

Abstract
ANK is a multipass transmembrane protein transporter thought to play a role in the export of intracellular inorganic pyrophosphate and so to contribute to the pathophysiology of chondrocalcinosis. As transforming growth factor-beta-1 (TGF-β1) was shown to favor calcium pyrophosphate dihydrate deposition, we investigated the contribution of ANK to the production of extracellular inorganic pyrophosphate (ePPi) by chondrocytes and the signaling pathways involved in the regulation of Ank expression by TGF-β1. Chondrocytes were exposed to 10 ng/mL of TGF-β1, and Ank expression was measured by quantitative polymerase chain reaction and Western blot. ePPi was quantified in cell supernatants. RNA silencing was used to define the respective roles of Ank and PC-1 in TGF-β1-induced ePPi generation. Finally, selective kinase inhibitors and dominant-negative/overexpression plasmid strategies were used to explore the contribution of several signaling pathways to Ank induction by TGF-β1. TGF-β1 strongly increased Ank expression at the mRNA and protein levels, as well as ePPi production. Using small interfering RNA technology, we showed that Ank contributed approximately 60% and PC-1 nearly 20% to TGF-β1-induced ePPi generation. Induction of Ank by TGF-β1 required activation of the extracellular signal-regulated kinase (ERK) pathway but not of p38-mitogen-activated protein kinase or of protein kinase A. In line with the general protein kinase C (PKC) inhibitor calphostin C, Gö6976 (a Ca2+-dependent PKC inhibitor) diminished TGF-β1-induced Ank expression by 60%, whereas a 10% inhibition was observed with rottlerin (a PKCδ inhibitor). These data suggest a regulatory role for calcium in TGF-β1-induced Ank expression. Finally, we demonstrated that the stimulatory effect of TGF-β1 on Ank expression was inhibited by the suppression of the Ras/Raf-1 pathway, while being enhanced by their constitutive activation. Transient overexpression of Smad 7, an inhibitory Smad, failed to affect the inducing effect of TGF-β1 on Ank mRNA level. These data show that TGF-β1 increases ePPi levels, mainly by the induction of the Ank gene, which requires activation of Ras, Raf-1, ERK, and Ca2+-dependent PKC pathways in chondrocytes.
Introduction
Chondrocalcinosis is a frequent human disease characterized by the deposition of calcium-containing crystals, mostly calcium pyrophosphate dihydrate (CPPD), within joints. CPPD crystals contribute to cartilage destruction by stimulating mitogenesis of synovial cells as well as synthesis and secretion of proteases, prostanoids, and proinflammatory cytokines that are implicated in cartilage matrix degradation [1]. Several forms of chondrocalcinosis have been described, including idiopathic ones, the frequency of which increases with aging, and familial forms. Some forms of familial chondrocalcinosis, typically inherited in an autosomal dominant manner, were reported to be linked to human chromosomes 8q (CCAL1) or 5p (CCAL2) [2]. Complementary genetic studies demonstrated the linkage between familial forms and the Ank gene, located on the CCAL 2 locus. More recently, mutations in the 5' untranslated region of Ank mRNA were also correlated with sporadic forms of chondrocalcinosis [3]. Mutations in the Ank gene were reported additionally in autosomal dominant craniometaphyseal dysplasia and ankylosing spondylitis [4,5], supporting a key role for the Ank gene in the field of mineralizing arthropathy.
It is generally recognized that a local buildup of excess extracellular inorganic pyrophosphate (ePPi), the anionic component of CPPD crystals, supports CPPD formation [6]. Intracellular inorganic pyrophosphate (iPPi) is a by-product of many synthetic intracellular reactions [7], but there is evidence that it is not able to diffuse across healthy cell membranes. As a consequence, ePPi generation by chondrocytes results from its de novo synthesis of ePPi by ecto-enzymes and/or from the contribution of a transport system allowing iPPi to reach the extracellular milieu where CPPD deposition takes place. Among ecto-enzymes, the ecto-nucleoside triphosphate pyrophosphohydrolase, also known as PC-1 (or NPP1), which is abundant in cell membrane [8], hydrolyzes extracellular nucleoside triphosphates into their monophosphate esters and ePPi [9]. On the other hand, the ANK protein was recently postulated to play a key role in the transport of iPPi across the cell membrane. ANK is a multipass transmembrane protein thought to serve either as an anion channel or as a regulator of such a channel [10]. Progressive ankylosis in (ank/ank) mice is an autosomal recessive form of joint destruction characterized by pathological mineralization in the articular surfaces and synovium [11]. This 'loss of function' mutation in the Ank gene increased iPPi concentration while reducing ePPi concentration in (ank/ank) mouse fibroblasts [10], and these alterations were reversed by overexpression of wild-type Ank. This correcting effect of Ank was blocked by probenecid, a general inhibitor of organic anion transport [10], which was also shown to inhibit transforming growth factor-beta-1 (TGF-β1)-induced inorganic pyrophosphate (PPi) elaboration by chondrocytes [12]. These data indicate an important role for ANK in the regulation of PPi export.
Finally, accumulation of ePPi in the extracellular milieu could also result from its reduced degradation in the pericellular matrix. Therefore, one must keep in mind that alkaline phosphatase (APase), also known as tissue-nonspecific alkaline phosphatase (TNAP), which is very abundant in chondrocytes adjacent to subchondral bone [13], can hydrolize ePPi into two molecules of extracellular inorganic phosphate. CPPD deposition is therefore highly dependent on the interplay among PC-1, ANK, and TNAP, which tightly regulate the balance between ePPi production and ePPi degradation.
TGF-β1 was shown to be the major growth factor that elevated the production of ePPi by normal chondrocytes [6]. Moreover, it was demonstrated that chondrocyte responsiveness to TGF-β1 increased with aging [14] and that TGF-β1 stimulated ePPi production by articular chondrocytes significantly more in old patients than in younger subjects [15]. These effects were closely related to the occurrence of sporadic chondrocalcinosis [16]. Previous studies showed that Ank mRNA level was higher in human chondrocytes exposed to TGF-β1 than in controls [17], as was also the case for murine cartilage and bone [18]. TGF-β1 was shown to induce two major signaling pathways, referred to as TGF-β1 Smad-dependent [19] or Smad-independent [20] signaling, in many cell types. However, intracellular pathways involved in the regulation of the Ank gene are not well documented in chondrocytes, and the contribution of ANK relative to other ePPi-regulating proteins remains unclear. Therefore, the identification of the signaling pathway implicated in Ank regulation and subsequent ePPi production by TGF-β1 warrants interest and could lead to insights in the therapy of sporadic chondrocalcinosis.
