

Abstract
Obesity-induced chronic inflammation is a key component in the pathogenesis of insulin resistance and the Metabolic syndrome. In this review, we focus on the interconnection between obesity, inflammation and insulin resistance. Pro-inflammatory cytokines can cause insulin resistance in adipose tissue, skeletal muscle and liver by inhibiting insulin signal transduction. The sources of cytokines in insulin resistant states are the insulin target tissue themselves, primarily fat and liver, but to a larger extent the activated tissue resident macrophages. While the initiating factors of this inflammatory response remain to be fully determined, chronic inflammation in these tissues could cause localized insulin resistance via autocrine/paracrine cytokine signaling and systemic insulin resistance via endocrine cytokine signaling all of which contribute to the abnormal metabolic state.
Keywords: Insulin resistance, inflammation, cytokines, macrophage
Insulin resistance and human disease
Insulin resistance is a characteristic feature of most patients with Type 2 diabetes mellitus (T2DM) and is one of the defining clinical features in the Metabolic Syndrome (MetS) or Syndrome X. MetS is a constellation of biochemical and physical derangements which can also include central obesity, atherogenic dyslipidemia (low HDL-C, elevated LDL-C and serum triglycerides), a prothrombotic state, a pro-inflammatory state and hypertension. The prevalence of MetS among adults 20 years or older is 23.7% in the United States according to the Third National Health and Nutrition Examination Survey (NHANES III). This would indicate that approximately 47 million Americans suffer from MetS when extrapolated to the 2000 US census data. As the epidemic of obesity shows no signs of reversing, this number is likely to rise dramatically in the future [1]. In the past, this syndrome has often been considered a disease associated with adulthood, but a recent epidemiologic study indicates that in the past decade there has been a 50% increase in the prevalence of MetS in adolescents [2]. While the clinical definition of MetS may vary between defining agencies, i.e. the World Health Organization (WHO), the National Cholesterol Education Program Adult Treatment Panel III (NCEP ATP III), and The International Diabetes Federation (IDF) the existence of it is indisputable and understanding its etiology is of high importance.
Insulin signaling
Insulin is a pleiotropic hormone which has diverse functions including stimulation of nutrient transport into cells, regulation of gene expression, modification of enzymatic activity and regulation of energy homeostasis via actions in the arcuate nucleus (reviewed in [3,4]). These functions of insulin are exerted across a variety of insulin target tissues through several intracellular signaling cascades. The main focus of this review with regard to insulin action will be glucose metabolism in the insulin target tissues: liver, adipose tissue and skeletal muscle. In skeletal muscle insulin promotes glucose uptake by stimulating translocation of the GLUT4 glucose transporter to the plasma membrane, and impaired skeletal muscle insulin signaling results in decreased glucose disposal. In the liver, insulin inhibits the expression of key gluconeogenic enzymes and, therefore, insulin resistance in liver leads to elevated hepatic glucose production. Adipose tissue insulin signaling results in decreased hormone sensitive lipase activity and this anti-lipolytic effect inhibits free fatty acid efflux out of adipocytes. Increased circulating free fatty acids can result in decreased insulin sensitivity in skeletal muscle due to an increase in intracellular lipid products, including fatty acyl-CoA and ceramide [5–7]. These lipid intermediates can activate the serine/threonine kinase, protein kinase C-θ (PKCθ), which then inhibits the insulin signaling cascade. Perturbations in glucose metabolism due to insulin resistance are further exacerbated when insulin production is compromised, as in patients with T2DM.
Insulin Receptor Substrate (IRS)-Mediated Insulin Signaling
Insulin signaling involves a complex signaling cascade downstream of the insulin receptor. This signaling cascade branches into two main pathways. The first is the phosphatidylinositol 3-kinase (PI3K)-AKT (also called protein kinase B (PKB)) pathway which is largely responsible for insulin action on glucose uptake, as well as other metabolic actions of insulin, including the suppression of gluconeogenesis. The second pathway is the Ras-mitogen-activated protein kinase (MAPK) pathway which mediates gene expression, but also interacts with the PI3K-AKT pathway to control cell growth and differentiation (reviewed in [8]). The common intermediate to these pathways is IRS, which include four distinct family members, IRS1-4. For the purpose of this review, we will focus on IRS1, as defects in insulin signaling typically involve this insulin receptor substrate. Activation of the insulin receptor leads to tyrosine phosphorylation of IRS1 thereby initiating signal transduction. When IRS1 is alternatively phosphorylated on serine 307, its downstream signaling ability is diminished [9]. Serine kinases that phosphorylate serine 307 include I kappa B kinase beta (Ikkb) in the NFκB pathway and C-Jun N-terminal kinase 1 (Jnk1) in the JNK/AP-1 pathway. Additional inflammation-related negative regulators of IRS proteins include the suppressor of cytokine signaling (Socs). Socs1 and Socs3, which are induced in inflammation, promote the ubiquitylation and subsequent degradation of IRS proteins[10]. (Figure 1)
Figure 1. Direct interaction of insulin signaling and inflammatory pathways.
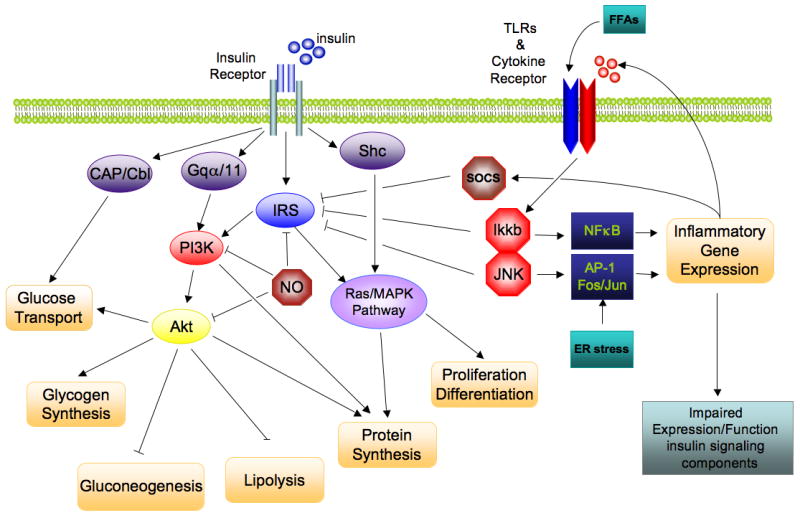
The insulin signaling cascade branches into two main pathways. The PI3K/AKT pathway mediates insulin action on nutrient metabolism including glucose uptake. The Ras/MAPK pathway mediates insulin’s effect on gene expression, but also interacts with the PI3K-AKT pathway to control cell growth and differentiation. Activation of the insulin receptor leads to tyrosine phosphorylation of IRS1 thereby initiating signal transduction. Stimulation of the NFκB and AP-1 Fos/Jun inflammatory pathways results in the activation of the serine kinases, Ikkb and Jnk1, which reduce the signaling ability of IRS1. Additional inflammation-related negative regulators of IRS proteins include the Socs proteins and NO, which are induced in inflammation, and promote IRS degradation. NO also reduces PI3K/Akt activity by s-nitrosylation of Akt.
