). It infects many wild and cultivated grass species and is an economically important pathogen of grain crops worldwide (D’arcy, 1995). The PAV and PAS species analyzed in this study are ecologically similar, sharing the same aphid vectors (Rhopalosiphum padi and Sitobion avenae) (Chay et al., 1996) and host species (many members of the family Poacea). PAV and PAS have approximately 90% and 78% nucleotide sequence identity in their capsid protein (Bisnieks et al., 2004) and polymerase genes (Hall, 2006), respectively. The effect of one virus species on the population growth of the other during mixed infections has not yet been reported, but plants infected with both species have been identified in natural plant populations in France (Mastari et al., 1998) and New York State (Hall, 2007).
For genetic variants that differ at few nucleotide sites it may be difficult or impossible to develop genotype specific primers, probes, or antibodies capable of differentiating between them in a mixed infection. RNase protection and restriction fragment analyses have been employed to discriminate between virus types that have small sequence differences (Kurath et al., 1993, Tenllado et al., 1997), but these techniques are generally not implemented in a quantitative manner. As such, they can only determine if the end result of competition is coexistence or complete exclusion. Measurement of the relative area or height of sequencing chromatogram peaks at polymorphic sites can result in quantitative data if a standard curve is constructed by mixing known amounts of viral template and performing PCR amplification followed by direct sequencing (Wilkening et al., 2005). Direct nucleotide sequencing has two major advantages over the previously listed methods. First, direct sequencing can differentiate templates that differ by as little as one nucleotide. Secondly, direct sequencing is less costly and requires less time to develop and optimize than antibody or other quantitative PCR based techniques.
Quantitative sequencing has previously been used to estimate allele frequency and mutation frequency in pooled DNA samples (Amos et al., 2000, Wilkening et al., 2005). The data are quantitative because the height or area of the peaks in a sequencing chromatogram at polymorphic nucleotide positions are proportional to the corresponding template concentration at the start of the sequencing reaction. Quantitative sequencing has never before been applied to RNA templates nor has it been used to measure the relative concentration of virus template within a host as is done in the present study. To facilitate the analysis of experimental samples we have developed a PERL script called polySNP that uses PHRED to automatically extract peak area and height data from multiple SNPs in sequence chromatograms. The objective of the present study is to evaluate the accuracy of quantitative sequencing when applied to mixtures of RNA templates. As an example, quantitative sequencing is used to determine if the presence of one BYDV species in the host inhibits the establishment of another species and if varying the time interval between inoculations affects the stability of the competitive hierarchy.
2. Materials and methods
2.1. Inoculation of experimental plants
Wheat (Triticum aestium) plants were grown from seed in six inch pots and randomly assigned to one of seven inoculation treatments: single inoculation with PAV, single inoculation with PAS, simultaneous inoculation with PAV and PAS, PAV challenged with PAS after a three day delay, PAV challenged with PAS after a fifteen day delay, PAS challenged with PAV after a three day delay, and PAS challenged with PAV after a fifteen day delay. To inoculate plants R. padi from laboratory maintained disease-free colonies were fed for 48 hours on detached leaves infected with the appropriate virus isolate. Aphids were then transferred to healthy plants and allowed an inoculation access period of five days. Inoculation with the protecting strain was carried out when plants were at the two-leaf stage. PAV and PAS isolates used in this study were collected from agricultural fields in central New York State. Isolate PAS-129 was obtained in 1992 from migrating alate R. padi alighting on winter wheat (Chay et al., 1996). PAV isolate Fa2k298 was obtained in 1998 from apterous R. padi collected in oat fields. Virus isolates were maintained in oat (Avena sativa) plants in the greenhouse since the initial isolation.