The present work aimed to investigate the molecular mechanisms underlying the induction of the Ank gene by TGF-β1 and to evaluate the relative contribution of ANK to ePPi production in chondrocytes. To that end, we characterized the kinetics of expression of Ank, PC-1, and TNAP in response to TGF-β1. Then, using small interfering RNA (siRNA), we evaluated the respective contributions of Ank and PC-1 in ePPi production. Finally, using selective kinase inhibitors and a dominant-negative/overexpression plasmid strategy, we distinguished the Smad from the non-Smad signaling events downstream of TGF-β1.
Materials and methods
Chondrocyte isolation and culture
Normal articular cartilage was obtained from 6-week-old male Wistar rats (130 to 150 g) killed under dissociative anesthesia (ketamine [Rhône-Mérieux, Lyon, France] and acepromazine [Sanofi Santé Animale, Libourne, France]) in accordance with local ethics committee and national animal care guidelines. Articular cartilage pieces were collected aseptically by joint surgery and were dissected from femoral head caps, and chondrocytes were obtained by sequential digestion with pronase and collagenase B (Roche Diagnostics, Meylan, France) as described previously [21]. Cells were washed twice in phosphate-buffered saline (PBS) and cultured to confluence in 75-cm2 flasks at 37°C in a humidified atmosphere containing 5% CO2. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM)/F-12 supplemented with L-glutamine (2 mM), gentamicin (50 μg/mL), amphotericin B (0.5 μg/mL), and heat-inactivated fetal calf serum (FCS) (10%) (Invitrogen Corporation, Cergy Pontoise, France). All experiments reported here were performed with first-passage chondrocytes plated at 4 × 105 cells per well in six-well plates.
Chemicals
All chemical reagents, including para-nitrophenylthymidine 5'-monophosphate (p-NPTMP), para-nitrophenyl phosphate (p-NPP), and para-nitrophenol (p-NP), were obtained from Sigma-Aldrich (Saint-Quentin Fallavier, France) unless otherwise indicated. Chemical kinase inhibitors were purchased from Calbiochem (now part of EMD Biosciences, Inc., San Diego, CA, USA): RcAMP (a protein kinase A [PKA] inhibitor), calphostin C (a general inhibitor of protein kinase C [PKC]), rottlerin (an inhibitor of PKCδ) or Gö6976 (an inhibitor of Ca2+-dependent PKCα and PKCβI isoenzymes), SB203580 (a p38-mitogen-activated protein kinase [MAPK] inhibitor), or PD98059 (a MAPK extracellular signal-regulated kinase [ERK] kinase 1 [MEK-1] inhibitor).
Study design
First, we controlled the cell phenotype by measuring the expression level of specific chondrocyte markers (type I, II, and X collagens as well as aggrecan) by real-time quantitative polymerase chain reaction (PCR). Second, we studied the kinetics of expression of Ank, PC-1, and TNAP in response to TGF-β1 (PeproTech France, Neuilly-sur-Seine, France) at the mRNA and protein levels. To that end, chondrocytes were incubated for 1, 3, 6, 12, 24, or 48 hours in the presence or absence of 10 ng/mL of TGF-β1 (prepared from a stock solution at 10 μg/mL in 2 mM citric acid containing 2 mg/mL bovine serum albumin) in DMEM/F-12 containing a final concentration of 1% FCS. Third, to investigate the respective contributions of Ank and PC-1 in ePPi production, we compared the response with TGF-β1 in chondrocytes transfected with 50 nM of siRNA for each gene. Fourth, to investigate signaling pathways implicated in Ank induction by TGF-β1, we performed the kinetics of activation (10 to 180 minutes) of PKC, MAPK, and Smad in cells stimulated with 10 ng/mL of TGF-β1. The contribution of each signaling pathway was assessed by pretreating chondrocytes for 1 hour with the following inhibitors before exposure to 10 ng/mL of TGF-β1 (12 hours): 10 μM of RcAMP (PKA), 1 μM of calphostin C, 5 μM of rottlerin or 5 μM of Gö6976 (PKC), 10 μM of SB203580 (p38-MAPK), or 10 μM of PD98059 (MEK-1). These final concentrations were chosen after preliminary experiments that demonstrated that inhibitors were active (Western blot assessing the phosphorylation of the concerned signaling pathway; data not shown) and not cytotoxic in the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) assay (data not shown). For these experiments, all inhibitors (MAPK, PKA, and PKC) were dissolved in dimethyl sulfoxide (0.1% final concentration). In the last set of experiments, Ank expression was studied in chondrocytes electroporated (Nucleofector®; amaxa AG, Cologne, Germany) with Ras, Raf-1 (wild-type, constitutively active, or dominant-negative forms) (Clontech-Takara Bio Europe, Saint-Germain-en-Laye, France) or wild-type Smad 7 (Addgene plasmid 11733 [22]) overexpressing plasmids before stimulation or not with 10 ng/mL of TGF-β1.
RNA extraction and reverse transcription-polymerase chain reaction analysis
Total RNA from cultured chondrocytes was isolated by the acidified guanidinium isothiocyanate method, using TRIZOL® reagent (Sigma-Aldrich). Two micrograms of total RNA was reverse-transcribed for 90 minutes at 37°C in a 20-μL reaction mixture containing 2.5 mM dNTPs, 5 μM random hexamer primers, 1.5 mM MgCl2, and 200 U Moloney murine leukemia virus reverse transcriptase (Sigma-Aldrich).
Real-time quantitative polymerase chain reaction
To quantify aggrecan, Ank, L27, PC-1, TNAP, type IA2, II, or X collagens, and S29 mRNA expression, a real-time quantitative PCR was performed using Lightcycler® (Roche Diagnostics) technology. PCR was performed with SYBR green master mix system (Qiagen S.A., Courtaboeuf, France). The gene-specific primer pairs are described in Table 1. Melting curve was performed to determine the melting temperature of each PCR product, as a control of its specificity, and after amplification, the product size was checked on a 2% agarose gel stained with ethidium bromide (0.5 μg/mL). Analyses depended upon a threshold cycle (Ct) that corresponded to the first clearly detectable increase in fluorescence secondary to SYBR green incorporation into double-stranded DNA. Briefly, the Ct was converted into picograms of DNA using calibration curves made of serial dilutions of known amounts of corresponding purified PCR products. Each run included positive standards and negative reaction controls. The mRNA levels of the gene of interest and of the housekeeping gene S29 were determined in parallel for each sample, and results were expressed as the ratio of mRNA level of each gene of interest over the S29 gene.
Table 1.