Non-IRS-Mediated Insulin Signaling
Several other substrates for mediating insulin action, in addition to IRS1-4, have been described. The activated insulin receptor can phosphorylate tyrosine residues on Src homology 2 containing protein (Shc) which then binds to the Son of sevenless (SOS)-growth factor receptor-bound protein 2 (Grb2) complex which then triggers the Ras-MAPK signaling cascade (reviewed in [8]). An additional mediator of insulin action is the heterotrimeric G protein, Gαq/11. Phosphorylation of Gαq/11 by the insulin receptor activates PI3K, thus, stimulating GLUT4-mediated glucose uptake. This signaling pathway is inhibited by G-protein-coupled receptor kinase-2 (Grk2) which binds and inhibits the Gα subunit of Gαq/11[11,12]. Finally, the proto-oncogene product, Cbl, is recruited to the insulin receptor by the adapter protein Cbl associated protein (CAP) and the CAP/Cbl complex can mediate cell surface translocation of GLUT4 independent of PI3K activity [13].
Inflammation and Insulin Signaling
As early as 1950’s, there was epidemiological evidence suggesting a correlation between inflammation and insulin resistant states such as obesity, but the mechanistic links were unknown. In the last decade, however, it has become increasingly evident that obesity and the concomitant development of inflammation are major components of insulin resistance. Studies in human obesity and insulin resistance have revealed a clear association between the chronic activation of pro-inflammatory signaling pathways and decreased insulin sensitivity. For example, elevated levels of tumor necrosis factor-α (TNF), interleukin-6 (IL-6) and interleukin (IL-8) have all been reported in various diabetic and insulin-resistant states [14–18]. In addition, the inflammatory marker C-reactive protein (CRP), a non-specific acute phase reactant, is commonly elevated in human insulin resistant states [19]. Also, experiments in naturally occurring rodent models of obesity, knockout and transgenic mice, as well as detailed studies of insulin signaling at the molecular level have begun to elucidate the mechanistic links between obesity-induced inflammation and insulin resistant states.
The initiation of inflammation in obesity
The precise physiological events leading to the initiation of the inflammatory response in obesity remain incompletely understood. One theory holds that the expansion of adipose tissue leads to adipocyte hypertrophy and hyperplasia and that large adipocytes outstrip the local oxygen supply leading to cell autonomous hypoxia with activation of cellular stress pathways. This causes cell autonomous inflammation and the release of cytokines and other pro-inflammatory signals. Adipokines such as resistin, leptin and adiponectin, which are secreted by adipocytes, can also affect inflammation and insulin sensitivity and this subject is reviewed by other contributors in this review series. As part of the chronic inflammatory process, locally secreted chemokines attract pro-inflammatory macrophages into the adipose tissue where they form crown-like structures around large dead or dying adipocytes. These tissue macrophages then release cytokines that further activate the inflammatory program in neighboring adipocytes, exacerbating inflammation and insulin resistance. Hepatic inflammation can also occur in obesity whereby inflammatory pathway activation could be the result of steatosis and/or increased hepatocyte stress pathway responses. This can result in hepatocyte-autonomous inflammation. Kupffer cells (liver-resident macrophage-like cells) can also become activated, releasing locally acting cytokines which further exacerbates inflammation and hepatic insulin resistance. In addition, overnutrition and obesity are often accompanied by elevations in tissue and circulating FFA concentrations, and saturated FFAs can directly activate pro-inflammatory responses in vascular endothelial cells, adipocytes and myeloid-derived cells [20]. The result of these obesity-induced physiologic events is the development of systemic inflammation.
The role of inflammation-related proteins in insulin target tissues and the development of insulin resistance
Tumor Necrosis Factor-alpha (Tnf)
Tnf (a.k.a. Tnfα) is a pro-inflammatory cytokine secreted predominantly by monocytes and macrophages and has wide ranging biological effects on lipid metabolism, coagulation and endothelial function. Activation of the Tnf receptor results in stimulation of NFκB signaling via Ikkb. An important early study on the interplay between obesity, inflammation and insulin resistance showed that Tnf expression was elevated in adipose tissue isolated from different obese rodent models. Hotamisligil et al also showed that immuno-neutralization of Tnf in obese fatty rats ameliorated insulin resistance [21]. Subsequently, Hotamisligil et al went on to show similar correlations between TNF levels, obesity and insulin resistance in man [18]. Corresponding in vitro experiments demonstrated that by activating Ikkb, Tnf stimulation leads to serine phosphorylation of Irs1 which attenuates its ability to transduce insulin mediated cellular events[22]. Mice genetically deficient in Tnf or the Tnf receptor 1 gene (Tnfr1) do not develop insulin resistance caused by high fat feeding or obesity[23].
Tnf can also affect insulin signaling independent of IRS1. Thus, treatment of cultured 3T3-L1 adipocytes with Tnf leads to reduced expression of the insulin receptor, IRS1 and Glut4 genes, as well as a decrease in insulin stimulated glucose uptake. [24]. Ruan et al also showed a Tnf-induced decrease in 3T3-L1 adipocyte genes, including GLUT4, Hormone Sensitive Lipase (HSL), long-chain fatty acyl CoA synthetase, adiponectin (ADIPOQ), the transcription factor CCAAT/enhancer binding protein-alpha (C/EBP), and the nuclear receptors Pparg and retinoic acid x receptor (RXR). These genes all contribute to glucose homeostasis, both directly and indirectly, and changes in adipocyte expression of these genes likely contribute to insulin resistance in obesity induced-chronic inflammation [25].
Jnk1 and Jnk2
Jnk1 (encoded by Mapk8) also contributes to the development of insulin resistance in obese and diabetic states. Hirosumi et al found elevated JNK activity in liver, adipose tissue and skeletal muscle of obese insulin resistant mice, and knockout of Jnk1 (Jnk1−/−) leads to amelioration of insulin resistance in high fat diet fed mice. At the cellular level, these workers also showed that Jnk1 knockout led to decreased IRS1 phospho-Ser307 in liver. Importantly, deletion of Jnk1 also caused resistance to the development of obesity so the improved insulin sensitivity in these animals could be a result of decreased adiposity and/or decreased Jnk1 activity in insulin target cells[26]. The role of Jnk2 was also assessed in these studies and did appear to play a significant role in the development of obesity-induced insulin resistance. More recent data, however, show that Jnk2 can be involved in metabolic regulation when Jnk1 is absent, since, Jnk1+/−Jnk2−/− mice phenocopy Jnk1−/− mice in their reduced adiposity and improved insulin sensitivity. This suggests functional in vivo interactions between these isoforms in the regulation of insulin action[27].
