

Abstract
Heparinase I from Flavobacterium heparinum has important uses for elucidating the complex sequence heterogeneity of heparin-like glycosaminoglycans (HLGAGs). Understanding the biological function of HLGAGs has been impaired by the limited methods for analysis of pure or mixed oligosaccharide fragments. Here, we use methodologies involving MS and capillary electrophoresis to investigate the sequence of events during heparinase I depolymerization of HLGAGs. In an initial step, heparinase I preferentially cleaves exolytically at the nonreducing terminal linkage of the HLGAG chain, although it also cleaves internal linkages at a detectable rate. In a second step, heparinase I has a strong preference for cleaving the same substrate molecule processively, i.e., to cleave the next site toward the reducing end of the HLGAG chain. Computer simulation showed that the experimental results presented here from analysis of oligosaccharide degradation were consistent with literature data for degradation of polymeric HLGAG by heparinase I. This study presents direct evidence for a predominantly exolytic and processive mechanism of depolymerization of HLGAG by heparinase I.
Heparin-like glycosaminoglycans (HLGAGs), such as heparin and heparan sulfate, are acidic polysaccharides, which are ubiquitous in the extracellular matrix and at cell surfaces (1). They are used extensively as anticoagulant drugs, and they play a central role in modulation of cell signals (2, 3). Commercial preparations of HLGAGs have 20–40 disaccharide repeat units (1). Individual disaccharides comprise an N-acetylated glucosamine and a glucuronic acid, which have been modified to a varying degree by deacetylation, sulfation, and isomerization of glucuronic acid to iduronic acid. These modifications control the specificity of HLGAG functions and make the molecular analysis of HLGAGs a daunting task (3, 4).
HLGAG-degrading enzymes are useful tools to investigate the composition and sequence of HLGAGs (4). Among the HLGAG-degrading enzymes, heparinase I from Flavobacterium heparinum has been studied most thoroughly biochemically and furthermore has been found to have important clinical applications (5). Heparinase I cleaves the glucosamine–uronic acid glycosidic bond of HLGAGs by an eliminative mechanism, leaving the uronic acid with an unsaturated C4–C5 bond (5). The substrate specificity of heparinase I, and of the similar heparinases II and III from F. heparinum, has been defined by a trisaccharide sequence containing the scissile glucosamine–uronic acid linkage; thus, heparinase I cleaves HNS,6X-I2S/G2S-HNS,6S,¶ heparinase II HNY,6X-I2X/G2X-HNY,6X, and heparinase III HNY,6X-I/G-HNY,6X, in which Y can be sulfated or acetylated and X can be sulfated or unsubstituted (6, 7).
Although the heparinase I substrate specificity is well understood based on the monosaccharides flanking the cleavable linkages, less is known about the sequence of events during enzymatic depolymerization of HLGAGs: The enzyme might cleave exolytically at either the reducing or the nonreducing end of the substrate. Alternatively, the enzyme might cleave each linkage that meets the above mentioned specificity criteria with equal probability (random endolytic cleavage) (8). Subsequent to the initial (exolytic or endolytic) cleavage of a substrate molecule, the enzyme may cleave processively, i.e., proceed to cleave the same substrate molecule several times before releasing it into solution, or the enzyme may cleave nonprocessively by releasing the substrate immediately after cleavage. For heparinase I, as well as for several other polysaccharide lyases, it has been difficult to unambiguously assign an endolytic or exolytic cleavage mechanism (5). It has been proposed that heparinase I acts by an entirely random endolytic mechanism (9, 10) or in a specific endolytic manner followed by processive cleavage (8). We suggested previously (5) that ambiguities in the exolytic/endolytic cleavage pattern might be explained by a subsite model in which the substrate binding domain of the enzyme would have a set of subsites, each of which interacts with a disaccharide repeat. Variations in the intrinsic binding energy for each subsite might explain different product profiles and degrees of processivity. This and similar models may provide a link between kinetic data (10, 11) and our biochemical studies of the enzyme active site (12, 13). However, as of yet, there has been no direct experimental investigation of any potential specificity of heparinase I for the reducing or nonreducing end of the substrate.