Gene-specific primer pairs used in real-time quantitative polymerase chain reaction
| Gene | Sense | Antisense | Amplicon length (base pair) | GenBank accession number |
| Aggrecan | 5'-ACA CCC CTA CCC TTG CTT CT-3' | 5'-AAA GTG TCC AAG GCA TCC AC-3' | 124 | NM_022190 |
| Ank | 5'-CAA GAG AGA CAG GGC CAA AG-3' | 5'-AAG GCA GCG AGA TAC AGG AA-3' | 173 | NM_053714 |
| L27 | 5'-TCC TGG CTG GAC GCT ACT C-3' | 5'-CCA CAG AGT ACC TTG TGG GC-3' | 227 | NM_022514 |
| PC-1 | 5'-TAT GCC CAA GAA AGG AAT GG-3' | 5'-GCA GCT GGT AAG CAC AAT GA-3' | 165 | NM_053535 |
| S29 | 5'-AAG ATG GGT CAC CAG CAG CTC TAC TG-3' | 5'-AGA CGC GGC AAG AGC GAG AA-3' | 67 | NM_012876 |
| TNAP | 5'-GAA CGT CAA TTA ACG GCT GA-3' | 5'-CAG ATG GGT GGG AAG AGG T-3' | 50 | NM_013059 |
| Type IA2 collagen | 5'-TTG ACC CTA ACC AAG GAT GC-3' | 5'-CAC CCC TTC TGC GTT GTA TT-3' | 197 | NM_053356 |
| Type II collagen | 5'-TCC CTC TGG TTC TGA TGG TC-3' | 5'-CTC TGT CTC CAG ATG CAC CA-3' | 161 | NM_012929 |
| Type X collagen | 5'-ATA TCC TGG GGA TCC AGG TC-3' | 5'-TGG GTC ACC CTT AGA TCC AG-3' | 241 | AJ131848 |
TNAP, tissue-nonspecific alkaline phosphatase.
Western blot analysis
Rat chondrocytes stimulated or not with TGF-β1 were harvested and lysed in 1× Laemmli buffer (2% SDS, 10% glycerol, 5% 2-β mercaptoethanol, 0.002% bromophenol blue, and 125 mM Tris HCl [pH 6.8]). Protein samples were run on SDS-polyacrylamide gels (10%) and transferred onto a polyvinylidene fluoride membrane (Immobilon; Sigma-Aldrich) as previously described [23]. After 1 hour in blocking buffer (Amersham Biosciences, now part of GE Healthcare, Little Chalfont, Buckinghamshire, UK), membranes were incubated overnight at 4°C with primary antibodies. ANK protein level was determined using rabbit antiserum Ab3 (1:5,000), kindly provided by David Kingsley, Stanford University School of Medicine, Stanford, CA, USA. PC-1 protein level was evaluated using antiserum (1:500) (designed by Eurogentec S.A. [Seraing, Belgium] using a keyhole limpet hemocyanin-coupled peptide of the following sequence: NH2-Glu-Arg-Asp-Gly-Glu-Gln-Ala-Gly-Gln-Gly-Pro-Arg-His-Gly-Pro-Cys-COOH) and a polyclonal antibody against β-actin (1:4,000) (Sigma-Aldrich). In signaling pathway experiments, incubation was carried out with anti-phospho-ERK 1/2, anti-phospho-p38-MAPK, anti-phospho-pan-PKC, and anti-phospho-Smad 3/1 (each at 1:500) (Cell Signaling Technology, Inc., Danvers, MA, USA) or β-actin (1:4,000). After three washings with Tris-buffered saline (TBS)-Tween, the blot was incubated with an anti-rabbit immunoglobulin G conjugated with horseradish peroxidase (Cell Signaling Technology, Inc.) diluted at 1:2,000 in blocking buffer for 1 hour at room temperature. After four washings with TBS-Tween, protein bands were detected by chemiluminescence with the Phototope Detection system (Cell Signaling Technology, Inc.) according to the manufacturer's recommendations. The band intensities were quantified by densitometry with a computerized image processing system (Geldoc 2000®; Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Nucleotide pyrophosphatase phosphodiesterase and alkaline phosphatase activity
Rat chondrocytes stimulated or not with TGF-β1 were harvested and lysed in a buffer containing 1% triton X-100, 1.6 mM MgCl2, and 0.2 M Tris Base (pH 8.1). Total protein extract (quantified by bicinchonic acid assay) was incubated for 15 minutes with 1 μmol of p-NPTMP for nucleotide pyrophosphatase phosphodiesterase (NPPase) activity or for 2.5 hours with 5 μmol of p-NPP for APase activity; these enzymatic activities both generate p-NP. At the end of incubation, the reaction was stopped by adding exactly 10 μmol of EDTA (ethylenediaminetetraacetic acid) and 200 μmol of NaOH and the absorbance was read at 410 nm. The standard concentrations, ranging from 0 to 0.2 mM p-NP, were included in each assay. Results were expressed as units per milligram of total cell proteins, which were quantified by bicinchonic acid assay [24].
Radiometric assay for extracellular inorganic pyrophosphate
ePPi levels were measured using the differential adsorption of UDP-(6-3H) glucose (GE Healthcare), and its reaction product 6-phospho-(6-3H) gluconate on activated charcoal, as previously described [25]. The standard concentrations, ranging from 10 to 400 pmol of PPi, were included in each assay. After adsorption of the reaction mixture on charcoal, and centrifugation at 14,000 rpm for 10 minutes, 100 μL of the supernatant was removed carefully and counted for radioactivity in 5 mL of Bio-Safe II (Research Products International Corp, Mt. Prospect, IL, USA). Results were expressed as picomole of ePPi per microgram of total cell proteins.
Silencing experiments with small interfering RNA
siRNA sequences (designed by Eurogentec S.A.) were Ank sense 5'-CUGGCCAACACGAACAACA-3' and antisense 5'-UGUUGUUCGUGUUGGCCAG-3' and PC-1 sense 5'-GAGGAUGUUUACUCUAUGA-3' and antisense 5'-UCAUAGAGUAAACAUCCUC-3' and were used at final concentrations of 50 nM. Transfections were carried out with one or the other siRNA using X-treme reagent (Roche Diagnostics). Briefly, siRNA and X-treme reagent were diluted separately in serum-free medium, and then diluted X-treme reagent was added to siRNA. After a short incubation at room temperature, cells were washed with PBS and incubated for 4 hours with siRNA-X-treme mix. After this time, medium containing 2% FCS was added to this mix. Stimulations with TGF-β1 (10 ng/mL) were performed the following day. L27 mRNA expression was used as a negative control to check for the specificity of siRNA effects.
