

Abstract
The intra-hippocampal administration of interleukin-1β (IL-1β) as well as the induction of elevated but physiological levels of IL-1β within the hippocampus interferes with the formation of long-term memory. There is evidence suggesting that the induction of prostaglandin (PG) formation by IL-1β is involved in impairments in working and spatial memory following IL-1β. The present experiments extend these findings by showing that PGs are responsible for memory deficits in contextual fear conditioning that occur following IL-1β injection into the dorsal hippocampus. Cyclooxygenase (COX) inhibition blocked the disruption in contextual fear conditioning produced by IL-1β and COX inhibition alone also disrupted contextual memory, suggesting an inverted U-shaped relationship between PG levels and memory. In addition to demonstrating the necessity of PGs in IL-1β mediated memory deficits, we also show that PGs injected directly into the dorsal hippocampus are sufficient to impair context memory and significantly reduce post-conditioning levels of BDNF within the hippocampus, suggesting a possible mechanism for the memory-impairing effects of PGs.
Keywords: memory, brain derived neurotrophic factor, cyclooxygenase, EP receptor
The pro-inflammatory cytokine interleukin-1β (IL-1β) exerts powerful central and peripheral actions. Recently, there has been considerable interest in its influence within the brain on learning and memory. This interest initially derived in part from findings that Alzheimer’s disease and AIDS-related dementia are associated with elevated brain levels of IL-1β (Griffen et al., 1989; Lombardi et al., 1999). More direct evidence implicating IL-1β in learning and memory has come from animal studies. Manipulations that increase levels of IL-1β within the hippocampus have been shown to impair memory formation. These manipulations include the injection of the gram-negative bacterial component lipopolysaccharide (LPS; Pugh et al. 1998), social isolation stress (Pugh et al., 1999), and aging (Gemma et al., 2005). The blockade of IL-1 receptors and/or the inhibition of IL-1 synthesis prevents these memory impairments, supporting the conclusion that they are mediated by IL-1β (Pugh et al. 1998; Pugh et al., 1999; Gemma et al., 2005). Moreover, direct injection of IL-1β into the dorsal hippocampus impairs memory (Barrientos et al., 2002). Consistent with these data, IL-1β potently inhibits long-term potentiation (LTP), a form of synaptic strengthening thought to underlie many forms of memory (Cunningham et al., 1996).
When released peripherally after an infectious or damaging event, IL-1β signals the brain via both blood-borne and neural routes, leading to de novo synthesis and release of IL-1β within the brain (Quan et al., 1999; Maier et al., 1998). IL-1β then binds to its receptors, which have a notably dense population within the hippocampus (Ban, 1994). One important action of IL-1β is to liberate arachidonic acid and induce the production of prostaglandins (PGs) via cyclooxygenase (COX), the rate-limiting enzyme in this pathway (Hoozemans et al., 2001). Indeed, circulating IL-1β, which has low diffusion across the blood brain barrier, has been shown to upregulate COX activity within brain vascular endothelium causing the release of PGs into the brain parenchyma (Monica et al., 2001). IL-1β within the brain parenchyma can also cause the release of PGs from brain glial cells (O’Banion et al., 1996). Thus, PGs are in a position to mediate some of the effects of IL-1β within the brain. Consistent with this possibility, PGs have been shown to mediate the physiological febrile response and several behavioral responses to IL-1β treatment (Hori et al., 1998; Dinarello et al., 1999; Avitsur and Yirmiya, 1999).
The foregoing suggests that PGs may be downstream mediators of the impact of IL-1β on memory processes. Indeed, a number of studies have shown that COX inhibitors can reverse memory impairments produced by LPS injection, immobilization stress, traumatic brain injury, and aging (Mesches et al., 2004; Shaw et al., 2005; Gopez et al., 2005; Bishnoi et al., 2005; Dhir et al., 2006), all of which increase brain levels of IL-1β. However, many pro-inflammatory processes other than elevated IL-1β levels are also engaged by the above manipulations. Few studies have examined the role of elevated PGs in regulating the specific effect of IL-1β within the hippocampus. Matsumoto et al. (2004) studied this relationship in a hippocampal-dependent working memory task. Animals injected with IL-1β into the hippocampus had impaired memory, which was reversed by coadministration of a COX inhibitor. Furthermore, direct application of a specific PG, prostaglandin E2 (PGE2), into the hippocampus caused memory deficits in this task. These results strongly implicate elevated PGs in mediating the effect of IL-1β on working memory and suggest that PGE2 alone is sufficient to impair working memory. Whether PGs are also critical in mediating the effects of IL-1β on hippocampal long-term memory formation is unknown.
The basal expression of PGs may also be important for learning and memory. For example, inhibition of COX activity under basal, non-inflammatory conditions can result in memory deficits, although deficits are not always observed (Shaw et al., 2005; Dhir et al., 2006; Sato et al., 2007). Similarly, the blockade of COX activity in vitro can impair LTP (Chen et al., 2002). Therefore, reducing PGs below some threshold level may have detrimental effects on memory.
The mechanism(s) by which elevated PGs may act to impair memory processes is largely unknown. A sizeable number of molecules are important in learning and memory processes, but brain derived neurotrophic factor (BDNF) is an intriguing candidate in the present context. BDNF is strongly upregulated following contextual fear conditioning and has been found critical in a number of memory tasks (Hall et al., 2000; Barrientos et al., 2004; Barrientos et al., 2003; Mu et al., 1999). Interestingly, BDNF appears to be involved in IL-1β induced memory impairments. Studies with IL-1β have shown that this cytokine negatively regulates BDNF. First, systemic injection of IL-1β, which elevates brain levels of IL-1β, as well as the induction of elevated but physiological levels of IL-1β within the hippocampus result in lowered BDNF levels (Lapchak et al., 1993; Barrientos et al., 2003). Furthermore, the direct intra-hippocampal administration of IL-1β reduces BDNF mRNA levels up to 6 hours after injection (Barrientos et al., 2004). In vitro studies have also shown that IL-1β reduces BDNF levels in cultures with neurons and astrocytes and that this reduction depends on PGs (Rage et al., 2006). Given the above data, it seems likely that IL-1β-induced reduction in BDNF in vivo also may be caused by PGs, and PGE2 may be sufficient to reduce BDNF levels.