2.2. Nucleic acid extraction, reverse transcription-PCR and sequencing
At 33 days post-inoculation (DPI) with the protecting strain leaves were harvested from at least three plants per inoculation treatment. Equal amounts of leaf tissue from plants singly inoculated with PAV or PAS (100 mg each) were combined into one sample prior to the nucleic acid extraction. For all other treatments the total nucleic acid extraction was performed on 200 mg of leaf tissue from a single plant. Leaf samples were homogenized with a glass rod in 1.5 mL microfuge tubes in 900 μL extraction buffer (7 M urea, 0.3 M NaCl, 50 mM tris pH 8.0, 20 mM EDTA and 1% SDS). The homogenate was incubated at 65° C for 10 minutes then extracted with 650 μl phenol:chloroform:isoamyl alcohol (25:24:1). Nucleic acids were precipitated by adding 700 μL of supernatant to a 700 μL solution containing isopropanol and ammonium acetate such that the final concentration of the solution was 0.7 M ammonium acetate pH 5.2. Single-step RT-PCR was carried out in a 50 μL reaction volume containing 25 mM tris pH 8.8, 10 mM KCl, 10 mM dTT, 10 mM (NH4)2SO4, 2.5 mM MgSO4, 1 μM each forward (5′-AGAGGCCACAGAATGTCCGG-3′) and reverse (5′- GTTCAGCTTCAACACCCAGC-3′) oligonucleotide primers, 200 μM dNTPs, 30 units RNase inhibitor, 50 units SuperScript II RNase H-reverse transcriptase and 5 units Taq polymerase (all enzymes Invitrogen, Carlsbad, CA). This primer combination amplifies a 106-nucleotide region of the viral RNA-dependent RNA-polymerase. Both virus species have exactly the same sequence in the primer binding region. Genbank accession numbers for Fa2k298 and PAS-129 sequences containing this gene region are DQ286379 and DQ286383, respectively. Thermocycling conditions were: reverse transcription 1 cycle of 40 min at 42° C, inactivation of reverse transcriptase and pre-denaturation 1 cycle of 2 min at 95° C, DNA amplification 35 cycles of 30 s at 94° C, 30 s at 60° C, 1 min at 72° C, and a final extension of 10 min at 72° C. RT-PCR products were purified using the Invitrogen PCR clean-up kit following the protocol prescribed by the manufacturer and then submitted to the Cornell University Bioresource Center for direct sequencing using an ABI 3730 sequencer with Big Dye Terminator chemistry and AmpliTaq-FS DNA polymerase. Sequencing was carried out with the forward amplification primer.
2.3. In vitro transcription of PAV and PAS polymerase gene template
PAS and PAV isolates were RT-PCR amplified with primers and under conditions described above. The reverse primer was modified by the addition of a T7 site at the 5’ end (5′-TAATACGACTCACTATAGGGTTCAGC-3′). Unincorporated primers and dNTPs were separated from the amplicon using the QIAquick PCR Purification kit (Qiagen Inc., Valencia, CA.) following the manufacturer’s instructions. Approximately 250 ng of purified DNA amplicon was used as a template for RNA synthesis using the MAXIscript in vitro RNA transcription kit (Ambion Inc., Austin, TX) and the supplied T7 polymerase. After 1 hour of transcription at 37° C, the DNA template was destroyed by the addition of DNase I (Roche Applied Science, Mannheim, Germany) followed by a 15 minute incubation at 37° C. The DNase was deactivated by a 10 minute incubation at 75° C (with 2.2 mM EDTA to protect the RNA). The transcribed RNA was further purified by ethanol precipitation in the presence of 45 mM ammonium acetate. The Nanodrop (Wilmington, DE) spectrophotometer was used to quantify the yield of RNA.
2.4. Construction of standard curves and analysis of experimental plants
In order to determine the relative concentration of PAV and PAS in experimental plants a standard curve was developed using in vitro transcribed RNA. PAV and PAS templates were mixed in eight ratios (from 1:9 to 9:1 PAV:PAS). Mixing was replicated three times for each ratio, after which samples were RT-PCR amplified (according to the protocol above) and submitted for direct sequencing. Within the 106-nucleotide amplicon nine SNPs differentiate virus species. Five sites that were consistently resolved in the chromatograms were chosen for further analysis. The area and height of the peaks for each of these SNPs was extracted from the sequence chromatograms by the PERL script polySNP (available at http://staging.nybg.org/polySNP.html). To extract data from the chromatograms, polySNP first produced .seq, .phd, and .poly files by running PHRED version 0.020425.c (Ewing et al., 1998) on each chromatogram file with a “-trim cutoff” value of 0.10. The pre-aligned reference sequences (DQ286379 and DQ286383) were aligned to the .seq file (a FASTA format file containing the called bases) using the “-profile” option of MUSCLE version 3.6. (Edgar, 2004). The alignment was used to locate dimorphic SNP positions in the .phd file (a text file containing the called bases, a quality assessment for each base, and location of the peak in the chromatogram) which was in turn used to find the corresponding line in the .poly file (a text file containing peak areas and heights for each chromatogram position). After confirming that the peaks correspond to the known variation in the SNP, the peak areas and heights were extracted and the relative peak areas or heights were calculated for each virus species. The relative proportions (PAV / (PAV + PAS)) were used instead of the ratio of the relative peak heights (PAV/PAS), as has been done in previous studies (Wilkening et al., 2005), because this study was interested in relative viral transcript concentration rather than the relative magnitude of separation between concentrations.