Plasmid electroporation
Chondrocytes were electroporated with plasmids encoding for Ras, Raf-1 (wild-type, constitutively active, or dominant-negative forms) or wild-type Smad 7 (overexpressing plasmid) using Human Chondrocyte Nucleofector® Kit (amaxa AG) according to the manufacturer's protocol. Briefly, trypsinized chondrocytes (1 × 106 cells) were gently mixed with 6 μg of either plasmid and then were electroporated using Nucleofector® program U-28. Immediately after transfection, cells were split equally into three wells containing 20% FCS-DMEM/F-12 culture medium and were left to recover for 24 hours. Cells were then stimulated or not with 10 ng/mL of TGF-β1 for 12 hours before mRNA (Ank, aggrecan) or 15 minutes before protein (ERK 1/2) extraction. Plasmid pmaxGFP™ (amaxa AG), encoding a green fluorescent protein, was used to determine the transfection efficiency.
Statistical analysis
Results were expressed as the mean ± standard deviation of at least three independent assays. Comparisons were made by analysis of variance, followed by Fisher t post hoc test using Statview™ 5.0 software (SAS Institute Inc., Cary, NC, USA). A p value of less than 0.05 was considered significant.
Results
Effect of TGF-β1 on the kinetics of proteins regulating inorganic pyrophosphate metabolism in chondrocytes
Our preliminary experiments confirmed that chondrocytes strongly expressed the cartilage-specific markers aggrecan and type II collagen, whereas the expression of type IA2 and X collagens was not detected (Figure 1a). This confirmed the mature phenotype of the articular chondrocytes used throughout the study, all the more so considering that experiments were carried out with first-passage cells.
Figure 1.
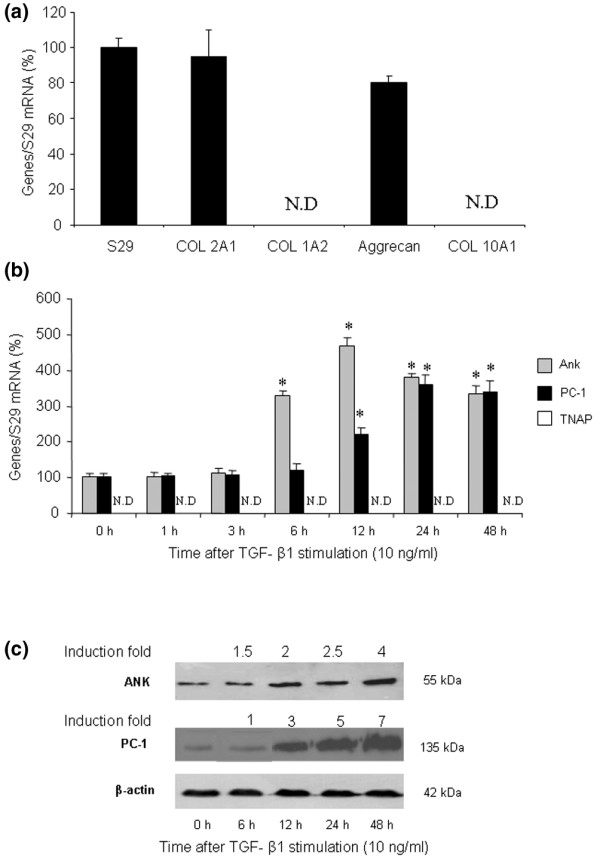
Effect of transforming growth factor-beta-1 (TGF-β1) on proteins regulating inorganic pyrophosphate metabolism. (a) Phenotypic characterization of chondrocytes. Total RNA was extracted from untreated rat chondrocytes and subjected to real-time polymerase chain reaction (PCR) analysis. The relative abundance of gene mRNAs was normalized to that of S29 mRNA. Results are presented in histograms as mean percentages (± standard deviation [SD]) over S29 value. (b) Effect of TGF-β1 on Ank, PC-1, and TNAP mRNA levels. Total RNA was extracted from rat chondrocytes exposed to 10 ng/mL of TGF-β1 from 1 to 48 hours and subjected to real-time PCR analysis. The relative abundance of gene mRNAs was normalized to that of S29 mRNA. Results are expressed as mean percentages (± SD) over control values. Statistically significant differences from the control are indicated as *p < 0.05. (c) Effect of TGF-β1 on ANK or PC-1 protein levels. Total proteins were extracted from rat chondrocytes exposed to 10 ng/mL of TGF-β1 from 6 to 48 hours and subjected to Western blotting using polyclonal anti-ANK and anti-PC-1 antibody. The protein band intensities were quantified by densitometry from enhanced chemiluminescence immunoblots. The relative abundance of these proteins was normalized to that of β-actin protein and expressed as induction folds over control value. N.D., not detected; TNAP, tissue-nonspecific alkaline phosphatase.
Then, we examined the time course of Ank, PC-1, and TNAP mRNA expression in TGF-β1-stimulated chondrocytes (Figure 1b). Ank was upregulated from 6 hours and reached a peak value of 4.5-fold at 12 hours after TGF-β1 exposure. In these conditions, PC-1 was induced approximately 2-fold after 12 hours and reached a maximal induction of approximately 3.5-fold after 24 hours of stimulation with TGF-β1. In contrast, we failed to detect any expression of TNAP in resting chondrocytes or after stimulation with TGF-β1.
Western blotting indicated that ANK protein was induced from 6 hours after TGF-β1 challenge, whereas PC-1 was upregulated after 12 hours (Figure 1c). Taken together, these data demonstrated that Ank was induced by TGF-β1 more rapidly than PC-1 in chondrocytes.
Effect of TGF-β1 on extracellular inorganic pyrophosphate
As shown in Figure 2a, ePPi level increased by 3-fold after 6 hours of stimulation with TGF-β1 and reached a plateau from 24 hours (5-fold). In these experimental conditions, TGF-β1 stimulated NPPase activity by 5-fold at 24 hours (Figure 2b). However, we failed to detect any APase activity in our experimental conditions, which is consistent with the lack of expression of type X collagen. These data suggested that TGF-β1 increased ePPi levels by activating ecto-enzymes and not by reducing TNAP activity, which remained undetectable.
Figure 2.