The findings reviewed above led us to explore whether, the impairments in long-term memory formation known to follow injection of IL-1β into the dorsal hippocampus are due to the actions of elevated PGs and whether inhibition of basal COX activity may be sufficient to impair long-term memory. To test these possibilities we 1) microinjected IL-1β either alone or with the non-selective COX inhibitor naproxen and 2) injected naproxen alone into the dorsal hippocampus following contextual fear conditioning and tested memory retention to the context. Contextual fear memory is known to depend on the hippocampus (Phillips and LeDoux, 1992). Furthermore, we determined whether direct injection of PGE2 into the dorsal hippocampus would be sufficient to impair context memory. We also assessed whether PGE2 would reduce BDNF mRNA levels post-conditioning.
EXPERIMENTAL PROCEDURES
Subjects
Animals were adult male Sprague-Dawley (Harlan, Indianapolis, IN, USA) rats weighing approximately 250g upon arrival. Rats were housed 2 to a cage at 25°C on a 12-h light/dark cycle (lights on at 07:00 h). Animals were allowed free access to food and water and were given 1 week to acclimate to colony conditions before experimentation began. All experiments were conducted in accordance with protocols approved by the University of Colorado Animal Care and Use Committee. All efforts were made to minimize the number of animals used and their suffering.
Surgery
Under halothane anesthesia, rats were placed into a Kopf stereotaxic apparatus and implanted with bilateral chronic stainless steel guide cannulae (Plastics One, Roanoke, VA) directed at the dorsal hippocampus. Relative to bregma, cannulae were placed at AP: −3.5 mm; ML: ±2.4 mm; DV: −3.0 mm. Cannulae were secured with dental acrylic and fitted with a dummy cannulae extending 1 mm beyond the tip of the guide cannulae (total length 4 mm) to maintain patency. Animals were allowed to recover for 4 weeks for Experiment 1 and 1–2 weeks for Experiments 2 and 3.
Apparatus
Conditioning chambers were 2 identical igloo coolers, as previously described (Barrientos et al., 2002). A 2-s, 1.5-mA shock was delivered through a removable floor of stainless steel rods 0.5 cm in diameter, spaced 1.75 cm center to center (Coulbourn Model E63-23-MOD001). Each rod was wired to a shock generator and scrambler (Colbourn Model H13-16). Chambers were cleaned with water before each animal was conditioned or tested.
Behavioral procedures
Experiment 1
Rats were taken two at a time from their home cage and each was placed in a conditioning chamber. Rats were allowed to explore the chamber for 2 min before the onset of a 2-s footshock (1.5 mA). Immediately after the footshock, animals were removed from the chamber. Rats then received a bilateral microinjection of vehicle, IL-1β, naproxen, or naproxen and IL-1β (see below) into the dorsal hippocampus. For the behavioral experiment, the rats were tested for fear of the conditioning context 48 h later as a measure of memory, as previously described (Barrientos et al., 2002). Briefly, rats were placed in the context and observed for 6 min, and scored as either moving or freezing every 10 s. Scoring was carried out by observers blind to experimental treatment and interrater reliability exceeded 97%. The following day, rats were placed into a novel context and freezing was observed every 10 s for 6 min to assess generalized fear. Animals displaying high levels of generalized fear (>50%) were excluded from analyses (n=4). In order to assess COX mRNA levels, a second group of animals was treated as described above but 2 h after conditioning were anesthetized with 0.5 ml of sodium pentobarbital (Abbott Labs, North Chicago, IL) and decapitated. Brains were rapidly removed, frozen in ice-cold isopentane, and stored at −80°C until further processing.
Experiments 2 and 3
Because PGE2 has a very short half-life in vivo (a few minutes), a single microinjection in the hippocampus as used in Experiment 1 for IL-1β is not sufficient to keep PGE2 levels elevated long enough to interfere with memory consolidation (data not shown). Therefore, we utilized a novel procedure to deliver prolonged injections of PGE2. A similar procedure is commonly used for microdialysis studies. We altered it by attaching microinjectors to the tubing and setting the flow rate to a very slow rate of 1 μl/h. In this way, animals received a steady flow of PGE2, keeping PGE2 levels elevated for an extended period of time while maintaining minimum overall injection volume.
The afternoon before conditioning, animals were transferred to large Plexiglas bowls (41 cm radius and 36 cm height) and their headcaps were attached to a spring to allow free movement within the bowl. Bowls contained bedding and rats had free access to food and water. Animals were habituated to this context for 1 h and then returned to their homecage. The following morning, context conditioning was carried out as described above. Immediately after removal from the conditioning chamber, animals were placed in the same Plexiglas bowl as the previous day and received bilateral microinjections of either vehicle or PGE2 (see below) into the dorsal hippocampus. For experiment 2, animals were returned to homecage after 6 h of microinjections. Rats were tested for fear of the conditioning context 72 h later and for generalized fear 96 h later as described above.
For experiment 3, microinjections were stopped after 2 h and animals were immediately anesthetized with 0.5 ml of sodium pentobarbital (Abbott Labs, North Chicago, IL) and decapitated. A group that was neither conditioned nor injected was also included to serve as basal controls. Brains were rapidly removed, frozen in cold isopentane, and stored at −80°C until further processing.
Microinjections
Experiment 1
Microinjections were carried out immediately following contextual fear conditioning as previously described (Barrientos et al., 2002). Briefly, a 33-gauge microinjector (Plastics One) attached to PE50 tubing was inserted through the indwelling guide cannula. The distal end of the PE50 tubing was attached to a 100-μl Hamilton syringe, which was placed into a Kopf microinjection unit (Model 5000) that accurately dispensed the desired volume. Injections were conducted over 1 min and injectors were left in place for an additional 2 min before removal and refitting of dummy cannulae.
Experiments 2 and 3
Microinjections were carried out as described above except that PE10 tubing was attached to 50-μl Hamilton syringes in a microdialysis pump (model CMA/102) to accurately dispense 1 μl per h. Flow rates were checked before each experiment. Animals were injected for 6 h in experiment 2 and for 2 h in experiment 3. Animals could freely move and explore during these prolonged injections. After the injections, dummy cannulae were refitted.