For each SNP the amount of virus template added to the RT-PCR reaction was regressed onto the height or area of the PAV peak as a proportion of the total peak height or area. This procedure minimized the error in our estimate of virus concentration (Hilsel and Hirsch, 2005). Given the shape of the plotted data, Minitab14 (Minitab Inc., State College, PA) was used to fit a linear, exponential, quadratic or cubic model. Following the same procedures described above, standard curves were also generated from mixtures of RT-PCR products amplified from plants singly infected with PAV and PAS.
To test their accuracy, standard curves were used to estimate relative virus concentration in a group of ten samples prepared by mixing in vitro transcribed PAS and PAV RNA. The proportion of PAV added to the sample was read from the first column of a random number table. Chi-square goodness-of-fit tests were used to test the null hypothesis: the quantity of PAV added to the reaction did not significantly differ from the quantity calculated from the standard curve for a given polymorphic site across the group of random samples. Paired t-tests were used to test the null hypothesis that for a given SNP there was no significant difference in virus concentration as calculated from peak area or height standard curves. A single factor analysis of variance was used to compare relative PAV or PAS concentration between inoculation treatments. Each of the five SNPs were treated as independent measures of virus concentration. Individual comparisons between treatments were carried out with Tukey’s procedure (α = 0.05). All statistical analyses were performed with Minitab 14.
3. Results
3.1. Standard curves
Relative concentration of PAV and PAS in doubly infected plants was determined by sequencing a portion of the virus coat protein gene using primers that can amplify both virus types. Chromatogram peak height or area measurements at five SNPs were compared to standard curves created by mixing known quantities of in vitro transcribed virus RNA in ratios from 1:9 to 9:1 PAV:PAS prior to RT-PCR and sequencing or RT-PCR amplified virus template prior to sequencing. In total, twenty standard curve plots were generated and regression lines (Table 1) were fit to each plot. Each of these was used to predict PAV and PAS template concentration in samples where a known quantity of viral RNA had been added. Virus concentration calculated from RNA and DNA standard curves did not significantly differ from the expected values (chi-square, α = 0.05; Table 2), except when virus concentration was estimated from the RNA standard curve for peak height at site 2088. Among those curves that where able to accurately predict the expected values, there was no significant difference in concentration calculated using peak area or height standard curves (P > 0.05 for all comparisons). Examples of standard curves generated from peak area measurements at sites 2074 and 2088 (full-length genome sequence of PAV-Aus used as reference; Genbank accession number M21347) are shown in Fig. 1. In general, peak area standard curves yielded the lowest sum of the squared differences across all SNPs (data not shown); therefore, these curves were used to calculate virus concentration in the experimental samples.
Table 1.