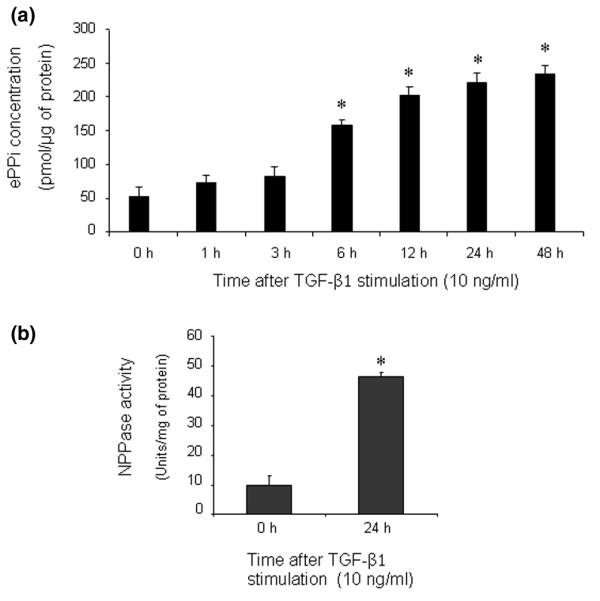
Effect of transforming growth factor-beta-1 (TGF-β1) on extracellular inorganic pyrophosphate (ePPi) and nucleotide pyrophosphatase phosphodiesterase (NPPase) activity. (a) Kinetics of ePPi levels in culture supernatant of rat chondrocytes stimulated with TGF-β1 (10 ng/mL). ePPi was assayed radiometrically and normalized to the amount of total cell proteins (n = 6). Data are expressed as mean (± standard deviation [SD]) in picomoles per microgram of protein. (b) NPPase activity in cultured rat chondrocytes stimulated with 10 ng/mL of TGF-β1. Enzyme activity was normalized to the amount of total cell proteins (n = 3). Results are expressed as mean (± SD) in micromoles of paranitrophenol per minute per milligram of protein. Statistically significant differences from the control are indicated as *p < 0.05.
Respective contributions of Ank and PC-1 to TGF-β1-induced increase in extracellular inorganic pyrophosphate levels in chondrocytes
The siRNA technology was used to investigate the respective contributions of Ank and PC-1 on TGF-β1-induced production of ePPi. Control experiments showed that siRNAs were efficient, as they reduced mRNA level by more than 80% in basal conditions (Figure 3a,b) and diminished the stimulating effect of TGF-β1 below control level (Figure 3a,b) at the time of maximal gene expression. No effect was observed on L27 mRNA level in either condition (Figure 3a,b), confirming that the concentration of 50 nM of siRNA was gene-specific for chondrocytes. When ePPi levels were measured in culture supernatant of chondrocytes transfected with siRNA, inhibition of PC-1 was ineffective on basal ePPi level and accounted for only a 16% decrease in ePPi level in TGF-β1-stimulated cells (Figure 3c). In contrast, transfection with Ank siRNA reduced the basal ePPi level by 33% and reduced the ePPi level by 60% in TGF-β1-stimulated cells (Figure 3c). These data demonstrated that Ank played a major role compared with PC-1 in the regulation of ePPi level in resting and TGF-β1-stimulated chondrocytes.
Figure 3.

Respective contributions of Ank and PC-1 to transforming growth factor-beta-1 (TGF-β1)-induced increase in extracellular inorganic pyrophosphate (ePPi) production. Effect of small interfering RNA (siRNA) on Ank (a) and PC-1 (b) mRNA levels. Rat chondrocytes were transfected with siRNA 24 hours before TGF-β1 stimulation. Total RNA was extracted from rat chondrocytes exposed to 10 ng/mL of TGF-β1 for 12 hours (Ank) (a) or 24 hours (PC-1) (b) and then subjected to real-time polymerase chain reaction analysis. The level of Ank, PC-1, and L27 mRNAs was normalized to that of S29 mRNA and expressed as mean percentages (± SD) over control values. (c) Effect of Ank or PC-1 siRNA on ePPi levels. Shown are levels of ePPi in culture supernatant of rat chondrocytes transfected with siRNA and then stimulated for 12 or 24 hours with 10 ng/mL of TGF-β1. ePPi levels were normalized to the amount of total cell proteins (n = 6) and are expressed as mean (± SD) in picomoles per microgram of protein. Statistically significant differences from the control are indicated as *p < 0.05 and from TGF-β1-treated cells as #p < 0.05.
Identification of TGF-β1-induced signaling pathways contributing to the regulation of Ank gene
As shown in Figure 4a, Western blotting revealed that TGF-β1 induced the phosphorylation of p38-MAPK and PKC as early as 10 minutes after stimulation. A major activation of ERK 1/2 was also observed from 10 to 15 minutes after TGF-β1 challenge, whereas a strong phosphorylation of Smad 3 was noted at 30 and 60 minutes. In our experimental conditions, the corresponding non-phosphorylated proteins remained unchanged (data not shown).
Figure 4.

Identification of several signaling pathways in transforming growth factor-beta-1 (TGF-β1)-induced expression of the Ank gene. (a) Kinetics of signaling events induced by TGF-β1. Total proteins were extracted from rat chondrocytes exposed to 10 ng/mL of TGF-β1 for 10 to 180 minutes and subjected to Western blotting using anti-phospho-ERK 1/2, anti-phospho-p38-MAPK, anti-phospho-pan-PKC, or anti-phospho-Smad 3/1. The relative abundance of these proteins was normalized to that of β-actin protein. (b) Effect of specific signaling inhibitors on TGF-β1-induced expression of Ank mRNA. Total RNA was extracted from rat chondrocytes stimulated with 10 ng/mL of TGF-β1 in the presence of 10 μM RcAMP (PKA inhibitor) or 10 μM SB203580 (a selective p38-MAPK inhibitor) or 10 μM PD98059 (a MEK-1 inhibitor) added 1 hour before TGF-β1. The mRNA level of Ank obtained from real-time polymerase chain reaction analysis was normalized to that of S29 mRNA and is expressed as mean percentages (± standard deviation) over control values from three independent experiments. Statistically significant differences from the control are indicated as *p < 0.05 and from TGF-β1-treated cells as #p < 0.05. ERK, extracellular signal-regulated kinase; MEK-1, mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1; p38-MAPK, p38-mitogen-activated protein kinase; PKA, protein kinase A; PKC, protein kinase C.
When chondrocytes were stimulated by TGF-β1 in the presence of SB203580, a selective p38-MAPK inhibitor, the increase in Ank mRNA level was not affected (Figure 4b). In contrast, PD98059, a selective MEK-1 inhibitor, reduced the stimulatory effect of TGF-β1 by 60% (Figure 4b), supporting a contribution of this MAPK to the induction of the Ank gene. RcAMP, a selective PKA inhibitor, was also ineffective in these experimental conditions (Figure 4b). These data showed that TGF-β1 induced multiple signaling pathways in chondrocytes but that neither p38-MAPK nor PKA contributed to its stimulating effect on the Ank gene.
Contribution of protein kinase C pathway to TGF-β1-induced Ank expression
As shown in Figure 5a, the stimulatory effect of TGF-β1 on Ank expression was suppressed almost completely by calphostin, a general PKC inhibitor. In these conditions, rottlerin, a specific inhibitor of PKCδ, decreased Ank induction by 10% whereas Gö6976, a specific inhibitor of Ca2+-dependent PKCs (PKCα and PKCβI), had a 60% inhibitory effect (Figure 5a). A subsequent dose-ranging study showed that inhibition of TGF-β1-induced expression of Ank by Gö6976 was clearly dose-dependent and reached the control level at 10 μM (Figure 5b). Taken together, these data supported a major role for PKCα and PKCβI isoenzymes in TGF-β1-stimulated expression of Ank.