Drugs
For experiment 1, 10 ng Human recombinant IL-1β (generously provided by the NIH Biological Response Modifier Program) and 1 μg of the nonselective COX inhibitor naproxen sodium (Sigma M1275) were microinjected in 0.5 μl per side of the hippocampus. Vehicle controls received equivolume saline (pH 7.4). For experiment 2, PGE2 (Cayman Chemical #14010) was dissolved in saline and 5% ethanol. To our knowledge, no one has performed similar PGE2 injections; therefore the optimal dose was unknown. As a result, we injected 2 doses of PGE2: 500ng/h or 250ng/h. Vehicle controls received equivolume of 5% ethanol in saline. Only the low dose of PGE2 was used in experiment 3.
Punches
For experiments 1 and 3, brains were coronally sectioned on a Leica cryostat until the hippocampus was reached. Cannulae placements were verified by visually inspecting cannulae tracts with all cannulae being accurately targeted to the dorsal hippocampus. Punches of the hippocampal subregions dentate gyrus (DG), CA1, and CA3 were taken for both dorsal and ventral hippocampus (Fig. 1) using a stainless steel punch needle of 1 mm diameter. Punches for each region were collected and stored at −80°C until processing.
Figure 1.

Photomicrographs of coronal brain slices. Circles indicate areas punched for the subregions DG, CA1, and CA3 of A) dorsal and B) ventral hippocampus. Cannulae tracks can be seen correctly targeted to dorsal hippocampus (A).
Real time RT-PCR
COX and BDNF mRNA levels in punches of hippocampal subregions were assessed using real time RT-PCR. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was also assessed as a house-keeping gene.
RNA isolation and enrichment
Total RNA was isolated from hippocampal tissue punches based on the method of Chomczynski and Sacchi (Chomczynski and Sacchi, 1987). Briefly, tissue was rapidly homogenized in 0.8 ml TRIzol reagent (Invitrogen, Carlsbad, CA). After incubation at room temperature for 5 min, 160 μl chloroform was added, tubes vortexed for 2 min, and incubated for 3 min. Samples were then centrifuged (11,900 × g) for 15 min at 4°C to achieve phase separation of nucleic acid. Glycogen (8 μl) and isopropyl alcohol (400 μl) were added to the aqueous phase to precipitate nucleic acid. Samples were vortexed briefly and incubated for 10 min followed by centrifugation (11,900 × g) for 10 min at 4°C. Nucleic acid precipitate was washed twice in 75% ethanol (1 ml) and centrifuged (7,500 × g) for 10 min at 4°C. Ethanol was decanted and samples air dried for 30 min. Nucleic acid pellet was resuspended in 8 μl nucleic acid and Rnase-free water. Samples were Dnase-treated (DNA-free kit, Ambion, Austin, TX) to remove contaminating DNA from total nucleic acid. Due to a small amount of RNA in samples, no quantification by spectrophotometric analysis was done.
cDNA synthesis
Detailed cDNA synthesis and quantitative real time PCR procedures are described elsewhere (Frank et al., 2006). Briefly, cDNA synthesis was completed for samples using the SuperScript II First Strand Synthesis System for RT-PCR (Invitrogen).
Primer specifications
cDNA sequences were obtained from Genbank at the National Center for Biotechnology Information (NCBI; www.ncbi.nlm.nih.gov). Primer sequences (Table 1) were designed using an online Oligo Analysis & Plotting Tool (Qiagen) and tested for sequence specificity using the Basic Local Alignment Search Tool at NCBI. Primer specificity (Proligo, Boulder, CO) has been previously verified in our laboratory by melt curve analysis.
Table 1.
RT-PCR primer sequences used for amplification of cDNA.
| Gene | Primer sequences 5′ → 3′ |
|---|---|
| GAPDH | F: gtttgtgatgggtgtgaacc
R: caaacactacccacacttgg |
| BDNF | F: atcccatgggttacacgaaggaag
R: agtaagggcccgaacatacgattg |
| COX-1 | F: aactggtctgcctcaacac
R: aacccacatcaaggactgtc |
| COX-2 | F: ctcgcctctttcaatgtgc
R: ggtcagtagactcttacagc |
Quantitative real time RT-PCR
PCR amplification of cDNA was performed using the Quantitect SYBR Green PCR Kit (Qiagen, Valencia, CA). cDNA (1 μl) was added to a reaction master mix (25 μl) containing 5 mM MgCl2, HotStar Taq DNA polymerase, SYBR Green I, dNTPs, fluorescein (10 nM) and gene specific primers (500 nM each of forward and reverse primer, Table 1). Samples were run in triplicate on a 96-well plate. Formation of PCR product was monitored in real time using the MyiQ Single-Color Real-Time PCR Detection System (BioRad). Threshold cycle (CT; number of cycles to reach threshold of detection) was determined for each reaction.
Relative quantitation of gene expression
Relative gene expression was determined using the 2−ΔΔCT method (Livak and Schmittgen, 2001; Pfaffl, 2001). The mean CT of triplicate measures was computed for each sample. The sample mean CT of GAPDH (internal control), whose expression did not vary as a result of treatment, was then subtracted from the sample mean CT of the gene of interest (ΔCT). The sample with the absolute highest mean ΔCT was selected as a calibrator and subtracted from the mean ΔCT of each experimental sample (ΔΔCT). 2−ΔΔCT thus yields fold change in gene expression of the gene of interest normalized to the internal control gene expression and relative to the calibrator sample.
Data Analysis
Data was analyzed using analyses of variance (ANOVA). When these were significant, Fisher’s protected least significant difference post-hoc tests were used to further differentiate groups. Differences were considered significant when p<0.05.
RESULTS
Experiment 1
The purpose of experiment 1 was to address two questions: (a) are PGs responsible for the long-term contextual fear memory impairment following IL-1β injection into the dorsal hippocampus? (b) does reducing basal levels of PGs impair contextual fear memory? To answer these questions, we injected vehicle, IL-1β, naproxen, or IL-1β and naproxen immediately after contextual fear conditioning and tested memory for the context 48 h later.
The data are shown in Fig. 2. A one-way ANOVA revealed a significant main effect for drug treatment [F(3,27) = 3.549, p<0.028]. Further post-hoc analyses show that we replicated the finding of Barrientos et al. (2004) that IL-1β injected into the dorsal hippocampus after contextual fear conditioning impairs memory for the context. In this experiment, animals injected with IL-1β (n=8) had reduced freezing scores compared to vehicle (n=8) injected animals (Fig. 2; p<0.033). When the COX inhibitor naproxen was coadministered with IL-1β (n=7), the reduction in freezing observed in animals injected with IL-1β alone was completely blocked (p<0.026). This finding indicates that the production of PGs within the hippocampus in IL-1β treated animals is necessary to impair context memory.