Ikkb
The use of the anti-inflammatory compounds, salicylate and its derivative aspirin, for treating symptoms of type 2 diabetes mellitus dates back over 100 years. While they are somewhat effective in achieving glucose control at high doses, side effects, such as gastrointestinal bleeding and tinnitus, prevent their widespread use[28]. These compounds are relatively weak inhibitors of Ikkb, thus preventing Irs1 serine307 phosphorylation and this presumably accounts for their insulin sensitizing effects [29]. Several approaches have been used to demonstrate Ikkb’s direct involvement in diet-induced chronic inflammation and insulin action.
Lipid infusion causes acute insulin resistance in rodents and Kim et al have shown that pretreatment of lipid infused rats with salicylates improves glucose utilization in skeletal muscle as measured during hyperinsulinemic-euglycemic clamp studies. They also performed lipid infusions in Ikkb heterozygous knockout mice (Ikkb+/−) and reported similar improvements in insulin sensitivity when compared to wildtype controls. [30].
A subsequent study by Yuan at al showed that Tnf treatment of 3T3L1 adipocytes induces insulin resistance and that this could be prevented by pretreatment of cells with aspirin. A parallel experiment was performed in adipocytes using okadaic acid to activate Ikkb independent of Tnf stimulation, and, again, aspirin prevented okadaic acid-induced insulin resistance. In vivo studies by these investigators have also demonstrated that salicylate treatment of obese rats and mice protects them from insulin resistance[31].
Mice transgenic for tissue specific activators and repressors of Ikkb have been used to determine the tissue-specific contribution of Ikkb to insulin resistance. Mice with a liver-specific constitutively active Ikkb transgene (LIKK) developed hyperglycemia and decreased hepatic insulin sensitivity with mild secondary systemic insulin resistance in skeletal muscle. These mice also displayed liver expression of the pro-inflammatory markers IL-6, IL-1β and Tnf similar to that found in the liver of obese mice. In rescue experiments, LIKK mice treated with sodium salicylate or IL-6 neutralizing antibodies had markedly improved insulin sensitivity. In addition, mice expressing the liver-specific IκBα super-repressor transgene (LISR), which prevents the activation of Ikkb, protected both LIKK and obese mice from hepatic insulin resistance. Another important finding in this study was the elevated expression of the macrophage specific markers, Emr1 (also known as F4/80) and Cd68, in the livers of LIKK and obese mice. Co-expression of LISR and LIKK in compound transgenic mice reduced both insulin resistance and the expression of these same macrophage markers. These data indicate that hepatic inflammation caused by high fat diet is mediated by both hepatocytes and Kupffer cells (liver macrophages) [32]. Taken together, these studies demonstrate the important role of Ikkb in the development of obesity and inflammation-induced insulin resistance.
Inducible Nitric Oxide Synthase (iNOS)
Nitric Oxide (NO) is an endogenous signaling molecule produced by nitric oxide synthase. NO acts as a signal transduction molecule for a number of physiological processes, such as vasodilation, and is also involved in many pathophysiologic states, including insulin resistance. Several inducers of insulin resistance, including FFAs, pro-inflammatory cytokines and oxidative stress, activate the expression of Nos2, the gene that encodes iNOS (reviewed in [33]). In the insulin signaling pathway, NO can reduce Akt activity by causing s-nitrosylation of a specific cysteine residue [34]. Increased iNOS activity also results in the degradation of Irs1 in cultured skeletal muscle cells [33]. Not surprisingly, Nos2 knockout mice are protected from obesity-induced skeletal muscle insulin resistance, and this is associated with improved PI3K-Akt activity [35]. In inflammation-induced insulin resistance, Nos2 is required for the development of sepsis-induced skeletal muscle insulin resistance which might also be mediated by the s-nitrosylation of the insulin receptor, Irs1 and Akt [36,37]. Collectively, these studies highlight the possibility that increased iNOS activity plays a direct role in the pathogenesis of insulin resistance.
Interleukin-10
Il-10 is an anti-inflammatory cytokine produced by macrophages and lymphocytes. Il-10 exerts its anti-inflammatory activity by inhibiting Tnf-induced NFκB activation by reducing IKK activity [38]. In human studies, it has been shown that there is a greater incidence of insulin resistance in subjects with reduced serum levels of IL-10 [39]. This raises the possibility that IL-10 might exert insulin-sensitizing effects. Consistent with this idea it has been shown that mice treated with Il-10 did not become insulin resistant when treated with either Il-6 or lipid infusions[40]. Furthermore, Lumeng et al have shown that Il-10 treated 3T3L1 adipocytes are protected from Tnf- induced cellular insulin resistance[41]. The treatment of inflammatory diseases such as psoriasis with recombinant IL-10 has been an effective therapy and therefore, modulation of Il-10 activity could serve as a potential treatment of insulin resistance[42].
There are many additional interleukins that have been studied with respect to their potential role in the development of insulin resitance. These include Il-6 and Il-1β which are downstream targets of IKKb/NFkB and JNK/AP1 activation. As such, they likely represent elements of a feed forward mechanism of inflammation.
The Macrophage
A major conceptual advance in the field of obesity-induced inflammation and insulin resistance was made by the discovery of bone marrow-derived macrophages in adipose tissue of obese mice and humans [43,44]. Weisberg et al performed microarray analyses comparing adipose tissue RNA profiles of various mouse models of obesity and identified a subset of genes consistently expressed in obese mice. Further analyses showed that this gene set, not typically expressed in adipocytes, was macrophage-derived. Immunohistochemical analyses confirmed the source of this expression to be adipose tissue-resident macrophages. This study showed that the percentage of macrophages in a given adipose tissue depot is positively correlated with adiposity and adipocyte size. They also found that adipose tissue macrophages are responsible for nearly all adipose tissue Tnf expression and a significant portion of Nos2 and Il6 expression. Weisberg et al also quantified the infiltration of macrophages in subcutaneous adipose tissue from obese human subjects and reported that as high as 50% of the total cell content consists of macrophages compared to 10% in lean controls [43]. Xu et al reported similar findings and also showed that thiazolidinedione (TZD) treatment could repress the expression of macrophage-specific genes, providing an additional mechanism by which TZD treatment improves insulin sensitivity [44].