Figure 5.

Contribution of protein kinase C (PKC) pathway to transforming growth factor-beta-1 (TGF-β1)-induced expression of the Ank gene. (a) Effect of PKC inhibitors on Ank expression. Total RNA was extracted from rat chondrocytes stimulated with 10 ng/mL of TGF-β1 in the presence of 10 μM of calphostin C (general PKC inhibitor) or 5 μM of rottlerin (PKCδ inhibitor) or 5 μM of Gö6976 (PKC α/β1 inhibitor) added 1 hour before TGF-β1. (b) Dose-response study of PKC-dependent induction of Ank by TGF-β1. Total RNA was extracted from rat chondrocytes stimulated with 10 ng/mL of TGF-β1 in the presence of 0, 1, 2.5, 5, or 10 μM of Gö6976 added 1 hour before TGF-β1. The mRNA level of Ank obtained from real-time polymerase chain reaction analysis was normalized to that of S29 mRNA and is expressed as mean percentages (± standard deviation) over control values from three independent experiments. Statistically significant differences from the control are indicated as *p < 0.05 and from TGF-β1-treated cells as #p < 0.05.
Contribution of Ras and Raf-1 pathways to TGF-β1-induced Ank expression
The experiments evaluating transfection efficiency did not show any significant difference among the wild-type, the constitutively active, or the dominant-negative forms of Ras and Raf-1 (data not shown). When chondrocytes were transfected to overexpress constitutively active Ras (Figure 6a) or Raf-1 (Figure 6b), the basal expression of Ank was increased by 2-fold and 1.5-fold, respectively. In these conditions, the induction of Ank by TGF-β1 was increased by 3-fold (Figure 6a) and 4.3-fold, respectively (Figure 6b), which remained similar to cells transfected either with an empty vector or with wild-type Ras or Raf-1 (Figure 6a,b). Transfection with dominant-negative forms of Ras (Figure 6a) or Raf-1 (Figure 6b) reduced both basal (by 75% and 45%, respectively) and TGF-β1-induced (by 3-fold and 2-fold, respectively) Ank expression. These data supported a major role for the Ras/Raf-1 pathway in TGF-β1-induced expression of Ank.
Figure 6.

Effect of Ras/Raf-1 modulation on transforming growth factor-beta-1 (TGF-β1)-induced expression of the Ank gene. Rat chondrocytes were electroporated with empty vector, wild-type, constitutively active, or dominant-negative plasmids for Ras (a) or Raf-1 (b) (2 μg/well of six-well plate) before stimulation with 10 ng/mL of TGF-β1 for 12 hours. Total RNA was extracted and subjected to real-time polymerase chain reaction analysis. The mRNA level of Ank was normalized to that of S29 mRNA and is expressed as mean percentages (± standard deviation) over control values from three independent experiments. Statistically significant differences from the control are indicated as *p < 0.05 and from TGF-β1-treated cells as #p < 0.05. (c) Effect of TGF-β1 on extracellular signal-regulated kinase (ERK) 1/2 phosphorylation in electroporated cells with wild-type, constitutively active, or dominant-negative plasmids for Ras (2 μg/well of six-well plate). Total proteins were extracted from rat chondrocytes exposed to 10 ng/mL of TGF-β1 for 15 minutes and subjected to Western blotting using anti-phospho- and anti-total-ERK 1/2 antibodies. The relative abundance of these proteins was normalized to that of β-actin protein.
Western blot analysis of ERK 1/2 phosphoproteins showed an activation of ERK 1/2 pathway by TGF-β1 in cells transfected with wild-type Ras (Figure 6c). More importantly, the phosphorylation of ERK 1/2 was suppressed almost completely in cells electroporated with a dominant-negative form of Ras, whereas overexpression of constitutively active Ras raised both basal and TGF-β1-induced levels of ERK 1/2 phosphorylation (Figure 6c). These results, similar to those obtained with equivalent Raf-1 constructs, demonstrated that activation of ERK 1/2 by TGF-β1 involved the Ras/Raf-1 pathway in chondrocytes.
Induction of Ank expression by TGF-β1 is a Smad-independent event
In chondrocytes transfected to overexpress wild-type Smad 7, an inhibitory Smad, control experiments showed that the basal expression of aggrecan, chosen as a specific Smad-dependent gene in chondrocytes, was reduced marginally (Figure 7a). In contrast, the stimulating effect of TGF-β1 on aggrecan was abolished, thus demonstrating the efficiency of the construct (Figure 7a). However, neither the basal expression of Ank nor its induction by TGF-β1 was affected by overexpression of Smad 7 in chondrocytes (Figure 7b). These data demonstrated that the Smad pathway did not play a major role in the induction of the Ank gene by TGF-β1.
Figure 7.
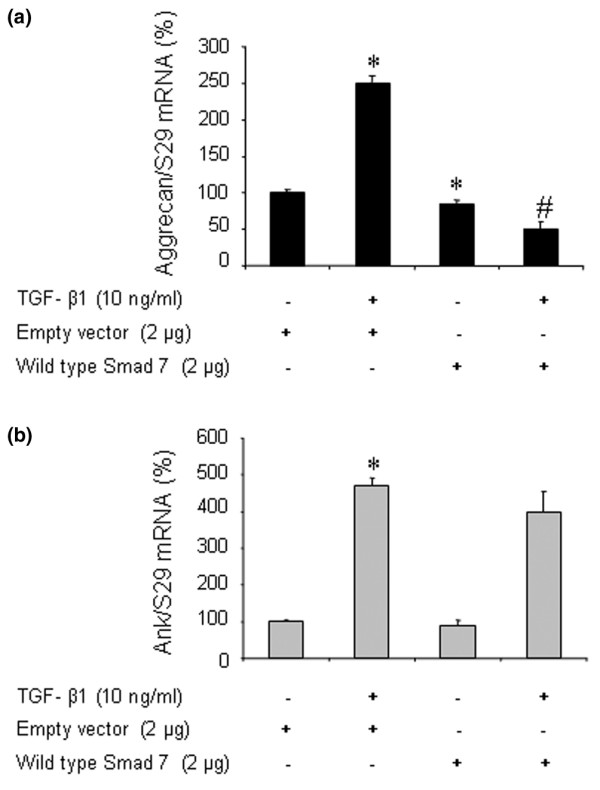
Effect of Smad 7 overexpression on transforming growth factor-beta-1 (TGF-β1)-induced responses in rat chondrocytes. Rat chondrocytes were electroporated with either empty vector or wild-type Smad 7 overexpressing plasmid (2 μg/well of six-well plate) and then treated for 12 hours with 10 ng/mL of TGF-β1. Total RNA was extracted and subjected to real-time polymerase chain reaction analysis. The mRNA level of aggrecan (a) and Ank (b) was normalized to that of S29 mRNA and is expressed as mean percentages (± standard deviation) over control values from three independent experiments. Statistically significant differences from the control are indicated as *p < 0.05 and from TGF-β1-treated cells as #p < 0.05.