Understanding the mechanisms of HLGAG depolymerization by heparinase I has been limited by the polydispersity and chemical heterogeneity of the substrates and by the scarcity of suitable analytical techniques for identification and quantitation of oligosaccharide intermediates larger than tetrasaccharides. In the accompanying paper by Rhomberg et al. (14), we report advances in the methodology for analyzing HLGAGs by matrix-assisted laser desorption ionization MS (MALDI MS) and capillary electrophoresis (CE), extending previous work (15). These techniques allow time-resolved determination of relative abundance of substrate and products during enzymatic depolymerization of homogeneous oligosaccharide substrates (up to at least decamers), which are typically available only in very small quantities. In the present study, we use results obtained by a combination of these MALDI MS and CE techniques to investigate heparinase I function. We use information on the formation and disappearance of specific substrates, intermediates, and end products to provide direct evidence for the enzymatic depolymerization mechanism of heparinase I.
MATERIALS AND METHODS
Materials.
Oligosaccharide H1 was a gift from D. Tyrrell of Glycomed (Alameda, CA). Substrates O2, D3, and D4 kindly were provided by R. J. Linhardt after repurification by R. Hileman of the University of Iowa. Derivatization of decasaccharide D3 with semicarbazide was carried out as described in the accompanying paper (14) and resulted in a mass change of 56.07 units. Heparinase I was derived from F. heparinum cultures.
Enzymatic and Analytical Methods.
Enzymatic reactions were carried out in ethylenediamine/acetic acid buffer as described (14). The enzyme-to-substrate ratio was ≈1:1000. For MALDI MS, analytes were mixed with a solution of (arg-gly)15 and caffeic acid and dried (14). For CE analysis, aliquots of the reaction mixture were sampled automatically at various time points, and the separation was monitored by UV detection (14).
Reaction Modeling.
The rates of the reactions involved in octasaccharide O2 digestion were approximated by first-order kinetics (16) because the substrate concentration was below the Michaelis–Menten constant (Km) of heparinase I for oligo- or polysaccharide substrates (11). The differential equations derived for the time rate of change of concentration of each of the five components were solved analytically by using standard methodologies. The parameters φp (processivity fraction), φint (internal cleavage fraction), k (rate of O2 digestion), and the initial and final concentration of O2 were fitted to experimental data by using Sigma Plot.
Computer Simulations.
A matlab algorithm was written that simulates the mass-average molecular weight (MWm) and cumulative product formation during degradation of polymeric HLGAGs. The progression of reaction is modeled iteratively. At the beginning of each iteration, the enzyme is considered associated to the nonreducing end of products of the previous iteration, which may become substrates of the next iteration. A fraction, φblock, of these substrates will have a noncleavable linkage next, and the enzyme will immediately release from them. Of the remaining enzyme-bound substrates, some (determined by φp for the appropriate substrate length) will cleave the next bond processively, whereas the others will be released from their substrates. The total amount of released enzyme will encounter and cleave the available substrate molecules according to the exolytic/endolytic specificity, expressed by φint (the probability of cleaving an internal linkage relative to the probability of cleaving the terminal linkage at the nonreducing end). Finally, the profile of mass-average molecular weight vs. cumulative product formation is calculated. Experimentally observed values for changes in solution viscosity and A232 absorbance during heparin degradation (10) were converted to changes in MWm and cumulative product formation by linear relationships as described (16, 17) and were compared with the simulated values.
Details of the algorithm are described elsewhere (16). In brief, a vector, c, records the concentration of individual oligosaccharides where the first entry corresponds to disaccharides, the second to tetrasaccharides, etc. Similarly, concentration of enzyme-associated oligosaccharides is described by the enzyme vector e. The kinetic information is contained in a processivity vector, p (each element corresponds to φp for a product of that length: 0 for disaccharides and tetrasaccharides, φp for all others) and in an encounter matrix, E, which, for the first step of enzymatic cleavage, gives the relative probabilities of cleaving a substrate (identified by the row number i) at a given bond (identified by the column number j). The reactions taking place during a given iteration are expressed as a matrix, R, with the elements Ri,j denoting the reaction rate for cleavage of substrate of length i at bond number j. These matrix elements are calculated by Eq. 1.
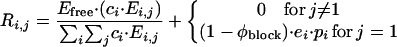 |
1 |
The first term represents primary encounters between free enzyme (Efree) and substrate (appropriately weighted by the concentration of substrates at the various lengths), and the second term represents processive reactions and applies only to cleavage at the first bond at the nonreducing terminus, i.e., when j = 1. Finally, the oligosaccharide concentration vector is updated by subtracting cleaved substrates and adding the two newly formed products. This updated concentration vector is used to calculate MWm and the cumulative product formation at that iteration. The full program accounts for oligosaccharides that are blocked at the nonreducing end by a second concentration vector and a second encounter matrix, which is identical to the first encounter matrix, except that it has zeros in the first column to reflect the blocked, nonreducing end of these substrates. These additional parameters are treated identically to the original concentration vector and encounter matrix, as described (16).