Figure 2.
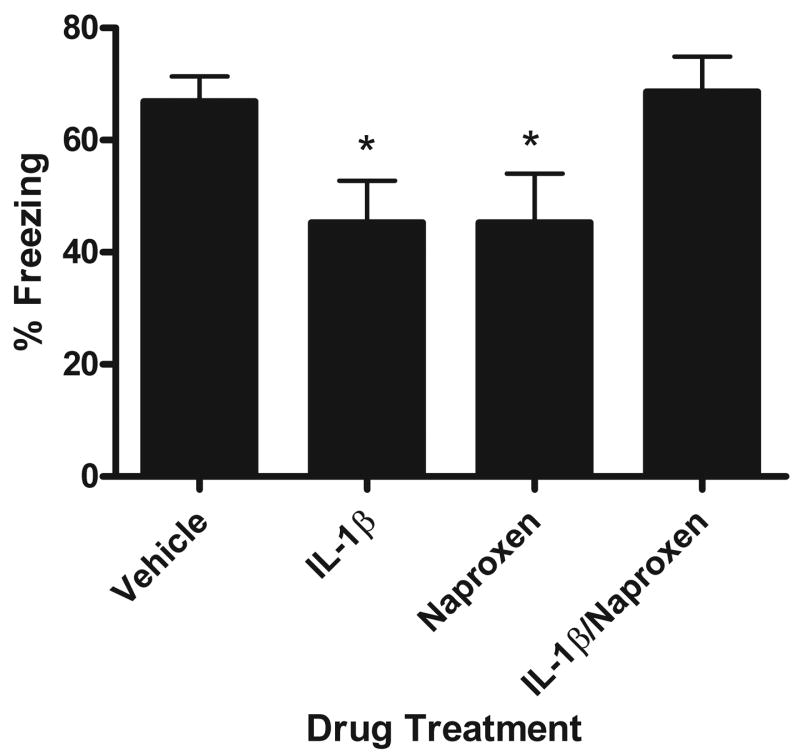
Mean percent freezing during contextual fear test. Animals were conditioned and then immediately injected with vehicle, IL-1β, naproxen, or IL-1β/naproxen. Animals treated with IL-1β or naproxen showed reduced freezing (* p<.03). When IL-1β and naproxen were administered together, animals showed similar freezing to vehicle injected animals. Error bars show S.E.M.
We confirmed that IL-1β treated animals had increased COX-2 mRNA within the hippocampus and that when naproxen was coadministered with IL-1β, COX-2 levels were similar to vehicle injected animals. Data is shown in Fig. 3. We found no subregion variability and so combined all hippocampal subregions. A one-way ANOVA revealed a main effect of drug [F(3,49)=3.825, p=.015] and post-hoc analyses show that IL-1β increased COX-2 mRNA above levels measured in vehicle, naproxen, and naproxen/IL-1β treated animals (p=.015, p=.017, and p=.003, respectively). COX-1 mRNA levels did not vary with drug treatment.
Figure 3.

Relative mean COX-1 and -2 mRNA values for hippocampus 2 h after injection of vehicle, IL-1β, naproxen, or IL-1β/naproxen. Intrahippocampal injection of IL-1β increases COX-2 but not COX-1 mRNA levels and coadministration of naproxen blocks this increase (* p<.02). Error bars show S.E.M.
Interestingly, reducing basal production of PGs within the hippocampus impaired context memory. Animals injected with only naproxen (n=8) immediately after context conditioning showed reduced freezing compared to vehicle and IL-1β/naproxen injected animals (p<0.033). The magnitude of their context memory impairment matched that of the IL-1β treated group.
Experiment 2
Experiment 1 demonstrated that PGs are necessary to produce memory impairments following a post-training intra-hippocampal IL-1β injection. Therefore, in Experiment 2 we tested whether PGE2 induces impairments in contextual fear memory. Fig. 4 shows that prolonged injection of PGE2 but not vehicle directly into the hippocampus following contextual fear conditioning is sufficient to impair context memory in a dose-dependent fashion. A one-way ANOVA revealed a significant effect of drug [F(2,34) = 6.014; p=0.0058]. Post-hoc analyses show that both low (n=15) and high (n=9) doses of PGE2 caused significant reductions in freezing to the conditioned context compared to vehicle injected (n=13) controls (p=0.023 and p=0.002 respectively). Therefore within the hippocampus, PGs appear necessary in the IL-1β-induced memory deficit and PGE2 itself is sufficient to impair context memory.
Figure 4.
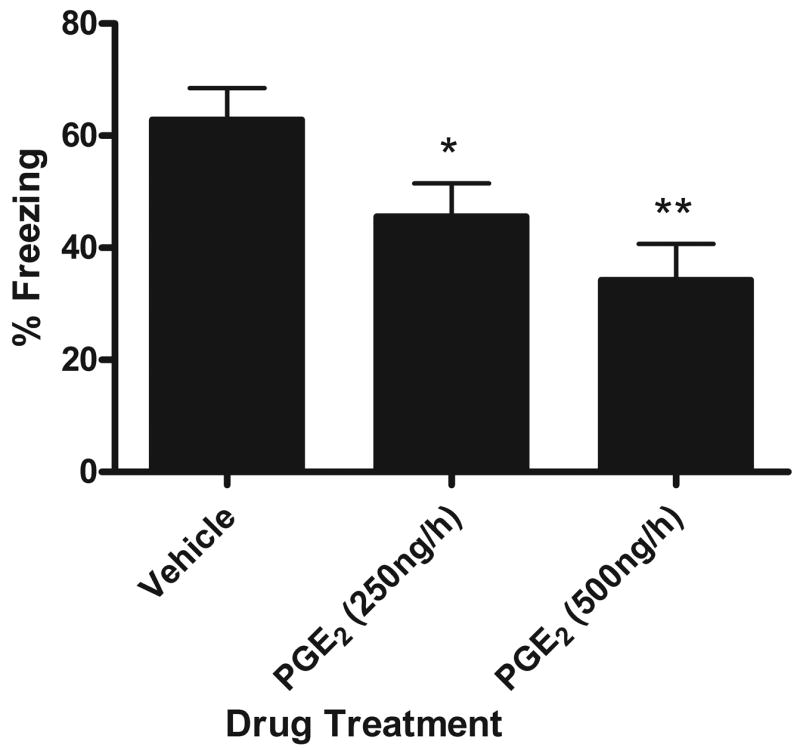
Mean percent freezing during contextual fear conditioning test. Immediately following conditioning, animals were injected with vehicle, 250ng/h PGE2, or 500ng/h PGE2 for 6 hours. Animals injected with PGE2 showed reduced freezing. This impairing effect increased with dose (* p<.03; ** p=.002 compared to vehicle injected animals). Error bars show S.E.M.