The best-fit line when the relative amount of PAV template in the standard sample was regressed onto PAV as a proportion of the total peak area or height [PAV / (PAV + PAS)] at five nucleotide sites that differentiate PAV and PAS.
| Sitea | templateb | peak
measurement |
Function |
|---|---|---|---|
| 2074 | area | - 0.097 + 15.95 area - 6.73 area2 | |
| RNA | height | 0.13 + 25.53 height - 31.9 height2 + 15.5 height3 | |
| area | - 0.006 + 15.24 area - 5.75 area2 | ||
| DNA | height | - 0.27 + 23.6 height - 29.3 height2 + 16.22 height3 | |
| 2080 | area | - 0.33 + 9.63 area | |
| RNA | height | - 0.021 + 9.6 height | |
| area | - 0.46 + 15.26 area - 5.96 area2 | ||
| DNA | height | - 1.14 + 14 height - 3.23 height2 | |
| 2083 | area | 1.6 + 8.05 area | |
| RNA | height | 0.49 + 9.25 height | |
| area | - 0.19 + 9.049 area | ||
| DNA | height | - 0.82 + 10.83 height | |
| 2086 | area | 5.92 - 20.92 area + 23.96 area2 | |
| RNA | height | 9.34 height 2.136 | |
| area | - 1.45 + 19.45 height - 46.86 area2 + 37.7 area3 | ||
| DNA | height | 3.96 - 17.24 height + 22.06 height2 | |
| 2088 | area | 1.95 + 11 area − 3.6 area2 | |
| RNA | height | - 3.1 + 37.9 height - 47.08 height2 + 22.12 height3 | |
| area | 1.95 + 11 area - 3.6 area2 | ||
| DNA | height | - 0.12 + 41.8 height - 77.32 height2 + 47 height3 |
complete genome of PAV-Aus (M21347) used as the reference sequence;
standard curves were created by mixing in vitro transcribed viral RNA or RT-PCR products in ratios from (1:9 to 9:1 PAV:PAS).
Table 2.
Mean difference between the expected and calculated PAV concentration in a group of ten PAV/PAS RNA template mixtures. The quantity of virus added to the sample was chosen randomly. Relative PAV concentration was calculated from standard curves created by mixing in vitro transcribed viral RNA or RT-PCR products in ratios from 1:9 to 9:1 (PAV:PAS).
| area | Height | |||
|---|---|---|---|---|
| Sitea | RNA | DNA | RNA | DNA |
| 2074 | 0.03 | 0.04 | 0.04 | 0.29 |
| 2080 | 0.64 | 0.07 | 0.44 | 0.44 |
| 2083 | 0.77 | 0.96 | 0.27 | 0.9 |
| 2086 | 0.6 | 0.1 | 1.1 | 0.24 |
| 2088 | 1.5 | 1.5 | 5b | 0.62 |
complete genome of PAV-Aus used as the reference sequence;
The null hypothesis of no difference between expected and calculated PAV concentration was rejected in a Chi-square goodness-of-fit test (α = 0.05). The chi-square failed to reject the null for all other calculated values.
Figure 1.

Standard curves for peak area measurements at polymorphic sites 2074 (A, B) and 2088 (C, D; complete genome of PAV-Aus used as reference sequence). Standard curves were created by mixing in vitro transcribed viral RNA (A, C) or RT-PCR products (B, D) in ratios from 1:9 to 9:1 (PAV:PAS).
3.2. Competitive interactions between PAV and PAS in mixed infections
For the virus competition study, wheat (Triticum aestium) plants were assigned to one of seven inoculation treatments differing in single or double infections, the order of inoculation, and the delay before the challenge inoculation. The relative proportion of PAV significantly differed between inoculation treatments (F = 15.73, P < 0.01; Fig. 2). The proportion of PAV to PAS in samples where leaf tissue from singly infected plants were combined prior to the nucleic acid extraction (‘one:one’) was less than that of plants simultaneously infected (P = 0.05). If there was no competitive interaction between species, one would expect to see the same relative concentration of PAV in both treatments. An inoculation delay of 15 days before challenge with PAS allowed significantly greater population growth of PAV when compared to other treatments. The relative proportion of PAS significantly differed between inoculation treatments (F = 11.66, P = 0.01) when PAS was inoculated to host first (Fig. 3). PAS concentration was greater in singly infected plants, than plants simultaneously inoculated with both virus species. A three-day delay before challenge with PAV did not allow for greater population growth of PAS. If, however, there was a fifteen-day delay before challenge with PAV, PAS concentration was significantly greater than in other treatments.
Figure 2.