RESULTS AND DISCUSSION
Framework for Heparinase I Mechanism.
The results of this study are interpreted in the framework of a two-step mechanism for each enzyme-substrate encounter during heparin depolymerization by heparinase I. First, the enzyme cleaves one of the cleavable linkages (HNS, 6X-I2S). The relative susceptibility of a linkage in this step may depend on its location relative to the termini of the substrate. In the next step, the enzyme either proceeds to cleave the same substrate processively (i.e., it moves along the HLGAG chain to the next site either toward the reducing or nonreducing direction relative to the site first cleaved), or the enzyme releases the substrate and becomes available for a new substrate encounter. To distinguish among these models, four different purified oligosaccharide substrates were chosen (Table 1). Three of these (O2, D3, and the derivatized decasaccharide D3d) contain one or more heparinase I-cleavable linkages at the nonreducing terminus. One substrate (D4) contains two cleavable linkages near the reducing terminus and two noncleavable linkages near the nonreducing terminus. The products of enzymatic cleavage (listed in Table 1) were identified and quantified to determine the heparinase I preference for exolytic cleavage at the reducing and nonreducing ends and for investigating the degree of processivity of heparinase I. Fig. 1 shows a schematic illustration of the substrates used in this study and the heparinase I-catalyzed depolymerization reactions that are discussed. Tetrasaccharides that contain cleavable linkages are degraded orders of magnitude slower than longer substrates (11, 14) and can be considered essentially uncleavable at the time scales of these experiments.
Table 1.
m/z of complexes of peptide and oligosaccharide
| Substrates (1-4) and products (5-13) | Underivatized | Derivatized | |
|---|---|---|---|
| H1 | I2S-HNS,6S-I2S-HNS,6S-I2S-Man6S | 4872.99 | NA |
| O2 | ΔU2S-HNS,6S-I2S-HNS,6S-I2S-HNS,6S-G-HNS,6S | 5447.47 | NA |
| D3 | ΔU2S-HNS,6S-[I2S-HNS,6S]4 | 6105.02 | 6161.09 |
| D4 | ΔU2S-HNS,6S-I-HNAc,6S-G-HNS,6S,3S-[I2S-HNS,6S]2 | 5986.93 | NA |
| T5 | ΔU2S-HNS,6S-I2S-HNS,6S | 4372.58 | 4428.65 |
| Di6 | ΔU2S-HNS,6S | 3795.10 | 3851.17 |
| T7 | ΔU2S-HNS,6S-I2S-Man6S | 4277.49 | NA |
| H8 | ΔU2S-HNS,6S-[I2S-HNS,6S]2 | 4950.06 | 5006.13 |
| T9 | ΔU2S-HNS,6S-G-HNS,6S | 4292.51 | NA |
| H10 | ΔU2S-HNS,6S-I2S-HNS,6S-G-HNS,6S | 4869.99 | NA |
| O11 | ΔU2S-HNS,6S-[I2S-HNS,6S]3 | 5527.54 | 5583.61 |
| H12 | ΔU2S-HNS,6S-I-HNAc,6S-G-HNS,6S,3S | 4831.97 | NA |
| O13 | ΔU2S-HNS,6S-I-HNAc,6S-G-HNS,6S,3S-I2S-HNS,6S | 5409.45 | NA |
Di, disaccharide; T, tetrasaccharide; H, hexasaccharide; O, octasaccharide; D, decasaccharide; NA, not applicable.
Figure 1.

Schematic representation of reactions for heparinase I digestion of the substrates octasaccharide O2 (Top), decasaccharide D3d (Middle), and decasaccharide D4 (Bottom). Filled symbols indicate linkages that are not cleavable by heparinase I, as specified. Substrate and product names refer to Table 1.
Processivity.
Substrates that contain multiple cleavable linkages adjacent to each other can be used to probe the processivity of an enzyme because all adjacent susceptible bonds should be cleaved before the substrate is released. If the enzyme cleaves only once before releasing the substrate, intermediates with only one of the linkages cleaved by heparinase I should be observable. O2, D3, and D3d represent such substrates [D3d is identical to D3, except that the reducing end has been derivatized to form a semicarbazone group (14)]. In the case of O2, nonprocessive cleavage at the external linkage (reaction A1.1) should lead to formation of hexasaccharide H10, and in the case of D3, nonprocessive heparinase I cleavage at a terminal linkage (reducing or nonreducing) should lead to formation of O11. Neither of these two heparinase I degradation products can be observed in the CE electropherogram for O2 (Fig. 2) nor in the MALDI MS spectra for O2 (14) and D3 (Fig. 3). These observations point to a strong preference for processive cleavage by heparinase I. This conclusion is supported further by the enzymatic cleavage products formed from the derivatized D3d (see below).