Discussion
Previous studies demonstrated a major contribution of ANK in the regulation of ePPi levels. ANK is a transporter able to export iPPi from the cells and is known to be upregulated in osteoarthritis [17,26]. Moreover, chondrocytes and cartilage extracts from patients with CPPD disease express high levels of Ank mRNA [17]. Besides, chondrocytes treated with TGF-β1 generated more ePPi than normal chondrocytes [15], and chondrocyte sensitivity to TGF-β1 increased with aging [14]. These data led us to suppose that Ank could be a major target of TGF-β1 in chondrocytes, likely contributing to its pathophysiological relevance to CPPD deposition.
In our experimental conditions, TGF-β1 increased both Ank mRNA and ANK protein levels, this induction beginning as early as 6 hours. PC-1 mRNA was also induced by TGF-β1 but in a more delayed fashion, whereas expression of TNAP could not be detected. Our data confirmed that TGF-β1 stimulated ePPi production [15] and demonstrated that this was concomitant with the increased expression of PC-1 and Ank. Moreover, similar experiments performed on cartilage explants showed an identical pattern of response, thus validating our monolayer culture system of chondrocytes (F. Cailotto, A. Bianchi, S. Sebillaud, N. Venkatesan, D. Moulin, J-Y. Jouzeau, P. Netter, unpublished data). Based on the higher levels of TGF-β1 found in the synovial fluid of osteoarthritis patients having developed CPPD deposition [27], our findings are in favor of a pathophysiological contribution of TGF-β1-induced dysregulation of Ank and PC-1 in sporadic chondrocalcinosis.
To date, no data are available on the relative contributions of ANK and PC-1 to TGF-β1-induced changes in ePPi production. Therefore, we developed an siRNA technology to clarify the respective roles of ANK and PC-1 since both genes differed rather in their kinetics of expression than in their extent of induction in response to TGF-β1. We demonstrated that, for a comparable knockdown of target genes by siRNA, Ank contributed 4-fold more than PC-1 to TGF-β1-induced increase in ePPi level. The minor effect of PC-1 siRNA on ePPi level could possibly be explained by a stronger basal expression of PC-1 (and, consequently, a residual enzymatic activity even after siRNA transfection) compared with ANK in our cell culture system. However, our data were not biased by the kinetics of induction of each gene since the consequence of their knockdown was studied at the time of their maximal expression in response to TGF-β1. Moreover, we also observed that Ank siRNA diminished TGF-β1-induced ePPi production by 65% at 24 hours (data not shown), confirming that the contribution of Ank was still the most significant at the time of maximal expression of PC-1 mRNA. Our results could be partly explained by the fact that ANK is a multipass transmembrane protein thought to serve either as an anion channel or as a regulator of such a channel [10]. As a consequence, an increase or a decrease of protein level could have a profound and rapid impact on PPi transport across the membrane. PC-1 has been shown to be strongly induced by TGF-β1, as well as NPPase activity in chondrocytes [28], and our data confirmed these observations. However, the contribution of PC-1 to ePPi generation could be estimated to be between 35% to 50% in osteoblasts [29], suggesting that PC-1 is an important contributor but not the major contributor of ePPi generation, which is consistent with our findings in chondrocytes. One important consideration is that, even though knockdown with siRNA was very efficient at the mRNA level, neither the repression of Ank nor that of PC-1 was sufficient to diminish ePPi levels below control level in TGF-β1-stimulated cells. This suggests that, although Ank seemed to play a major role, the generation of ePPi could require a coordinated contribution of Ank and PC-1, as suggested by others [26].
When we studied the regulation of Ank expression by TGF-β1, we demonstrated firstly that ERK 1/2 and p38-MAPKs were activated in our cell culture system. These results agree well with their contribution to the inducing effect of TGF-β1 on aggrecan [30] or TIMP-3 [31] expression in chondrogenic cells. Complementary experiments with specific kinase inhibitors showed that inhibition of MEK-1 by PD98059 reduced the stimulating effect of TGF-β1 by 2-fold. In contrast, the lack of efficiency of SB203580 demonstrated that p38-MAPK was not involved in TGF-β1-induced expression of Ank. Our results are in accordance with other studies reporting that the contribution of the p38-MAPK pathway to the TGF-β1 effect in cartilage was influenced greatly by the species [32] and cell culture system [33].
To investigate further the contribution of the MEK-1 pathway, we modulated the expression of Ras and Raf-1 proteins, which are known to trigger MEK-1 activation in chondrocytes [34]. Our results showed that Ras and, to a lesser extent, Raf-1 were implicated in TGF-β1-induced expression of Ank in chondrocytes, a finding consistent with data demonstrating the activation of Ras and Raf-1 by TGF-β1 in several cell systems [35,36].
Furthermore, we demonstrated that the activation of ERK 1/2 by TGF-β1 was strongly dependent on the activation of Ras and Raf-1 since transfection with dominant-negative forms of these proteins completely inhibited the stimulating effect of TGF-β1. Moreover, constitutively active forms raised both basal and TGF-β1-induced ERK 1/2 phosphorylation. Taken together, these findings confirmed the linkage between activation of Ras/Raf-1, MEK-1, and ERK pathways [34-36] and highlighted their contribution to the induction of Ank by TGF-β1 in chondrocytes.
Ryan and colleagues [37] demonstrated that, in addition to MAPKs, adenylate cyclase activation and generation of cAMP reduced ePPi production in chondrocytes. The PKA pathway was also described to be induced by TGF-β1 in chondrocytes [38,39]. However, using RcAMP as a PKA inhibitor, we failed to demonstrate any contribution of PKA to the induction of Ank by TGF-β1.