The discovery of adipose tissue macrophages has introduced a new target tissue for potential therapies to treat insulin resistance. This advance has driven the field to examine the importance of macrophage-inflammatory cytokines, as well as proteins regulating macrophage migration, tissue infiltration and pathogen sensing in the development of insulin resistance. This has been achieved by genetic manipulation in mice. These studies have been critical to our understanding of the relationship between insulin resistance, inflammation, and obesity.
Myeloid-IkkB
Initial studies of the role of macrophages in insulin signaling showed that inhibition of the macrophage inflammatory pathway protects mice from obesity-induced insulin resistance [45]. In this study, the investigators generated both a myeloid-specific deletion of Ikkb (IkkbΔmye) and liver-specific deletion of Ikkb (IkkbΔhep). They found that IkkbΔhep mice are protected from HFD-induced hepatic insulin resistance, but that this was a tissue autonomous effect, since these mice still developed insulin resistance in muscle and fat. They also showed a significant reduction in expression of inflammatory markers in the liver supporting the hypothesis that inactivation of inflammation can prevent HFD-induced insulin resistance. Importantly, these studies showed that tissue-specific deletion of Ikkb in myeloid cells (IkkbΔmye mice), led to systemic insulin sensitivity with globally improved insulin action in muscle, liver and fat. As such, these results showed that inactivation of myeloid- Ikkb activity prevents systemic insulin resistance, most likely by interrupting local paracrine effects between resident macrophages and insulin target tissues. [45].
Ccr2/Mcp1
Monocyte chemoattractant protein-1 (Mcp-1, also known as chemokine ligand 2 (Ccl2)) and its cognate receptor chemokine receptor 2 (Ccr2) are also major components of insulin resistance in obese mice. Mcp-1 is a chemokine secreted primarily by macrophages and endothelial cells which promotes the recruitment of monocytes to damaged or inflamed tissues. Ccr2, the receptor for Mcp1, is expressed in monocytes but also in the lung, spleen and thymus [46]. Weisberg et al examined the role of Ccr2 and found that obesity-matched Ccr2−/− mice displayed reduced adipose tissue macrophage infiltration, reduced hepatic steatosis, decreased inflammatory profiles and improved systemic insulin sensitivity. Ccr2 deficiency also attenuated high fat diet-induced weight gain by causing a reduction in caloric intake, highlighting the possible involvement of Ccr2 in the control of eating behavior. Furthermore, treatment of obese mice with a pharmacological antagonist of Ccr2 led to decreased adipose tissue macrophage infiltration and improved insulin sensitivity [47].
Complementary studies on the Ccr2 ligand, Mcp1, have shown that its expression is increased in obese mice, suggesting that changes in Mcp1 levels promote the recruitment of macrophages to adipose tissue which then causes inflammation and insulin resistance. This hypothesis has been tested in transgenic mice that overexpress Mcp1 under the control of the adipose tissue-specific AP2 promoter. This study found that overexpression of Mcp1 in adipose tissue can enhance macrophage infiltration and cause insulin resistance [48,49]. Kanda et al went further to show that the onset of these abnormalities in obese mice could be prevented by genetic deletion of Mcp1 [48]. Recently, Inouye et al suggest that Mcp1 may also have a role in energy metabolism. However, in contrast to previous studies, they showed that HFD-fed Mcp1 KO mice developed hyperinsulinemia and increased adiposity independent of adipose tissue macrophage levels, which were unchanged. Differences in experimental approaches as well as the complexity/redundancy of chemokine signaling may account for these conflicting conclusions. [50]. Taken together, the bulk of the evidence suggests that the Mcp1/Ccr2 axis could provide one mechanistic link between obesity, adipose tissue inflammation and insulin resistance.
The dynamic inflammatory properties of adipose tissue macrophages
Recent studies by Lumeng et al showed that adipose tissue macrophages have heterogeneous inflammatory properties that are influenced by diet and obesity. They showed differences in the inflammatory properties of resident adipose tissue macrophage versus macrophages that are recruited to adipose tissue in obesity. In vivo macrophage pulse labeling studies showed that newly recruited adipose tissue macrophages are pro-inflammatory, as indicated by increased expression of Il6, Nos2 and Ccr2, while resident adipose tissue macrophages are anti-inflammatory. This difference in inflammatory phenotype requires an intact Mcp1/Ccr2 axis because resident macrophages in lean adipose tissue originate from a CCR2−CX3CR1hi monocyte pool, whereas recruited pro-inflammatory adipose tissue macrophages originate from a pool of CCR2+CX3CR1low circulating monocytes [51]. Chemokine (C-X3-C motif) receptor 1 (CX3CR1), is a fractalkine receptor that mediates cell-adhesive and migratory functions on monocytes in inflammation.
Further studies showed that macrophages are also capable of undergoing a phenotypic switch from an M1 state, which was defined as the “classically activated” pro-inflammatory macrophage, to the M2 state or the “alternatively activated” non-inflammatory cell. The M2 classification involves cells that express arginase and the anti-inflammatory cytokine Il-10 and have a high capacity to repair damaged tissue [52]. M1 cells express inflammatory cytokines and the cell surface marker Cd11c [41,53]. Adipose tissue from obese mice contains proportionately more M1 macrophages, whereas, lean adipose tissue contains more M2 macrophages, and increased M1 content positively correlates with inflammation, macrophage infiltration and insulin resistance. The authors propose a mechanism for how a macrophage phenotypic switch occurs in obesity. In their model, obesity causes changes in circulating monocytes that become Ly-6ChiCCR2+ “inflammation-primed” monocytes predisposing them toward an M1 conversion in tissue. When these macrophages enter obese adipose tissue, local environmental cues cause these pre-M1 macrophages to fully convert to the M1 state [41]. It is probable that local concentrations of inflammatory mediators, such as FFAs, Tnf or other cytokines/adipokines contribute to this polarity switch.
Cbl-associated protein
Recent findings have shown that Cbl-associated protein (CAP), known as a positive regulator of glucose transport into cells, plays a role in obesity-related inflammation due to its function in maintaining normal macrophage activity [13]. Cap knockout mice (Sorbs1−/−) are protected from high fat diet-induced insulin resistance because of decreased adipose tissue macrophage content and reduced inflammation. Sorbs1−/− mice show reduced activity of both Ikkb and Jnk, decreased Mcp1 protein levels, reduced macrophage infiltration and lower white blood cell counts. Furthermore, elicited macrophages from Sorbs1−/− mice exhibit decreased mobility when measured in vitro. By using bone marrow transplantation (BMT) to achieve adoptive transfer of myeloid cells, these investigators showed that this insulin sensitive phenotype was transferred to wildtype obese mice that were transplanted with bone marrow from Sorbs1−/− mice. This study further highlights the importance of macrophages in the development of obesity-induced inflammation and insulin resistance, showing that normal macrophage function is necessary for the development of HFD/obesity-induced insulin resistance [54].