Figure 2.

CE analysis of octasaccharide O2 digestion mixture. A full CE electropherogram is shown in the accompanying paper by Rhomberg et al. (14). O2 (hollow diamond) is degraded to form tetrasaccharides T5 (solid square) and T9 (solid diamond) and disaccharide Di6 (hollow square). Lines indicate best fit to reaction model (see text). The curve corresponding to O2 approaches a nonzero value due to the presence of an uncleavable impurity, also observed in the MS analysis of this substrate, as described (14).
Figure 3.

MALDI MS spectrum of a decasaccharide D3 digestion mixture after a 270-min incubation with heparinase I. P refers to the protonated peptide (arg-gly)15. SOS, T5, H8, O11, and D3 refer to protonated complexes of peptide with sucrose octasulfate and with the oligosaccharide substrates and products (Table 1). Complexes shown in brackets were not detected. Adducts are indicated by asterisks as in the accompanying paper by Rhomberg et al. (14).
Exolytic or Endolytic Cleavage?
O2 contains two heparinase I-cleavable linkages, one internal and one terminal. Heparinase I cleavage at the terminal linkage, if fully processive (reaction A1.2), would lead to only disaccharide Di6 and tetrasaccharide T9, whereas cleavage at the internal linkage (reaction A2) would lead to T9 and T5. The substrate and these three products were identified and quantitated by using CE (Fig. 2). As stated above, heparinase I cleavage of this substrate is mainly processive (reaction A1 immediately proceeds to reaction A1.2). The ratio of T5 to T9 (RT5/T9) can be used to determine the relative rates of cleavage by reactions A1 and A2. For exclusive cleavage at the internal linkage, RT5/T9 would approach 1.0, whereas for exclusive exolytic cleavage, RT5/T9 = 0. From the data in Fig. 2, RT5/T9 = 0.05 is calculated. Thus, the rate of internal cleavage in the initial step is ≈5% of the rate of terminal cleavage, indicating that heparinase I is predominantly exolytic but that it has a small probability of endolytic cleavage.
Which End Is Most Susceptible to Exolytic Cleavage?
Two of the substrates contain heparinase I-cleavable linkages at the reducing end (D4 and D3) and can be used to determine whether heparinase I cleavage at the reducing end is preferentially exolytic, as it was shown above to be at the nonreducing end. D4 contains two heparinase I-cleavable linkages at the reducing end and two uncleavable linkages at the nonreducing end. Digestion with heparinase I leads to formation of the heparinase I-uncleavable H12 and T5, whereas no O13 is observed (Fig. 4). Cleavage at the internal linkage (reaction C1) would result in a product ratio of H12-to-T5 of R12/T5 = 1, whereas cleavage at the terminal linkage at the reducing end followed by cleavage of the resulting O13 (reaction C2) would lead to R12/T5 = 0. Taking into account that the ionization efficiency of H12 is approximately twice as high as that of T5 and assuming that the de-sulfated tetrasaccharide observed in Fig. 4 arises from the reducing terminus of a lesser sulfated decasaccharide impurity of the substrate, it can be concluded that the ratio of formation of H12 and T5 is close to 1. Hence, the rate of cleavage at the internal linkage relative to the rate of cleavage at the terminal linkage at the reducing end is very high.
Figure 4.

MALDI MS spectrum of a decasaccharide D4 digestion mixture after a 50-min incubation with heparinase I. Two tetrasaccharides are observed with a difference in m/z of 80.06, corresponding to one sulfate group. Most likely, the lesser sulfated tetrasaccharide arises from the reducing end of a substrate impurity. Notation similar to Fig. 3.
D3d has four heparinase I-cleavable linkages and can be used to assess the relative frequency of exolytic cleavage at the nonreducing (leading to nonderivatized Di6) and reducing (leading to derivatized Di6d) ends. With D3d as the substrate, no derivatized disaccharide was formed (Fig. 5), indicating a strong preference for exolytic cleavage at the nonreducing end.
Figure 5.

MALDI MS spectrum of a decasaccharide D3d digestion mixture after a 60-min incubation with heparinase I. Notation similar to Fig. 3.