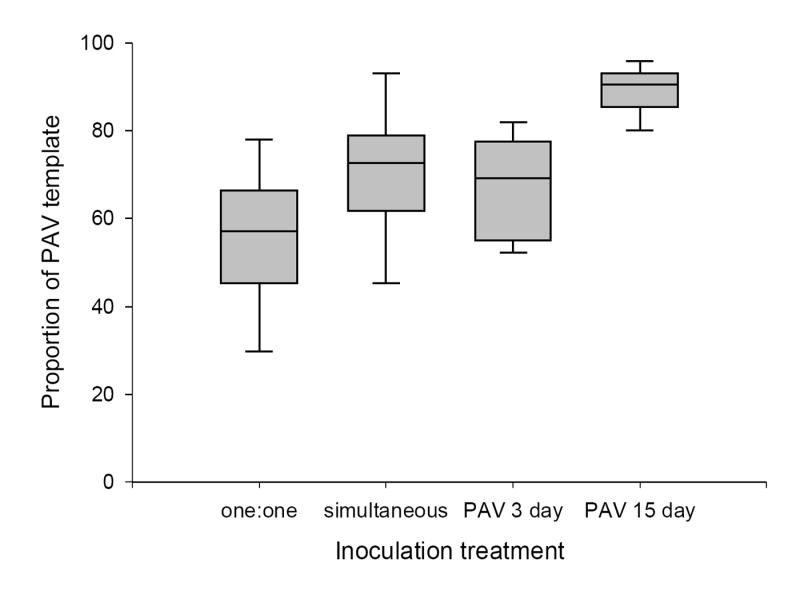
Proportion of PAV template in plants harvested 33 days post primary inoculation with PAV. Inoculation treatments were: equal amounts of leaf tissue from plants singly infected with PAV and PAS isolates paired prior to nucleic acid extraction (one:one), simultaneous inoculation with PAV and PAS, PAV challenged with PAS three days later (PAV 3 day) and PAV challenged with PAS fifteen days later (PAV 15 day). Box plots show the median, 5th, 25th, 75th and 95th percentiles.
Figure 3.
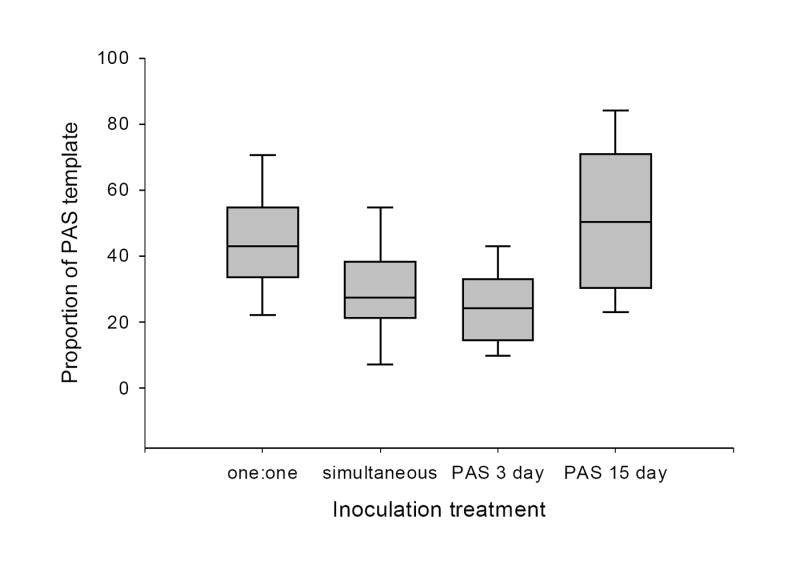
Proportion of PAS template in plants harvested 33 days post primary inoculation with PAS. Inoculation treatments were: equal amounts of leaf tissue from plants singly infected with PAV and PAS paired prior to nucleic acid extraction (one:one), simultaneous inoculation with PAV and PAS, PAS challenged with PAV three days later (PAS 3 day) and PAS challenged with PAV 15 days later (PAS 15 day). Box plots show the median, 5th, 25th, 75th and 95th percentiles.