Macrophage-Peroxisome Proliferator Activated Receptor-gamma (Pparg)
Pparg is a nuclear receptor required for adipogenesis. It is also the target for the insulin sensitizing TZD class of drugs, which promote adipogenesis and cause systemic insulin sensitivity. In addition to adipocytes, Pparg is also expressed in macrophages where it negatively regulates a large set of inflammatory pathway genes by a unique transrepression mechanism. Based on this concept, deletion of Pparg from macrophages should derepress the inflammatory program, and indeed Hevener et al showed that myeloid-specific deletion of Pparg (Mac-Pparg-KO) results in inflammatory pathway activation with the development of skeletal muscle and hepatic insulin resistance in lean C57/Bl6 mice fed a normal diet. The relative degree of insulin resistance became more severe when these mice where subjected to HFD compared to HFD-fed controls. Interestingly, the ability of TZD treatment to improve insulin sensitivity was attenuated in these animals, indicating that the anti-inflammatory properties of TZDs in tissue-resident macrophages are a component of their insulin sensitizing effects [55].
In a similar study, myeloid deletion of Pparg in BALB/c mice resulted in insulin resistance and glucose intolerance. It was also shown that myeloid Pparg is required for the alternative activation of macrophages. As a result, these mice had a reduction in the total number of adipose tissue macrophages but increased local inflammation due to the absence of M2 macrophages. This study suggests a potential beneficial role of alternatively activated macrophages in protecting against obesity-induced insulin resistance [52].
Macrophage Jnk1
As discussed previously, Jnk1 is an important component of obesity-induced insulin resistance. Due to its apparent role in the development of obesity, it is necessary to separate the roles of Jnk1 in adiposity versus inflammation. In a recently completed study, Solinas et al generated mice with Jnk1-deficient macrophages by transplanting donor Jnk1−/− bone marrow into Jnk1+/+ recipient mice. These mice became obese when placed on HFD but were more insulin sensitive and had reduced inflammatory markers compared to control, Jnk1+/+ mice transplanted with Jnk1+/+ bone marrow. Chemotaxis assays showed that Jnk1−/− primary macrophages exhibited an impaired ability to migrate down a chemotactic gradient. Additionally, “reverse chimeras”, i.e. wildtype donor bone marrow transplanted into recipient Jnk1−/− mice, manifested an improvement in glucose homeostasis as well as resistance to diet-induced obesity [56]. Taken together, these findings demonstrate that: 1) Jnk1 signaling in macrophages is a key component of macrophage function and a mediator of the macrophage inflammatory response which ultimately leads to insulin resistance, and 2) Jnk1 also plays an important role in non-hematopoietic cells in the development of obesity and the associated insulin resistance.
Obesity-induced cellular events leading to inflammation
The above studies have largely focused on the effectors of the inflammatory program, but not on the underlying causes, which initiate the pro-inflammatory state. For example, Tnf and other cytokines/chemokines are symptomatic of inflammation, and while they propagate and/or maintain the inflammatory state, they are not the initial cause(s) of inflammation. Determining the local environmental cues that ignite the initial inflammatory response is important to our understanding of the root mechanisms of obesity-induced inflammation and insulin resistance. The next section focus on advances made to identify causes of chronic inflammation in metabolic disorders.
FFA stimulation of Toll-like Receptor 4
Tlr4 belongs to the family of Toll-like receptors that function as pattern recognition receptors that guard against microorganismal infections as part of the innate immune system. These receptors recognize/sense bacterial and viral components and respond by activating innate immunity pathways. Tlr4, in particular, is stimulated by lipopolysaccharide (LPS), an endotoxin released by gram-negative bacteria. Tlr4 stimulation results in the activation of both Ikkb/NFκB and JNK/AP-1 signaling, culminating in the expression and secretion of pro-inflammatory cytokines/chemokines, including, Il1b, IL-6, Tnf, Mcp1, etc. (reviewed in [57]).
The role of Tlr4 as a key component in fatty acid-induced inflammation was first identified in studies examining the ability of dietary fatty acids to activate the inflammatory response via Tlr4 signaling in cultured macrophages (reviewed in [58]).
The link between Tlr4 and fatty acid and obesity-induced insulin resistance was recently further assessed by Shi et al. This study found that free fatty acids (FFA) activate the NFkB signaling pathway in primary macrophages from wildtype mice but not in macrophages derived from Tlr4 deficient (Tlr4−/−) mice. In addition, they demonstrated that saturated fatty acids are the most potent inducers of this inflammatory response. In in vivo studies, they found that obese mice have increased Tlr4 expression in adipose tissue compared to lean controls. Furthermore, male Tlr4−/− mice are protected from lipid infusion-induced insulin resistance, presumably due to decreased FFA-induced NFκB transcriptional activity, as well as reduced expression of Tnf, Il6 and Mcp1 expression in adipose tissue. They also show that obese female Tlr4−/− mice are partially protected from insulin resistance due to decreased inflammation in both liver and adipose tissue. This work implicates Tlr4 in the development of lipid and obesity-induced insulin resistance and suggests that Tlr4 can act as a potential sensor of elevated tissue and/or serum free fatty acid concentrations that are commonly associated with obesity [59].
The study by Shi et al implicates Tlr4 in the inflammation/insulin resistance process, but do not elucidate the specific tissues in which Tlr4 depletion functions to protect mice from insulin resistance. Recent progress has been made on the latter question by use of bone marrow transplantation to create chimeric mice in which Tlr4 is depleted from the myeloid compartment. Thus, wt mice reconstituted with bone marrow from Tlr4−/− mice represent a functional macrophage knockout model, and these animals are protected from the effects of HFD and obesity to produce insulin resistance. Expression analysis in these mice showed a significant reduction in hepatic Ccl5 (also known as RANTES) and Tnf levels, indicating reduced inflammation, despite pronounced hepatic steatosis [60]. This suggests that Kupffer cells, known to be activated in hepatic steatosis, can mediate inflammatory responses via Tlr4 signaling (reviewed [61]). These results indicate that Tlr4 in macrophages and Kupffer cells participates in a sensing mechanism facilitating fatty acid-induced inflammation and insulin resistance.