Taken together with the above mentioned results for cleavage of O2, which has two cleavable linkages at the nonreducing end of the substrate, we can conclude that, at the nonreducing end, heparinase I initiates cleavage in a highly exolytic manner (with only 5% probability of endolytic cleavage at each internal linkage), while at the reducing end, heparinase I cleaves in a preferentially endolytic manner at the first internal bond. Given a substrate with cleavable linkages at both ends, heparinase I cleaves the nonreducing end exolytically much faster than it cleaves at the reducing end. This result is consistent with previous results for degradation of the substrate H1, which was cleaved preferentially at the exolytic linkage toward the nonreducing end (14).
Digestion of Internal Linkages in Decasaccharides D3 and D3d.
The products formed from digestion of the homogeneous D3 (and its derivatized counterpart D3d) reveal subtleties with respect to cleavage of internal linkages in HLGAG oligosaccharides. For the underivatized D3, the MALDI MS spectrum shows a H8 peak of approximately one-third the height of the T5 peak (Fig. 3). Given the two-fold difference in ionization efficiency (14), this corresponds to an approximate 1:6 ratio of H8-to-T5 in the reaction mixture. H8 could arise from either of two pathways: It could be generated by nonprocessive cleavage at either of the first two linkages at the nonreducing end (reactions B1 or B2 not completed), or it could arise from nonprocessive internal cleavage at the third bond (reaction B3), with immediate release of the products. The first pathway involves lack of processivity in the direction of the reducing end, whereas the second pathway involves lack of processivity in the direction of the nonreducing end. Hence, the latter pathway is consistent with the results so far, which have suggested high preference for processive cleavage in the direction of the reducing end but have shown no evidence for processivity in the direction of the nonreducing end. Thus, these data suggest that the main cleavage pattern is initially exolytic at the nonreducing end and then processive toward the reducing end; however, some side reactions take place at internal bonds in the initial step.
This assessment also is supported by results for the derivatized D3d. For this substrate, the ratio of derivatized to underivatized decasaccharide was Rderiv./underiv. = 3.1 (±0.3) (14). The main products from degradation, which can be observed by MS, are T5 and T5d (Fig. 5). Throughout the reaction, Rderiv./underiv. for these tetrasaccharides was constant at 2.0 (±0.2). Thus, a relative enrichment of the underivatized tetrasaccharide by digestion had taken place, indicating some endolytic cleavage. Each of the two initial endolytic cleavage reactions (reactions B2 and B3) will lead to the ultimate formation of two tetrasaccharides, one from the nonreducing end (which will always be underivatized) and one from the reducing end (which will have Rderiv./underiv. = 3.1 just like the original substrate). Thus, to achieve a final ratio of derivatized tetrasaccharide Rderiv./underiv. = 2.0, the rate of initial endolytic cleavage must have been 0.14 (±0.06) times the rate of initial exolytic cleavage [because 2.0 = 3.1/(1 + 0.14⋅(3.1 + 1)]. This number is the aggregate rate of endolytic cleavage at both internal sites, which may explain why it is larger than the 5% probability of cleavage at the single susceptible internal site of O2, determined above.
Finally, minor peaks of H8d and H8 were observed. These hexasaccharides could either be formed by nonprocessive cleavage at the second linkage (reaction B2), which would result in a 76% derivatized H8d, or by cleavage at the third linkage (reaction B3), which would result in underivatized H8. Significantly, the underivatized H8 was consistently more abundant than the derivatized H8d, indicating that the hexasaccharide is formed predominantly by cleavage at the third linkage with immediate release of H8 at the nonreducing side of the cleaved bond. Furthermore, cleavage at the second linkage (reaction B2), according to the previous results, would be followed by processive cleavage at the third linkage, leading to an underivatized T5, an underivatized Di6, and a derivatized T5d.
Taken together, the observations are consistent with the mechanism being initially exolytic cleavage at the nonreducing end (with ≈5% endolytic cleavage at each internal bond) followed by processive cleavage in the direction of the reducing end. However, the ratio (RT5/H8) of formation of T5 to formation of H8 from the underivatized, homogeneous D3 substrate is ≈6 (Fig. 3). RT5/H8 should be ≈20 if H8 was generated exclusively by cleavage at the third bond (reaction B3) and the mechanism was strictly processive toward the reducing end. Thus, a value of RT5/H8Å 6 indicates that the processivity is only ≈90% complete. This conclusion is consistent with the other data. For example, our experiences with the MALDI MS methodology indicates that small amounts of side product (for instance 10% relative abundance of a hexasaccharide from degradation of O2) may fall below the analytical detection limit (14). Also, the fact that oligosaccharides that are isolated from heparinase I reaction mixtures may still contain heparinase I-cleavable linkages at the nonreducing end (like substrates O2 and D3) indicates that the mechanism involves endolytic and nonprocessive side reactions.