4. Discussion
Standard curves generated from in vitro transcribed RNA and RT-PCR products were able to accurately estimate template concentration using peak area or peak height from sequence chromatograms. Standard curves derived from RT-PCR products performed as well as the RNA standard curves, thus in vitro transcription of the RNA template appears to be unnecessary for conducting a quantitative sequencing analysis. This suggests that the reverse-transcription step is completely quantitative. For SNP 2088 there is a strong incorporation bias for the base in the PAS template (T versus A). The same incorporation bias was observed in the standard curves constructed from RT-PCR products. Thus, the sequencing reaction or the normalizing function of PHRED, not the reverse transcription process preferentially selected the T in this case.
In applying quantitative sequencing to plants singly and doubly infected with PAS and PAV, it was found that the ratio of PAV to PAS template was 55:45 in singly infected plants at 33 days post-inoculation (DPI). However, when both species are inoculated to plants at the same time PAV dominates the virus population (70:30 PAV to PAS). When either virus was the primary infection, a three-day delay before challenge with the other species did not change the outcome of competition, i.e. the relative template ratio was 70:30 PAV to PAS in both treatments. It appears that three days was not enough establishment time to significantly enhance or ameliorate the negative effects of competition on PAS. A similar result was found when BYDV-MAV and PAV were inoculated to the same host (Wen et al., 1991). A three day interval between the first (MAV) and second (PAV) inoculation inhibits population growth of the challenge virus at early infection stages, but by 30 DPI PAV dominates the virus population. If closely related variants of MAV are inoculated to the same host, a three day interval before challenge inoculation lead to almost complete exclusion of the challenging strain. In the present study, a fifteen-day inoculation interval led to a stronger inhibition in accumulation of the challenge virus. This effect was more pronounced for PAV which is clearly the stronger competitor.
A limitation of the quantitative sequencing method in viral competition studies is that total virus population size in singly and doubly infected plants must be independently measured in order to understand the mechanism of competition. For example, the dominance of PAV regardless of the order of inoculation or the starting concentrations of each of the competitors does not necessarily indicate that there was direct resource competition between PAV and PAS. It may be that PAV exploits some part of the host cell machinery that is unused by PAS or the presence of PAS complements the growth of PAV but PAV does not have a reciprocal effect on PAS. Thus, the population size of PAV may be greater than that of PAS in relative terms but it has no direct negative impact on the accumulation of PAS. It is also possible that the population size of one or both viruses is reduced by a host defense mechanism, such as virus-induced gene silencing, which would spuriously appear as competition between viruses. Measuring total virus population size (PAV + PAS) in singly and doubly infected plants would allow one to determine if there is synergy, mutual inhibition or exploitation competition in mixed infections.
A previous study found that genetic variants of satellite tobacco mosaic virus that differ at only five nucleotide sites across their entire genome can restrict host colonization and replication of the other variant depending on which is inoculated to the host first (Kurath & Dodds, 1994). One can speculate that in every replicating virus population there are competitive interactions between the progeny genomes produced early and those produced later in infection. As a result competition might enhance the effect of stochastic processes (such as transmission bottlenecks and founder effects) on genetic diversity and structure in the virus population. For the reasons described above none of the methods most commonly used to measure virus concentration in hosts could be applied to test such a hypothesis. As shown in the present study, quantitative sequencing can accurately and efficiently address questions related to the within-host dynamics of closely related pathogen genotypes.