Endoplasmic Reticulum Stress
Another potential cause of inflammation in obesity is endoplasmic reticulum (ER) stress. This idea holds that overnutrition causes mechanical stress, excess lipid accumulation and protein synthesis, and abnormal energy metabolism all of which lead to an overburdened ER. This heightened synthetic state in the ER disrupts the normal folding of proteins and activates the unfolded protein response (UPR) that is known to induce stress response pathways. For example, Özcan et al have shown the induction of ER stress induces insulin resistance via JNK-mediated serine phosphorylation of IRS1 in cultured liver cells. Furthermore they demonstrated that obese mice deficient for one allele of X-box binding protein-1 (XBP1), a transcription factor that promotes expression of molecular chaperones in response to ER stress, are more severely insulin resistant compared to obese controls. These mice exhibit ER stress, increased JNK activity and IRS1 serine phosphorylation [62]. This same group has also reported that oral administration of active chemical chaperones reduce ER stress and improves glucose homeostasis in obese mice [63].
Conclusion
Insulin resistance is a complex metabolic defect that most likely has several etiologies dependent on the pathophysiologic state. In humans, there is a genetic component to decreased insulin sensitivity in patients with T2DM and it has been suggested that inherited defects in mitochondrial function with a decreased capacity to oxidize fatty acids is a contributory factor [64]. In addition, obesity is a major cause of insulin resistance in T2DM and recent information demonstrates that chronic, low-grade inflammation associated with obesity is an important etiologic mechanism in decreasing insulin signaling. In this regard, inflammation causes cell-autonomous insulin resistance in muscle, liver and adipose cells and activated tissue macrophages can underlie tissue-autonomous inflammatory processes. What remains to be defined, however, is the sequence of events by which obesity initiates this cascade, and which tissue(s) are the primary effectors in this process versus those that are secondarily involved. Based on the evidence summarized in this review, we highlight a process of gradual progression to insulin resistance in obese states. In this model, obesity causes excessive growth of adipose depots with adipocyte hypertrophy and hyperplasia. This fat overload results in the activation of stress/inflammatory pathways and subsequent paracrine/autocrine-mediated cellular insulin resistance. This in turn increases adipocyte FFA release which stimulates resident macrophages and adipocytes, at least partly through Toll-like receptors, causing increased expression of Tnf, Mcp1 and other pro-inflammatory cytokines. Additionally, these hypertrophied unstable adipocytes eventually die and release their lipid content causing additional migration of macrophages to clear dead or dying adipocytes. The resulting internal environment of this adipose tissue is lipotoxic and pro-inflammatory, favoring the M2 to M1 conversion of resident macrophages or of newly recruited Ly-6ChiCCR2+ “inflammation-primed” monocytes. All of these events culminate in adipose tissue inflammation and insulin resistance. This can be envisioned as a two step process in which the inflammatory pathway is activated in a tissue resident macrophage which leads to paracrine activation of inflammatory pathway components in neighboring insulin target cells, causing impaired insulin signaling. The resulting efflux of FFA and increased secretion of cytokines can lead to secondary insulin resistance in other tissues.
In addition to inflammation and insulin resistance within adipose tissue, the same process most likely occurs in the liver. Thus, the liver contains a large pool of tissue-resident macrophages, (i.e. Kupffer cells) and disabling the inflammatory pathway within these cells prevents hepatic insulin resistance [65]. There may be similarities in the triggers for hepatic inflammation, since increased lipid content and steatosis are common findings in insulin resistant states. Such inflammation in the liver, besides causing localized insulin resistance, would also contribute to systemic inflammation and secondary insulin resistance in other tissues, such as skeletal muscle.
Although chronic inflammation can cause skeletal muscle insulin resistance, the precise sequence and the connections between inflammation and decreased insulin signaling in myocytes remains poorly elucidated. It is possible that increased circulating FFAs or altered cytokine/adipokine levels cause secondary insulin resistance in muscle. Extramyocellular adipose deposits could also contribute to this process in a paracrine fashion. Finally, increased macrophage content has been reported in insulin resistant muscle, raising the possibility of macrophage-mediated tissue autonomous insulin resistance, as in liver and fat [66,67]. Of course, these possibilities are not mutually exclusive.
Clearly, much progress has been made in this area in recent years, and it is now clear that chronic low-grade tissue inflammation is an important contributor to the etiology of obesity and high fat diet-induced insulin resistance. Resident tissue macrophages play a key role in orchestrating this tissue inflammatory response, and future studies will undoubtedly shed much light on the underlying mechanisms.
Figure 2. Obesity and the development of inflammation and insulin resistance.
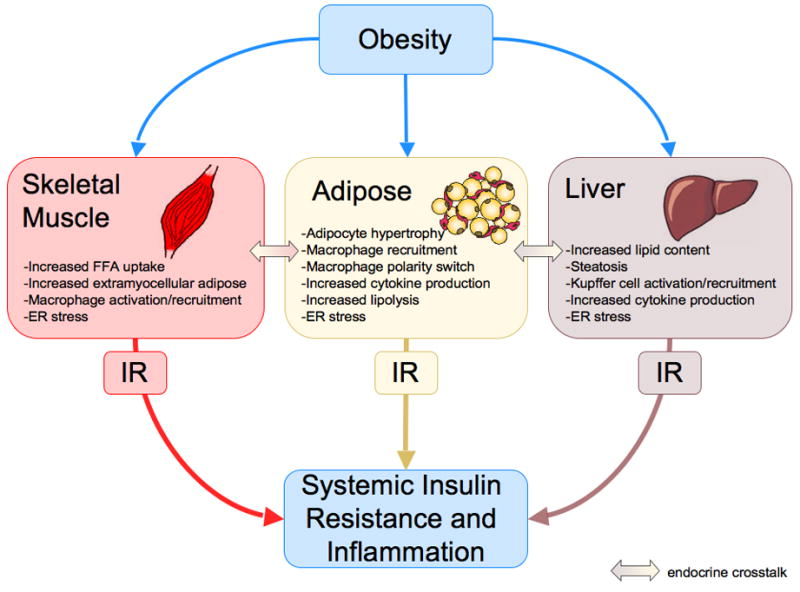
Obesity-induced changes in skeletal muscle, adipose tissue and the liver result in localized inflammation and insulin resistance (IR) through autocrine and paracrine signaling. Endocrine-mediated cross-talk between insulin target tissues contributes to insulin resistance in distant tissues. Systemic inflammation and insulin resistance are the net effect of these changes.
Acknowledgments
Funded by grants from the National Institutes of Health: NIHDK033651 and NIHDK074868 (J.M.O), NIHDK007494 (C.D.)