Digestion of Polymeric Heparin.
The results from oligosaccharide studies have pointed to a heparinase I mechanism, which is primarily exolytic and processive. It is of interest to investigate whether this result is general for cleavage of polymeric HLGAGs, such as commercial heparin; however, the analytical techniques used here are limited by the substrate size (14).
The shape of a plot of mass-average molecular weight (MWm) (measured as viscosity) vs. cumulative product formation (measured as A232) for degradation of polymeric heparin depends on the mechanism of cleavage and can be simulated computationally (8, 9). An initial steep drop in MWm relative to product formation indicates endolytic cleavage, and a decreasing curve indicates exolytic cleavage. To probe how well the results obtained for degradation of oligosaccharide substrates can be extrapolated to degradation of polymeric heparin, we constructed a mathematical model of polysaccharide degradation that can be compared with viscosity and A232 data obtained during heparin depolymerization.
The inputs to the model are the starting MWm and size distribution of the substrate, the relative rates of initial cleavage as a function of linkage position relative to the nonreducing end, the degree of processivity, and the frequency and distribution of noncleavable linkages in heparin. Given the semi-quantitative nature of the MS data, only approximate numbers were used. The purpose of this simulation was to establish whether the general conclusions from the oligosaccharide studies are consistent with data for polymeric heparin degradation, not to optimize the fit to data nor to validate the exact values of the parameters we determined. In Fig. 6, experimental data (10) are compared with our simulation by using the following values, which represent best estimates based on the oligosaccharide studies reported above: The starting material is heparin of MWm 12,000 with a size distribution similar to that reported by Laurent et al. (18); the relative rate of endolytic to exolytic cleavage is 0.05; the relative rate of exolytic cleavage at the reducing end to exolytic cleavage at the nonreducing end is 0.0; the degree of processivity is 0.9; and the fraction of noncleavable linkages in heparin was calculated to be 0.1 from the detailed data provided in Table 1 of Linhardt et al. (19). Given only these assumptions and no fitting to the experimental data, Fig. 6 shows good agreement of the model to the data. Hence, the conclusions of the oligosaccharide experiments are indeed consistent with the viscosity and UV data from degradation of polymeric heparin with heparinase I (10).
Figure 6.

Changes in mass-average molecular weight (MWm) and cumulative product formation during heparin digestion with heparinase I. Results from a computer simulation (see text) are plotted (line) and compared with the experimental data (stars) of Jandik et al. (10). MWm was determined from viscosity measurements, as described (16).
Implications: Subsite Model of Active Site Structure.
The observed product formation profiles that we determined for defined oligosaccharide substrates have led to a heuristic two-step model for heparinase I degradation of substrate, which is consistent with data for degradation of polymeric heparin. To corroborate these observations with the structure–function relationships that we have determined previously for heparinase I (12, 13, 20), we propose a subsite model for the molecular interactions between heparinase I and its substrate (Fig. 7). The active site of heparinase I (Fig. 7, black) is shown to interact with the substrate at a number of sites positioned either to the reducing side of the scissile bond (S1′, S2′, and S3′) or to the nonreducing side of the scissile bond (S1 and S2). Each subsite interacts with a disaccharide repeat of HLGAG and has an intrinsic binding energy ΔGj (free energy/residue), as defined by Jencks (21). This intrinsic binding energy for site j corresponds to the difference in binding free energy between the enzyme and an oligosaccharide that covers site j with a terminal disaccharide, compared with the binding energy of enzyme and an oligosaccharide that is one disaccharide shorter and hence does not cover site j. Such an analysis of polysaccharide-degrading enzymes previously has been applied to α-amylases (22).
Figure 7.

Schematic representations of the proposed depolymerization mechanism. HLGAG–heparinase I interactions that determine the exolytic preference of initial cleavage of a substrate polymer are described by a subsite model (Upper). The significance of ΔG values for the individual subsites is explained in the text. The processive cleavage of several linkages of the same substrate molecule is shown (Lower). The relative position of heparinase I (large molecule) and substrate (copolymer of uronic acid (circles) and glucosamine (squares)) shifts from a position during previous bond cleavage (hatched) to the current position (gray). Uronic acid residues that make the H–U linkages resistant to heparinase I cleavage are shown in black. Arrows indicate the linkages cleaved.