Experiment 3
In Experiment 3, we examined BDNF levels as one possible mechanism for PGE2’s memory impairing effect. The data are shown in Fig. 5. As has been shown previously, conditioning induced an increase in BDNF mRNA within the hippocampus (Hall et al., 2000; Barrientos et al., 2004). Vehicle injected animals sacrificed 2 h post-conditioning (n=9) had significantly increased BDNF mRNA compared to unconditioned, uninjected animals (n=7) within all subregions of the hippocampus [F(1,83)=45; p<0.0001].
Figure 5.

Relative mean BDNF mRNA values for dorsal and ventral DG, CA1, and CA3 regions of the hippocampus after 2 h of vehicle or PGE2 (250ng/h) injections post-conditioning. Unconditioned and uninjected animals served as baseline controls. Conditioning increased BDNF mRNA and PGE2 reduced this increase. Error bars show S.E.M.
PGE2 injected during the 2 h post-conditioning (n=8) reduced the conditioning-induced increase in BDNF. A 2×6 ANOVA revealed a significant main effect of Drug [F(1,91)=6.6; p=0.012] and of Hippocampal Subregion [F(5,91)=4.5, p=0.001]. The Drug × Subregion interaction was not significant, indicating that the drug effect was similar throughout the entire hippocampus.
As discussed earlier, IL-1β treatment can impair memory and reduce BDNF mRNA within the hippocampus (Barrientos et al., 2004). Although PGE2 is not known to induce IL-1β, we measured IL-1β levels in vehicle and PGE2 injected animals. No differences in IL-1β protein or mRNA were observed between vehicle and PGE2 injected animals after injections for 6 or 2 h post-conditioning, respectively (data not shown).
DISCUSSION
The present experiments demonstrate that PGs are necessary for IL-1β to impair context memory and that PGE2 injected into the dorsal hippocampus is sufficient to produce similar memory impairments, possibly via reducing post-conditioning BDNF levels. It is possible to conclude that these effects involve memory formation rather than conditioning because the manipulations did not occur until after the conditioning session, not before.
In Experiment 1, we show that a nonselective COX inhibitor, naproxen, coadministered with IL-1β is able to completely block the detrimental effect of IL-1β on memory. Furthermore, animals treated with only naproxen exhibit memory deficits of the same magnitude as IL-1β treated animals, suggesting an inverted U-shaped relationship between PGs and memory. This inverted U-shaped relationship, also known as hormesis, is also present for IL-1β and memory (Goshen and Yirmiya, 2006). Thus, for both IL-1β and PGs elevations above and reductions below a basal, threshold level have deleterious effects on memory. This hormetic relationship for PGs has been suggested elsewhere and is supported by a large body of research (see Goshen and Yirmiya, 2006).
In this experiment, we did not examine specific types of PGs that might be involved in memory impairments. It can only be concluded that the formation of PGs in general by COX enzymes is required for the IL-1β effect. However, PGE2 is a likely candidate for influencing memory. The present research, along with that of Matsumoto et al. (2004), demonstrates that the actions of PGE2 are sufficient to impair memory. To our knowledge, other specific PGs have not been examined for effects on learning and memory processes. PGs of the D, E, F, and I forms are all expressed within the brain. PGD2 has a sleep-promoting function, the alpha isoform of PGF has been found to induce fever in rabbits although it seems to be primarily involved in reproduction, and PGI2 is largely involved in pain pathways (Narumiya et al., 1999). Based on this evidence, we suspect that PGE2 may be the most important PG in modulating learning and memory processes, but specific pharmacologic studies need to be conducted to test this hypothesis directly.
The induction of COX-2 but not COX-1 by IL-1β in experiment 1 suggests that COX-2 is the isoform responsible for producing the elevated PG levels necessary to impair memory. This data is congruent with previous studies suggesting that COX-2 is involved. Classically, COX-2 is considered the inducible form of COX and IL-1β strongly upregulates COX-2 mRNA within brain glial cells (O’Banion et al., 1996). Furthermore, selective COX-2 inhibitors prevent impairments from LPS, immobilization stress, traumatic brain injury, and aging, although to our knowledge selective COX-1 inhibitors have never been examined (Jain et al., 2002; Mesches et al., 2004; Gopez et al., 2005; Bishnoi et al., 2005; Dhir et al., 2006). Therefore, COX-2 seems critically involved in memory impairments from elevated PG levels, however a role for COX-1 cannot be ruled out.
Following naproxen injection, we found no difference in basal COX-1 or -2 mRNA expression. However, previous data would again suggest that COX-2 is the likely isoform involved in the basal expression of PGs necessary for memory. Although COX-2 is typically induced by inflammatory events, it is also constitutively expressed within the brain and a few other tissues (Yamagata et al., 1993). Of note, COX-2 is enriched within the cortex and hippocampus. Within these regions, COX-2 is often localized to postsynaptic dendrites and is regulated by synaptic activity and upregulated by high frequency stimulation (Yamagata et al., 1993; Kaufmann et al., 1996). In terms of the necessary basal expression of PGs for memory, COX-2 also seems responsible. Sato et al. (2007) found that i.p. injection of a selective COX-2 inhibitor impaired memory in a passive avoidance task, while a selective COX-1 inhibitor had no effect. LTP studies have also shown that selective COX-2 inhibitors block LTP while COX-1 inhibitors have no effect (Chen et al., 2002). Therefore, it is likely that inhibition of COX-2 in particular led to the reversal of memory impairments in IL-1β treated animals and caused the memory deficits in animals treated with naproxen.