Abbreviations
- T2DM
type 2 diabetes mellitus
- MetS
Metabolic Syndrome
- TNF
tumor necrosis factor
- CRP
C-reactive protein
- DAG
diacylglyceride
- PKC
protein kinase C
- PI3K
phosphatidylinositol 3-kinase
- PKB
protein kinase B
- MAPK
mitogen activated protein kinase
- IRS
insulin receptor substrate
- IKKb
I kappa B kinase beta
- JNK1
C-jun N-terminal kinase 1
- DIO
diet-induced obesity
- HFD
high fat diet
- ER
endoplasmic reticulum
- Mcp1
monocyte chemoattractant protein 1
- Ccr2
chemokine receptor 2
- FFA
free fatty acids
- NO
nitric oxide
- iNOS
inducible nitric oxide synthase
- Pparg
peroxisome proliferator activated receptor gamma
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. Jama. 2002;287:356–9. doi: 10.1001/jama.287.3.356. [DOI] [PubMed] [Google Scholar]
- 2.Weiss R, et al. Obesity and the metabolic syndrome in children and adolescents. N Engl J Med. 2004;350:2362–74. doi: 10.1056/NEJMoa031049. [DOI] [PubMed] [Google Scholar]
- 3.Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571–8. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- 4.de Luca C, Olefsky JM. Stressed out about obesity and insulin resistance. Nat Med. 2006;12:41–2. doi: 10.1038/nm0106-41. discussion 42. [DOI] [PubMed] [Google Scholar]
- 5.Yu C, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277:50230–6. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- 6.Pickersgill L, Litherland GJ, Greenberg AS, Walker M, Yeaman SJ. Key role for ceramides in mediating insulin resistance in human muscle cells. J Biol Chem. 2007 doi: 10.1074/jbc.M611157200. [DOI] [PubMed] [Google Scholar]
- 7.Lee JS, et al. Saturated, but not n-6 polyunsaturated, fatty acids induce insulin resistance: role of intramuscular accumulation of lipid metabolites. J Appl Physiol. 2006;100:1467–74. doi: 10.1152/japplphysiol.01438.2005. [DOI] [PubMed] [Google Scholar]
- 8.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 9.Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser307 in Insulin Receptor Substrate-1 Blocks Interactions with the Insulin Receptor and Inhibits Insulin Action. J Biol Chem. 2002;277:1531–1537. doi: 10.1074/jbc.M101521200. [DOI] [PubMed] [Google Scholar]
- 10.Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem. 2002;277:42394–8. doi: 10.1074/jbc.C200444200. [DOI] [PubMed] [Google Scholar]
- 11.Imamura T, et al. G alpha-q/11 protein plays a key role in insulin-induced glucose transport in 3T3-L1 adipocytes. Mol Cell Biol. 1999;19:6765–74. doi: 10.1128/mcb.19.10.6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Usui I, Imamura T, Huang J, Satoh H, Olefsky JM. Cdc42 is a Rho GTPase family member that can mediate insulin signaling to glucose transport in 3T3-L1 adipocytes. J Biol Chem. 2003;278:13765–74. doi: 10.1074/jbc.M208904200. [DOI] [PubMed] [Google Scholar]
- 13.Baumann CA, et al. CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature. 2000;407:202–7. doi: 10.1038/35025089. [DOI] [PubMed] [Google Scholar]
- 14.Roytblat L, Rachinsky M, Fisher A, Greemberg L, Shapira Y, Douvdevani A, Gelman S. Raised Interleukin-6 Levels in Obese Patients. Obes Res. 2000;8:673–675. doi: 10.1038/oby.2000.86. [DOI] [PubMed] [Google Scholar]
- 15.Straczkowski M, Dzienis-Straczkowska S, Stepien A, Kowalska I, Szelachowska M, Kinalska I. Plasma Interleukin-8 Concentrations Are Increased in Obese Subjects and Related to Fat Mass and Tumor Necrosis Factor-{alpha} System. J Clin Endocrinol Metab. 2002;87:4602–4606. doi: 10.1210/jc.2002-020135. [DOI] [PubMed] [Google Scholar]
- 16.Hotamisligil GS, Spiegelman BM. Tumor necrosis factor alpha: a key component of the obesity-diabetes link. Diabetes. 1994;43:1271–1278. doi: 10.2337/diab.43.11.1271. [DOI] [PubMed] [Google Scholar]
- 17.Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. PNAS. 2003;100:7265–7270. doi: 10.1073/pnas.1133870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995;95:2409–15. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. Elevated C-Reactive Protein Levels in Overweight and Obese Adults. JAMA. 1999;282:2131–2135. doi: 10.1001/jama.282.22.2131. [DOI] [PubMed] [Google Scholar]
- 20.Kim F, et al. Free fatty acid impairment of nitric oxide production in endothelial cells is mediated by IKKbeta. Arterioscler Thromb Vasc Biol. 2005;25:989–94. doi: 10.1161/01.ATV.0000160549.60980.a8. [DOI] [PubMed] [Google Scholar]
- 21.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 22.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271:665–8. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 23.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389:610–4. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- 24.Stephens JM, Lee J, Pilch PF. Tumor necrosis factor-alpha-induced insulin resistance in 3T3-L1 adipocytes is accompanied by a loss of insulin receptor substrate-1 and GLUT4 expression without a loss of insulin receptor-mediated signal transduction. J Biol Chem. 1997;272:971–6. doi: 10.1074/jbc.272.2.971. [DOI] [PubMed] [Google Scholar]
- 25.Ruan H, Hacohen N, Golub TR, Van Parijs L, Lodish HF. Tumor necrosis factor-alpha suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3T3-L1 adipocytes: nuclear factor-kappaB activation by TNF-alpha is obligatory. Diabetes. 2002;51:1319–36. doi: 10.2337/diabetes.51.5.1319. [DOI] [PubMed] [Google Scholar]
- 26.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 27.Tuncman G, Hirosumi J, Solinas G, Chang L, Karin M, Hotamisligil GS. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc Natl Acad Sci U S A. 2006;103:10741–6. doi: 10.1073/pnas.0603509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williamson RT, Lond MD. On the treatment of glycosuria and diabetes mellitus with sodium salicylate. British Med J. 1901;1:760–762. doi: 10.1136/bmj.1.2100.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 1998;396:77–80. doi: 10.1038/23948. [DOI] [PubMed] [Google Scholar]
- 30.Kim JK, et al. Prevention of fat-induced insulin resistance by salicylate. J Clin Invest. 2001;108:437–46. doi: 10.1172/JCI11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, Shoelson SE. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293:1673–7. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
- 32.Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–90. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sugita H, Fujimoto M, Yasukawa T, Shimizu N, Sugita M, Yasuhara S, Martyn JA, Kaneki M. Inducible nitric-oxide synthase and NO donor induce insulin receptor substrate-1 degradation in skeletal muscle cells. J Biol Chem. 2005;280:14203–11. doi: 10.1074/jbc.M411226200. [DOI] [PubMed] [Google Scholar]