The model we propose, based on the data reported in this paper, has the following characteristics: The major part of the binding energy is contributed by a hexasaccharide (i.e., most of the binding energy is attributed to three sites), which explains the low dependence of reaction rate on substrate size for hexasaccharides and larger substrates. The scissile bond is the first bond at the nonreducing side (i.e., the three sites with negative ΔGj are sites S1, S1′, and S2′), consistent with the observation that a major product from cleavage of several substrates is T5. Occupation of the second site to the nonreducing side of the scissile bond (site S2) imposes a moderate penalty (i.e., ΔG2 is positive), which explains the exolytic preference as well as the nonzero rate of initial internal cleavage. Finally, the major end-product from processive cleavage is the tetrasaccharide at the reducing end, indicating that site S2′ contributes favorably and that site S3′ does not contribute. In general, we have not made any observations that indicate that there should be significant contributions to the binding energy from sites further to the reducing side than site S2′ (i.e., ΔG3′ is close to 0). For simplicity, the favorable binding interactions (S1, S1′, and S2′) are shown as complementary shapes, and unfavorable binding interactions at subsite S2 are shown as a steric barrier. The contributions to binding may, of course, just as well arise from ionic or other energetic interactions between enzyme and substrate.
Site-directed mutagenesis and biochemical studies have shown that His-203 of heparinase I is involved directly in catalysis (13) and that the primary heparin binding region (residues 195–221) encompasses basic residues on either side of this histidine (12). This idea is consistent with the subsite model, presuming that His-203 is located immediately adjacent to the scissile bond and that residues of the substrate on both the reducing and nonreducing sides of the scissile bond are primarily involved in enzyme binding. It is important to note that this subsite model points to the possible existence of a discrete group of amino acid residues located at subsite S2 that control the exolytic preference of heparinase I. If this hypothesis is true, it may be possible to alter the exo/endolytic specificity by site-directed mutagenesis of those amino acids.
Significance.
The elucidation of the individual steps involved in heparin depolymerization by heparinases may increase the utility of heparinase I in biological systems and as tools for structural analysis of HLGAGs. For example, addition of heparinase in cell culture and in vivo experiments has been used to interpret the function of extracellular HLGAGs, which presumably are degraded by the heparinase (23). However, fragments of HLGAGs may substitute the effect of full length HLGAGs in some situations (24). Furthermore, the length and sequence of HLGAG fragments are strong determinants for their function (25). Hence, it is important to understand the mechanism of enzymatic HLGAG depolymerization and product formation to interpret the effect of heparinase-mediated HLGAG degradation in biological systems.
MALDI MS and CE analysis of the products from heparinase I digestion of HLGAG oligosaccharides helped elucidate the two-step mechanism of this enzyme. The initial step of the heparinase I mechanism involves a strong preference for exolytic cleavage at the nonreducing end of the substrate, although some endolytic cleavage takes place at ≈5% probability at each internal linkage. The second step is highly processive in the direction toward the reducing end, such that the enzyme never fully releases the product on the reducing side of the broken linkage but instead allows the substrate to slide along the active site, until the next cleavable bond is correctly positioned for catalysis (Fig. 7). This study represents the direct determination of the individual steps in heparinase enzyme mechanism.
Acknowledgments
We thank Drs. R. Hileman, R. J. Linhardt, and S. Tyrrell for oligosaccharides and the National Science Foundation (to S.E. through the Biotechnology Process Engineering Center at MIT), National Institutes of Health (GM57073 to R.S. and GM05472 to K.B.), and the Austrian-American Educational Commission (to A.R.) for funding.
ABBREVIATIONS
- HLGAG
heparin-like glycosaminoglycans
- MALDI
matrix-assisted laser desorption ionization
- CE
capillary electrophoresis
Footnotes
HLGAG oligosaccharides are abbreviated as follows: I, α-l-iduronic acid; G, β-d-glucuronic acid; ΔU, I or G with an unsaturated C4–C5 bond; and H, α-d-2-deoxy 2-aminoglucose. 2S and 6S, 2-O and 6-O sulfation; NS and NAc, N-sulfation and N-acetylation of the glucosamine.