In experiments 2 and 3, we extended the findings of the first experiment by showing that not only are PGs necessary for IL-1β induced memory impairments, but PGE2 in particular impairs context memory and reduces post-conditioning BDNF levels. These are the first direct studies implicating PGE2 in impairing context memory and reducing BDNF levels. The paucity of previous direct research may largely be due to PGE2’s short half-life, which hinders usual microinjection studies. Our procedure for slow, prolonged microinjections circumvented this problem and could be employed to examine other biological molecules and drugs that have similarly short half-lives.
In experiment 3, PGE2 did not differentially affect BDNF mRNA levels in various hippocampal subregions despite cannulae directed at dorsal hippocampus. This result may be due to drug diffusion from prolonged microinjections and a large, overall volume injected into the dorsal hippocampus.
BDNF has been implicated in many aspects of learning and memory processes. In particular, we have found BDNF to be maximally induced 2 h following contextual fear conditioning (Barrientos et al., 2004) and Alonso et al. (2002) has shown that BDNF is necessary for at least 4 h after training to form a long-term memory. Since PGE2 reduces post-conditioning BDNF mRNA at 2 h, it is likely that this may be a mechanism for PGE2’s memory impairing effects. Clearly, the present BDNF data are only correlational and further work will be required.
Moreover, it is unlikely that that reduced BDNF is the sole mechanism for the memory impairments observed. PGE2 injections may affect other processes that interfere with memory, such as the hypothalamic-pituitary-adrenal (HPA) axis. PGE2 is known to induce secretion of adrenocorticotropin releasing hormone when injected into the preoptic area of the hypothalamus or given intravenously and COX-2 inhibition can reduce physiologically elevated corticosterone (CORT) levels (Zaretsky et al., 2006; Nasushita et al., 1997; Casolini et al., 2002). Therefore, the PGE2 microinjections used here may have elevated CORT, which could interfere with memory. However, there is no evidence to indicate that PGE2 within the hippocampus increases CORT. In fact, we have shown that physiologically elevated PGE2 within the hippocampus, such as after an IL-1β injection, does not change plasma CORT levels (Barrientos et al., 2002). Therefore, it is unlikely that the reductions in BDNF mRNA following intra-hippocampal PGE2 microinjection are due to activation of the HPA axis and elevated CORT levels. However, other products of PGE2 could certainly be involved.
The mechanism(s) by which IL-1β reduces BDNF is largely unknown. We have previously suggested that IL-1β could interfere with CREB, a transcription factor for BDNF, by inhibiting calcium currents or competitively usurping NFk-B needed for CREB signaling (Barrientos et al., 2004). The present data suggest, however, that IL-1β may reduce BDNF indirectly through PGE2. This conclusion leads to a simpler mechanistic explanation.
PGE2 binds to EP receptors, of which there are 4 known subtypes, 1–4, in the rat brain (Sugimoto and Narumiya, 2007). Binding of PGE2 to these G protein coupled receptors leads to activation of different signaling pathways. Activation of the EP1 receptor induces calcium mobilization and EP2 and EP4 are linked to Gs molecules mediating a rise in intracellular cAMP (Narumiya and FitzGerald, 2001). Conversely, activation of the EP3 receptor reduces cAMP through Gi signaling and is therefore termed the “inhibitory” receptor. Of these EP receptors, EP3 has the highest affinity for PGE2 and is most abundantly expressed within the brain (Narumiya et al., 1999). Therefore, PGE2 injected into the hippocampus likely binds mainly to EP3 receptors and results in reduced intracellular cAMP levels. This signaling would result in a reduction in CREB phosphorylation and reduced BDNF transcription. Moreover, a recent report by Rage et al. (2006) showed that neuron cultures treated with astrocyte-conditioned media downregulated BDNF in a PG dependent manner and upregulated EP3 receptors. Therefore, the careful regulation of EP receptor levels may be an important factor in vulnerabilities to immune-related memory impairments.
From these studies it cannot be claimed that PGE2 is the only PG involved in learning and memory. PGE2 is sufficient to impair memory and reduce BDNF levels, but other PGs may have similar effects as discussed above. Similarly, binding to the EP3 receptor is a likely candidate for PGE2’s effect on BDNF but we did not study this relationship directly. Future studies using specific EP receptor agonists and antagonists need to be done to determine their role in learning and memory processes.
The findings of these studies may have broader implications for human populations vulnerable to immune-mediated memory impairments. For example, not only are IL-1β levels elevated in Alzheimer’s disease, but PGs are also oftentimes increased (Montine et al., 1999; Casadesus et al., 2007). Therefore, COX inhibitors may be useful drugs to treat or retard the progression of Alzheimer’s disease. Numerous clinical trials have examined the therapeutic benefit of COX inhibitors in Alzheimer’s patients with the best success in patients treated early during disease progression and when used for more than 24 months (Stewart et al., 1997; in’t Veld et al., 2001). Similarly, animal studies with aged rats demonstrate that PGs can be elevated with normal aging and COX inhibitors can reverse age-related memory impairments (Casolini et al., 2002; Mesches et al., 2004). These data suggest that COX inhibitors may be useful drugs in normal aging to prevent dementias associated with normal aging, although these effects have not been studied in humans. A key factor in these studies may be the regulation of the EP receptors themselves. Future research into these EP receptors and the use of COX inhibitors for preventing memory impairments due to pro-inflammatory processes are needed to elucidate their role in human populations.
Acknowledgments
This work is supported by NIH grants AG028271.