- 34.Yasukawa T, Tokunaga E, Ota H, Sugita H, Martyn JA, Kaneki M. S-nitrosylation-dependent inactivation of Akt/protein kinase B in insulin resistance. J Biol Chem. 2005;280:7511–8. doi: 10.1074/jbc.M411871200. [DOI] [PubMed] [Google Scholar]
- 35.Perreault M, Marette A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat Med. 2001;7:1138–43. doi: 10.1038/nm1001-1138. [DOI] [PubMed] [Google Scholar]
- 36.Carvalho-Filho MA, Ueno M, Carvalheira JB, Velloso LA, Saad MJ. Targeted disruption of iNOS prevents LPS-induced S-nitrosation of IRbeta/IRS-1 and Akt and insulin resistance in muscle of mice. Am J Physiol Endocrinol Metab. 2006;291:E476–82. doi: 10.1152/ajpendo.00422.2005. [DOI] [PubMed] [Google Scholar]
- 37.Carvalho-Filho MA, et al. S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: a novel mechanism of insulin resistance. Diabetes. 2005;54:959–67. doi: 10.2337/diabetes.54.4.959. [DOI] [PubMed] [Google Scholar]
- 38.Schottelius AJ, Mayo MW, Sartor RB, Baldwin AS., Jr Interleukin-10 signaling blocks inhibitor of kappaB kinase activity and nuclear factor kappaB DNA binding. J Biol Chem. 1999;274:31868–74. doi: 10.1074/jbc.274.45.31868. [DOI] [PubMed] [Google Scholar]
- 39.van Exel E, Gussekloo J, de Craen AJ, Frolich M, Bootsma-Van Der Wiel A, Westendorp RG. Low production capacity of interleukin-10 associates with the metabolic syndrome and type 2 diabetes: the Leiden 85-Plus Study. Diabetes. 2002;51:1088–92. doi: 10.2337/diabetes.51.4.1088. [DOI] [PubMed] [Google Scholar]
- 40.Kim HJ, et al. Differential effects of interleukin-6 and -10 on skeletal muscle and liver insulin action in vivo. Diabetes. 2004;53:1060–7. doi: 10.2337/diabetes.53.4.1060. [DOI] [PubMed] [Google Scholar]
- 41.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–84. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Asadullah K, Sterry W, Volk HD. Interleukin-10 therapy--review of a new approach. Pharmacol Rev. 2003;55:241–69. doi: 10.1124/pr.55.2.4. [DOI] [PubMed] [Google Scholar]
- 43.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu H, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arkan MC, et al. IKK-[beta] links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 46.Kurihara T, Bravo R. Cloning and functional expression of mCCR2, a murine receptor for the C-C chemokines JE and FIC. J Biol Chem. 1996;271:11603–7. doi: 10.1074/jbc.271.20.11603. [DOI] [PubMed] [Google Scholar]
- 47.Weisberg SP, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116:115–24. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kamei N, et al. Overexpression of Monocyte Chemoattractant Protein-1 in Adipose Tissues Causes Macrophage Recruitment and Insulin Resistance. Journal of Biological Chemistry. 2006;281:26602–26614. doi: 10.1074/jbc.M601284200. [DOI] [PubMed] [Google Scholar]
- 49.Kanda H, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. Journal of Clinical Investigation. 2006;116:1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inouye KE, Shi H, Howard JK, Daly CH, Lord GM, Rollins BJ, Flier JS. Absence of CC Chemokine Ligand 2 Does Not Limit Obesity-Associated Infiltration of Macrophages into Adipose Tissue. Diabetes. 2007 doi: 10.2337/db07-0425. [DOI] [PubMed] [Google Scholar]
- 51.Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. 2007;56:16–23. doi: 10.2337/db06-1076. [DOI] [PubMed] [Google Scholar]
- 52.Odegaard JI, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007 doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23:344–6. doi: 10.1016/j.immuni.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 54.Lesniewski LA, et al. Bone marrow-specific Cap gene deletion protects against high-fat diet-induced insulin resistance. Nat Med. 2007;13:455–62. doi: 10.1038/nm1550. [DOI] [PubMed] [Google Scholar]
- 55.Hevener AL, et al. Macrophage PPAR(gamma) is Required for Normal Skeletal Muscle and Hepatic Insulin Sensitivity and Full Anti-diabetic Effects of TZDs. J Clin Invest. 2007 doi: 10.1172/JCI31561. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Solinas G, Vilcu C, Neels JG, Luo J-L, Naugler W, Olefsky JM, Karin M. Keystone Symposium: Metabolic Syndrome and Cardiovascular Disease. Steamboat Springs, CO; 2007. JNK in Macrophages Plays a Key Role in the Development of Obesity-Induced Insulin Resitance. [Google Scholar]
- 57.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–95. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 58.Lee JY, Hwang DH. The modulation of inflammatory gene expression by lipids: mediation through Toll-like receptors. Mol Cells. 2006;21:174–85. [PubMed] [Google Scholar]
- 59.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. Journal of Clinical Investigation. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.deLuca C, Saberi M, Schenk S, Lu J-C, Castorena C, Greenberg AW, Olefsky JM. Keystone Symposium: Metabolic Syndrome and Cardiovascular Disease. Steamboat Springs, CO; 2007. Myeloid-derived Toll-like receptor 4 (Tlr4) is required for high fat diet induced hepatic insulin resitance. [Google Scholar]
- 61.Diehl AM. Nonalcoholic steatosis and steatohepatitis IV. Nonalcoholic fatty liver disease abnormalities in macrophage function and cytokines. Am J Physiol Gastrointest Liver Physiol. 2002;282:G1–5. doi: 10.1152/ajpgi.00384.2001. [DOI] [PubMed] [Google Scholar]
- 62.Ozcan U, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–61. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 63.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–40. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–71. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Laskin DL, Weinberger B, Laskin JD. Functional heterogeneity in liver and lung macrophages. J Leukoc Biol. 2001;70:163–70. [PubMed] [Google Scholar]
- 66.Bruun JM, Helge JW, Richelsen B, Stallknecht B. Diet and exercise reduce low-grade inflammation and macrophage infiltration in adipose tissue but not in skeletal muscle in severely obese subjects. Am J Physiol Endocrinol Metab. 2006;290:E961–7. doi: 10.1152/ajpendo.00506.2005. [DOI] [PubMed] [Google Scholar]
- 67.Di Gregorio GB, et al. Expression of CD68 and macrophage chemoattractant protein-1 genes in human adipose and muscle tissues: association with cytokine expression, insulin resistance, and reduction by pioglitazone. Diabetes. 2005;54:2305–13. doi: 10.2337/diabetes.54.8.2305. [DOI] [PubMed] [Google Scholar]