References
- 1.Kjellèn L, Lindahl U. Annu Rev Biochem. 1991;60:443–475. doi: 10.1146/annurev.bi.60.070191.002303. [DOI] [PubMed] [Google Scholar]
- 2.Bourin M-C, Lindahl U. Biochem J. 1993;289:313–330. doi: 10.1042/bj2890313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindahl U, Lidholt K, Spillmann D, Kjellén L. Thromb Res. 1994;75:1–32. doi: 10.1016/0049-3848(94)90136-8. [DOI] [PubMed] [Google Scholar]
- 4.Linhardt R J, Wang H M, Ampofo S A. In: Heparin and Related Polysaccharides. Lane D A, Björk I, Lindahl U, editors. New York: Plenum; 1992. pp. 37–47. [Google Scholar]
- 5.Ernst S, Langer R, Cooney C L, Sasisekharan R. CRC Crit Rev Biochem Mol Biol. 1995;30:387–444. doi: 10.3109/10409239509083490. [DOI] [PubMed] [Google Scholar]
- 6.Desai U R, Wang H, Linhardt R J. Arch Biochem Biophys. 1993;306:461–468. doi: 10.1006/abbi.1993.1538. [DOI] [PubMed] [Google Scholar]
- 7.Yamada S, Murakami T, Tsuda H, Yoshida K, Sugahara K. J Biol Chem. 1995;270:8696–8705. [PubMed] [Google Scholar]
- 8.Cohen D M, Linhardt R J. Biopolymers. 1990;30:733–741. doi: 10.1002/bip.360300708. [DOI] [PubMed] [Google Scholar]
- 9.Linhardt R J, Fitzgerald G L, Cooney C L, Langer R. Biochim Biophys Acta. 1982;702:197–203. doi: 10.1016/0167-4838(82)90503-9. [DOI] [PubMed] [Google Scholar]
- 10.Jandik K A, Gu K, Linhardt R J. Glycobiology. 1994;4:289–296. doi: 10.1093/glycob/4.3.289. [DOI] [PubMed] [Google Scholar]
- 11.Rice K G, Linhardt R J. Carbohydr Res. 1989;190:219–233. doi: 10.1016/0008-6215(89)84127-8. [DOI] [PubMed] [Google Scholar]
- 12.Sasisekharan R, Venkataraman G, Godavarti R, Ernst S, Cooney C L, Langer R. J Biol Chem. 1996;271:3124–3131. doi: 10.1074/jbc.271.6.3124. [DOI] [PubMed] [Google Scholar]
- 13.Godavarti R, Cooney C L, Langer R, Sasisekharan R. Biochemistry. 1996;35:6846–6852. doi: 10.1021/bi960356g. [DOI] [PubMed] [Google Scholar]
- 14.Rhomberg A J, Ernst S, Sasisekharan R, Biemann K. Proc Natl Acad Sci USA. 1998;95:4176–4181. doi: 10.1073/pnas.95.8.4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Juhasz P, Biemann K. Proc Natl Acad, Sci USA. 1994;91:4333–4337. doi: 10.1073/pnas.91.10.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ernst S. Ph.D. thesis. Cambridge, MA: Massachusetts Institute of Technology; 1998. [Google Scholar]
- 17.Lasker S E, Stivala S S. Arch Biochem Biophys. 1966;115:360–372. doi: 10.1016/0003-9861(66)90286-4. [DOI] [PubMed] [Google Scholar]
- 18.Laurent T C, Tengblad A, Thunberg L, Höök M, Lindahl U. Biochem J. 1978;175:691–701. doi: 10.1042/bj1750691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Linhardt R J, Ampofo S A, Fareed J, Hoppensteadt D, Mulliken J B, Folkman J. Biochemistry. 1992;31:12441–12445. doi: 10.1021/bi00164a020. [DOI] [PubMed] [Google Scholar]
- 20.Sasisekharan R, Leckband D, Godavarti R, Venkataraman G, Cooney C L, Langer R. Biochemistry. 1995;34:14441–14448. doi: 10.1021/bi00044a022. [DOI] [PubMed] [Google Scholar]
- 21.Jencks W P. Proc Natl Acad Sci USA. 1981;78:4046–4050. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allen J D. Methods Enzymol. 1980;64:248–277. doi: 10.1016/s0076-6879(80)64012-9. [DOI] [PubMed] [Google Scholar]
- 23.Sasisekharan R, Moses M A, Nugent M A, Cooney C L, Langer R. Proc Natl Acad Sci USA. 1994;91:1524–1528. doi: 10.1073/pnas.91.4.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ornitz D M, Herr A B, Nilsson M, Westman J, Svahn C-M, Waksman G. Science. 1995;268:432–436. doi: 10.1126/science.7536345. [DOI] [PubMed] [Google Scholar]
- 25.Sasisekharan R, Ernst S, Venkataraman G. Angiogenesis. 1997;1:45–54. doi: 10.1023/A:1018318914258. [DOI] [PubMed] [Google Scholar]