- BDNF
Brain derived neurotrophic factor
- CORT
Corticosterone
- COX
Cyclooxygenase
- DG
Dentate Gyrus
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- HPA
Hypothalamic-Pituitary-Adrenal
- IL-1β
Interleukin-1β
- PG
Prostaglandin
- PGE2
Prostaglandin E2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alonso M, Vianna MR, Depino AM, Mello e Souza T, Pereira P, Szapiro G, Viola H, Pitossi F, Izquierdo I, Medina JH. BDNF-triggered events in the rat hippocampus are required for both short- and long-term memory formation. Hippocampus. 2002;12:551–560. doi: 10.1002/hipo.10035. [DOI] [PubMed] [Google Scholar]
- Akaneya Y, Tsumoto T, Kinoshita S, Hatanaka H. Brain-derived neurotrophic factor enhances long-term potnetiation in rat visual cortex. J Neurosci. 1997;17(17):6707–6716. doi: 10.1523/JNEUROSCI.17-17-06707.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avitsur R, Yirmiya R. Cytokines inhibit sexual behavior in female rats: I. Synergistic effects of tumor necrosis factor alpha and interleukin-1. Brain Behav Immun. 1999;13(1):14–32. doi: 10.1006/brbi.1999.0555. [DOI] [PubMed] [Google Scholar]
- Ban EM. Interleukin-1 receptors in the brain: characterization by quantitative in situ autoradiography. Immunmethods. 1994;5(1):31–40. doi: 10.1006/immu.1994.1035. [DOI] [PubMed] [Google Scholar]
- Barrientos RM, Higgins EA, Sprunger DB, Watkins LR, Rudy JW, Maier SF. Memory for context is impaired by a post context exposure injection of interleukin-1 beta into dorsal hippocampus. Behav Brain Res. 2002;134:291–298. doi: 10.1016/s0166-4328(02)00043-8. [DOI] [PubMed] [Google Scholar]
- Barrientos RM, Sprunger DB, Campeau S, Higgins EA, Watkins LR, Rudy JW, Maier SF. Brain-derived neurotrophic factor mRNA downregulation produced by social isolation is blocked by intrahippocampal interleukin-1 receptor antagonist. Neuroscience. 2003;121(4):847–853. doi: 10.1016/s0306-4522(03)00564-5. [DOI] [PubMed] [Google Scholar]
- Barrientos RM, Sprunger DB, Campeau S, Watkins LR, Rudy JW, Maier SF. BDNF mRNA expression in rat hippocampus following contextual learning is blocked by intrahippocampal IL-1 beta administration. J Neuroimmunol. 2004;155:119–126. doi: 10.1016/j.jneuroim.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Bishnoi M, Patil CS, Kumar A, Kulkarni SK. Protective effects of nimesulide (COX inhibitor), AKBA (5-LOX inhibitor), and their combination in aging-associated abnormalities in mice. Methods Find Exp Clin Pharmacol. 2005;27(7):465–470. doi: 10.1358/mf.2005.27.7.920929. [DOI] [PubMed] [Google Scholar]
- Casadesus G, Smith MA, Basu S, Hua J, Capobianco DE, Siedlak SL, Zhu X, Perry G. Increased isoprostane and prostaglandin are prominent in neurons in Alzheimer’s disease. Mol Neurodegener. 2007;2:2. doi: 10.1186/1750-1326-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casolini P, Catalani A, Zuena AR, Angelucci L. Inhibition of COX-2 reduces the age-dependent increase of hippocampal inflammatory markers, corticosterone secretion, and behavioral impairments in the rat. J Neurosci Res. 2002;68(3):337–343. doi: 10.1002/jnr.10192. [DOI] [PubMed] [Google Scholar]
- Chen G, Kolbeck R, Barde YA, Bonhoeffer T, Kossel A. Relative contribution of endogenous neurotrophins in hippocampal long-term potentiation. J Neurosci. 1999;19(18):7983–7990. doi: 10.1523/JNEUROSCI.19-18-07983.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Magee JC, Bazan NG. Cyclooxygenase-2 regulates prostaglandin E2 signaling in hippocampal long-term synaptic plasticity. J Neurophysiol. 2002;87(6):2851–2857. doi: 10.1152/jn.2002.87.6.2851. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162(1):156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Cunningham AJ, Murray CA, O’Neil LAJ, Lynch MA, O’Connor JJ. Interleukin-1 (IL-1) and tumor necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci Lett. 1996;203:17–20. doi: 10.1016/0304-3940(95)12252-4. [DOI] [PubMed] [Google Scholar]
- Dhir A, Padi SS, Naidu PS, Kulkarni SK. Protective effect of naproxen (non-selective COX-inhibitor) or rofecoxib (selective COX-2 inhibitor) on immobilization stress-induced behavioral and biochemical alterations in mice. 2006;535(1–3):192–198. doi: 10.1016/j.ejphar.2006.01.064. [DOI] [PubMed] [Google Scholar]
- Dinarello CA, Gatti S, Bartfai T. Fever: links with an ancient receptor. Curr Biol. 1999;9(4):R147–R150. doi: 10.1016/s0960-9822(99)80085-2. [DOI] [PubMed] [Google Scholar]
- Gemma C, Fister M, Hudson C, Bickford PC. Improvement of memory for context by inhibition of caspase-1 in aged rats. Eur J Neurosci. 2005;22(7):1751–1756. doi: 10.1111/j.1460-9568.2005.04334.x. [DOI] [PubMed] [Google Scholar]
- Goshen I, Yirmiya R. The role of pro-inflammatory cytokines in memory processes and neural plasticity. In: Ader R, Dantzer R, Glaser R, Heijnen C, Irwin M, Padgett D, Sheridan J, editors. Psychoneuroimmunology. 4. Elsevier; NY: 2006. pp. 337–378. [Google Scholar]
- Gopez JJ, Yue H, Vasudevan R, Malik AS, Fogelsanger LN, Lewis S, Panikashvili D, Shohami E, Jansen SA, Narayan RK, Strauss KI. Cyclooxygenase-2-specific inhibitor improves functional outcomes, provides neuroprotection, and reduces inflammation in a rat model of traumatic brain injury. Neurosurgery. 2005;56(3):590–604. doi: 10.1227/01.NEU.0000154060.14900.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J, Thomas KL, Everitt BJ. Rapid and selective induction of BDNF expression in the hippocampus during contextual learning. Nat Neurosci. 2000;3:533–535. doi: 10.1038/75698. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, Veerhuis R, Janssen I, Rozemuller AJ, Eikelenboom P. Interleukin-1beta induced cyclooxygenase 2 expression and prostaglandin E2 secretion by human neuroblastoma cells: implications for Alzheimer’s disease. Exp Gerontol. 2001;36(3):559–570. doi: 10.1016/s0531-5565(00)00226-6. [DOI] [PubMed] [Google Scholar]
- Hori T, Oka T, Hosoi M, Aou S. Pain modulatory actions of cytokines and prostaglandin E2 in the brain. Ann NY Acad Sci. 1998;840:269–281. doi: 10.1111/j.1749-6632.1998.tb09567.x. [DOI] [PubMed] [Google Scholar]