Acknowledgments
I thank Alison Power, Micheal Milgroom, and John Losey for reviewing previous drafts of this manuscript. Research support was provided by the National Institutes of Health Ruth L. Kirschstein individual research fellowship. All laboratory work was performed in the Evolutionary Genetics Core Facility in the Department of Ecology and Evolutionary Biology at Cornell University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Gerod S. Hall, New York State Department of Health/Arbovirus Laboratory, 5668 State Farm Rd., Slingerland, NY 12159, Office phone 518-869-4592, Fax 518-869-4530, gsh01@health.state.ny.us
Damon P. Little, The New York Botanical Garden, Bronx, New York 10458, Office phone 718-817-8130, Fax 718-817-8101, dlittle@nybg.org
References
- Amos CI, Frazier ML, Wang W. DNA pooling in mutation detection with reference to sequence analysis. Am J Hum Gen. 2000;66:1689–1692. doi: 10.1086/302894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisnieks M, Kvarnheden A, Sigvald R. Molecular diversity of the coat protein-encoding region of Barley yellow dwarf virus-PAV and Barley yellow dwarf virus-MAV from Latvia and Sweden. Arch Virol. 2004;149:843–853. doi: 10.1007/s00705-003-0242-2. [DOI] [PubMed] [Google Scholar]
- Chay C, Smith DM, Vaughan R, Gray SM. Diversity among isolates within the PAV serotype of barley yellow dwarf virus. Phytopathology. 1996;86:370–377. [Google Scholar]
- D’arcy CJ. Symptomology and host range of barley yellow dwarf. In: D’arcy CJ, Burnett PA, editors. Barley Yellow Dwarf 40 Years of Progress. The American Phyotopathological Society; St. Paul, MN: 1995. pp. 9–28. [Google Scholar]
- Hall G. Dissertation. Cornell University; Ithaca, NY: 2007. Vector dynamics in relation to host phenology will influence the disease prevalence and population structure of Barley yellow dwarf virus; pp. 46–65. [Google Scholar]
- Helsel DR, Hirsch RM. Statistical methods in water resources. US Geological Survey. 2005 URL: http://pubs.water.usgs.gov/twri4a3.
- Edgar RC. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendi M, Green P. Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Kong P, Rubio L, Polek M, Falk BW. Population structure and genetic diversity within California Citrus tristeza virus (CTV) isolates. Virus Gen. 2000;21:139–145. doi: 10.1023/a:1008198311398. [DOI] [PubMed] [Google Scholar]
- Kurath G, Dodds JA. Satellite tobacco mosaic virus sequence variants with only five nucleotide differences can interfere with each other in a cross protection-like phenomenon in plants. Virology. 1994;202:1065–1069. doi: 10.1006/viro.1994.1441. [DOI] [PubMed] [Google Scholar]
- Kurath G, Heick JA, Dodds JA. Rnase Protection Analyses Show High Genetic Diversity among Field Isolates of Satellite Tobacco Mosaic-Virus. Virology. 1993;194:414–418. doi: 10.1006/viro.1993.1278. [DOI] [PubMed] [Google Scholar]
- Levin S, Pimentel D. Selection of intermediate rates of increase in parasite-host systems. Amer Nat. 1981;117:308–315. [Google Scholar]
- Mastari J, Lapierre H, Dessens JT. Asymmetrical distribution of barley yellow dwarf virus PAV variants between host plant species. Phytopathology. 1998;88:818–821. doi: 10.1094/PHYTO.1998.88.8.818. [DOI] [PubMed] [Google Scholar]
- Miller WA, Liu S, Beckett R. Barley yellow dwarf virus: Luteoviridae or Tombusviridae? Mol Plant Pathol. 2002;3:177–183. doi: 10.1046/j.1364-3703.2002.00112.x. [DOI] [PubMed] [Google Scholar]
- Nowak MA, May RM. Superinfection and the Evolution of Parasite Virulence. Proc R Soc Lond [Biol] 1994;255:81–89. doi: 10.1098/rspb.1994.0012. [DOI] [PubMed] [Google Scholar]
- Schneider WL, Roossinck MJ. Evolutionarily related Sindbis-like plant viruses maintain different levels of population diversity in a common host. J Virol. 2000;74:3130–3134. doi: 10.1128/jvi.74.7.3130-3134.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenllado F, Garcia-Luque I, Serra MT, Diaz-Ruiz JR. Pepper resistance-breaking tobamoviruses: Can they co-exist in single pepper plants? Eur J Plant Pathol. 1997;103:235–243. [Google Scholar]
- Tsompana M, Abad J, Purugganan M, Moyer JW. The molecular population genetics of Tomato spotted wilt virus (TSWV) genome. Mol Ecol. 2005;14:53–66. doi: 10.1111/j.1365-294X.2004.02392.x. [DOI] [PubMed] [Google Scholar]
- Wilkening S, Hemminki K, Thirumaran J, Bonn S, Forsti A, Rajiv K. Determination of allele frequency in pooled DNA: Comparison of three PCR-based methods. Biotechniques. 2005;39:853–857. doi: 10.2144/000112027. [DOI] [PubMed] [Google Scholar]