- In’t Veld BA, Ruitenberg A, Horman A, Launer LJ, van Duijn CM, Stijnen T, et al. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. New Engl J Med. 2001;345:1515–1521. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- Jain NK, Patil CS, Kulkarni SK, Singh A. Modulatory role of cyclooxygenase inhibitors in aging- and scopolamine or lipopolysaccharide-induced cognitive dysfunction in mice. Behav Brain Res. 2002;133(2):369–376. doi: 10.1016/s0166-4328(02)00025-6. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Worley PF, Pegg J, Bremer M, Isakson P. COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc Natl Acad Sci USA. 1996;93(6):2317–2321. doi: 10.1073/pnas.93.6.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapchak PA, Araujo D, Hefti F. Systemic interleukin-1 beta decreases brain-derived neurotrophic factor messenger RNA expression in the rat hippocampal formation. Neurosci. 1993;53:297–301. doi: 10.1016/0306-4522(93)90196-m. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lombardi VR, Garcia M, Rey L, Cacabelos R. Characterization of cytokine production, screening of lymphocyte subset patterns and in vitro apoptosis in healthy and Alzheimer’s disease (AD) individuals. J Neuroimmunol. 1999;97:163–171. doi: 10.1016/s0165-5728(99)00046-6. [DOI] [PubMed] [Google Scholar]
- Maier SF, Goehler LE, Fleshner M, Watkins LR. The role of the vagus nerve in cytokine-to-brain communication. Ann New York Acad Sci. 1998;840:289–300. doi: 10.1111/j.1749-6632.1998.tb09569.x. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Yamaguchi T, Watanabe S, Yamamoto T. Involvement of arachidonic acid cascade in working memory impairment induced by interleukin-1 beta. Neuropharmacology. 2004;46:1195–1200. doi: 10.1016/j.neuropharm.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Mesches MH, Gemma C, Veng LM, Allgeier C, Young DA, Browning MD, Bickford PC. Sulindac improves memory and increases NMDA receptor subunits in aged Fischer 344 rats. Neurobiol Aging. 2004;25(3):315–324. doi: 10.1016/S0197-4580(03)00116-7. [DOI] [PubMed] [Google Scholar]
- Monica EK, Engblom D, Saha Sipra Blomqvist A, Jakobsson PJ, Ericsson-Dahlstrand A. Inflammatory response: Pathway across the blood-brain barrier. Nature. 2001;410:430–431. doi: 10.1038/35068632. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Sidell KR, Crews BC, Markesbery WR, Marnett LJ, Roberts LJ. Elevated CSF prostaglandin E2 levels in patients with probable AD. In: Morrow JD, editor. Neurology. 2. 7. Vol. 53. 1999. pp. 1495–1498. [DOI] [PubMed] [Google Scholar]
- Mu JS, Li WP, Yao ZB, Zhou XF. Deprivation of endogenous brain-derived neurotrophic factor results in impairment of spatial learning and memory in adult rats. Brain Res. 1999;835(2):259–265. doi: 10.1016/s0006-8993(99)01592-9. [DOI] [PubMed] [Google Scholar]
- Narumiya S, FitzGerald GA. Genetic and pharmacological analysis of prostanoid receptor function. J Clin Invest. 2001;108(1):25–30. doi: 10.1172/JCI13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- Nasushita R, Watanobe H, Takebe K. A comparative study of adrenocorticotropin-releasing activity of prostaglandins E1, E2, F2α, and D2 in the rat. Prostaglandins Leukot Essent Fatty Acids. 1997;56(2):165–168. doi: 10.1016/s0952-3278(97)90515-9. [DOI] [PubMed] [Google Scholar]
- O’Banion MK, Miller JC, Chang JW, Kaplan MD, Coleman PD. Interleukin-1 beta induces prostaglandin G/H synthase-2 (cyclooxygenase-2) in primary murine astrocyte cultures. J Neurochem. 1996;66(6):2532–2540. doi: 10.1046/j.1471-4159.1996.66062532.x. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan N, Stern EL, Whiteside MB, Herkenham M. Induction of pro-inflammatory cytokine mRNAs in the brain after peripheral injection of subseptic doses of lipopolysaccharide in the rat. J Neuroimmunol. 1999;93:72–80. doi: 10.1016/s0165-5728(98)00193-3. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdale and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106(2):274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Pugh CR, Kumagawa K, Fleshner M, Watkins LR, Maier SF, Rudy JW. Selective effects of peripheral lipopolysaccharide administration on contextual and auditory-cue fear conditioning. Brain Behav Immun. 1998;12(3):212–229. doi: 10.1006/brbi.1998.0524. [DOI] [PubMed] [Google Scholar]
- Pugh CR, Nguyen KT, Gonyea JL, Fleshner M, Watkins LR, Maier SF, Rudy JW. Role of interleukin-1 beta in impairment of contextual fear conditioning caused by social isolation. Behav Brain Res. 1999;106(1–2):109–118. doi: 10.1016/s0166-4328(99)00098-4. [DOI] [PubMed] [Google Scholar]
- Rage F, Silhol M, Tapia-Arancibia L. IL-1 beta regulation of BDNF expression in rat cultured hypothalamic neurons depends on the presence of glial cells. Neurochem Int. 2006;49(5):433–441. doi: 10.1016/j.neuint.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Sato T, Ishida T, Irifune M, Tanaka K, Hirate K, Nakamura N, Nishikawa T. Effect of NC-1900, an active fragment analog of arginine vasopressin, and inhibitors of arachidonic acid metabolism on performance of a passive avoidance task in mice. Eur J Pharmacol. 2007;560(1):36–41. doi: 10.1016/j.ejphar.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Shaw KN, Commins S, O’Mara SM. Cyclooxygenase inhibition attenuates endotoxin-induced spatial learning deficits, but not an endotoxin-induced blockade of long-term potentiation. Brain Res. 2005;1038(2):231–237. doi: 10.1016/j.brainres.2005.01.035. [DOI] [PubMed] [Google Scholar]
- Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology. 1997;48:626–632. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282(16):11613–11617. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neuron: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11(2):371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- Zaretsky DV, Hunt JL, Zaretskaia MV, DiMicco JA. Microinjection of prostaglandin E2 and muscimol into the preoptic area in conscious rats: comparison of effects on plasma adrenocorticotrophic hormone (ACTH), body temperature, locomotor activity, and cardiovascular function. Neurosci Lett. 2006;397(3):291–296. doi: 10.1016/j.neulet.2005.12.032. [DOI] [PubMed] [Google Scholar]