In chondrocytes, the production of ePPi was also reported to be stimulated by PKC-dependent pathways [37], which are known to be activated by TGF-β1 [40]. We showed that the stimulation of Ank expression depended on PKC activation but with a variable effect of the inhibitors depending on their specificity for PKC isoenzymes. Thus, when cells were pretreated with calphostin C (a broad PKC inhibitor), a strong effect on Ank mRNA expression was observed whereas rottlerin (a PKCδ inhibitor) was weakly effective. These findings suggested that PKCδ, alongside the other members of the novel PKC family (PKCε, PKCη, and PKCθ [41]), was not implicated in the regulation of Ank expression by TGF-β1. In contrast, we demonstrated that Gö6976 (a Ca2+-dependent PKCα and PKCβI isoenzyme inhibitor) was strongly inhibitory. Since the only difference between novel PKC and conventional PKC (for example, Ca2+-dependent PKCα and PKCβI) activation is the dependence on calcium [41], our findings support a possible regulatory role for calcium in the induction of Ank expression by TGF-β1.
Some genes induced by TGF-β1 are partly or totally regulated by Smad proteins. We demonstrated that, in our experimental conditions, TGF-β1 was able to induce the phosphorylation of Smad 3, known to modulate the expression of chondrocyte-specific genes, such as aggrecan [42]. Smad 7 is a natural inhibitor of the Smad pathway, preventing Smad 3 phosphorylation in the cytosol and therefore suppressing Smad signaling [19]. Scharstuhl and colleagues [42] demonstrated that Smad 7 overexpression was able to counteract TGF-β1-induced expression of aggrecan in chondrocytes, as was the case in our experiments. However, we demonstrated that overexpression of wild-type Smad 7 failed to reduce TGF-β1-induced expression of Ank in chondrocytes, thus demonstrating that it was a non-Smad signaling event.
Taken as a whole, these observations suggest that, contrary to degenerative joint pathologies [43], targeting the non-Smad TGF-β1 signaling events may lead to insights in the field of sporadic chondrocalcinosis since the repression of Ank would translate into reduced ePPi levels in synovial fluid. Thus, selective conventional PKC inhibitors should be powerful agents, considering that some of them, such as Gö6976 [44,45], are currently in clinical development for cardiovascular diseases and cancer. Moreover, probenecid was shown to inhibit Ank function [10] and TGF-β1-induced PPi elaboration by chondrocytes [12]. Therefore, the effect of the combination of probenecid and Gö6976 on CPPD formation could be worthy of study.
Conclusion
To summarize, the present study shows that TGF-β1 increases ePPi levels, with a major contribution of Ank, despite its similar inducing effect on Ank and PC-1. These results are in favor of a main role of Ank as an effector of TGF-β1 in CPPD formation. Induction of Ank is mediated by the dual activation of Ras, Raf-1, and MEK-1/ERK cascade and Ca2+-dependent PKC but is independent of the Smad signaling pathway. Our data support the strong contribution of Ank to the pathogenesis of sporadic chondrocalcinosis, in addition to familial chondrocalcinosis, and identify signaling pathways involved in TGF-β1-induced Ank expression which could lead to new therapies for CPPD disease.
Abbreviations
APase = alkaline phosphatase; CPPD = calcium pyrophosphate dihydrate; Ct = threshold cycle; DMEM = Dulbecco's modified Eagle's medium; ePPi = extracellular inorganic pyrophosphate; ERK = extracellular signal-regulated kinase; FCS = fetal calf serum; iPPi = intracellular inorganic pyrophosphate; MAPK = mitogen-activated protein kinase; MEK-1 = mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1; NPPase = nucleotide pyrophosphatase phosphodiesterase; PBS = phosphate-buffered saline; PCR = polymerase chain reaction; PKA = protein kinase A; PKC = protein kinase C; p-NP = para-nitrophenol; p-NPP = para-nitrophenyl phosphate; p-NPTMP = para-nitrophenylthymidine 5'-monophosphate; PPi = inorganic pyrophosphate; siRNA = small interfering RNA; TBS = Tris-buffered saline; TGF-β1 = transforming growth factor-beta-1; TNAP = tissue-nonspecific alkaline phosphatase.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
FC performed cell culture, RNA extraction, real-time quantitative PCR, ePPi assays, and siRNA assays and was involved in drafting the manuscript. AB carried out the inhibitor experiments, drafted the manuscript, and contributed to the study design. SS performed chondrocyte isolation, cell culture, Western blot analysis, and real-time quantitative PCR. NV participated in revising the manuscript and in the interpretation of data. DM carried out RNA extraction and participated in revising the manuscript. J-YJ and PN contributed to the study design and revised the manuscript for intellectual content. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
This work was supported by grants from the CPRC/PHRC, the CHU Nancy, and the Communauté Urbaine du Grand Nancy.
Contributor Information
Frederic Cailotto, Email: frederic.cailotto@hotmail.fr.
Arnaud Bianchi, Email: arnaud.bianchi@medecine.uhp-nancy.fr.
Sylvie Sebillaud, Email: sylvie.sebillaud@medecine.uhp-nancy.fr.
Narayanan Venkatesan, Email: vnar12@yahoo.com.
David Moulin, Email: moulin@medecine.uhp-nancy.fr.
Jean-Yves Jouzeau, Email: jouzeau@medecine.uhp-nancy.fr.
Patrick Netter, Email: netter@medecine.uhp-nancy.fr.
References
- Liu-Bryan R, Liote F. Monosodium urate and calcium pyrophosphate dihydrate (CPPD) crystals, inflammation, and cellular signaling. Joint Bone Spine. 2005;72:295–302. doi: 10.1016/j.jbspin.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Pendleton A, Johnson MD, Hughes A, Gurley KA, Ho AM, Doherty M, Dixey J, Gillet P, Loeuille D, McGrath R, et al. Mutations in ANKH cause chondrocalcinosis. Am J Hum Genet. 2002;71:933–940. doi: 10.1086/343054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Johnson K, Russell RG, Wordsworth BP, Carr AJ, Terkeltaub RA, Brown MA. Association of sporadic chondrocalcinosis with a – 4-basepair G-to-A transition in the 5'-untranslated region of ANKH that promotes enhanced expression of ANKH protein and excess generation of extracellular inorganic pyrophosphate. Arthritis Rheum. 2005;52:1110–1117. doi: 10.1002/art.20978. [DOI] [PubMed] [Google Scholar]
- Timms AE, Zhang Y, Bradbury L, Wordsworth BP, Brown MA. Investigation of the role of ANKH in ankylosing spondylitis. Arthritis Rheum. 2003;48:2898–2902. doi: 10.1002/art.11258. [DOI] [PubMed] [Google Scholar]
- Reichenberger E, Tiziani V, Watanabe S, Park L, Ueki Y, Santanna C, Baur ST, Shiang R, Grange DK, Beighton P, et al. Autosomal dominant craniometaphyseal dysplasia is caused by mutations in the transmembrane protein ANK. Am J Hum Genet. 2001;68:1321–1326. doi: 10.1086/320612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal AK, McCarty BA, Cheung HS, Ryan LM. A comparison of the effect of transforming growth factor beta 1 on pyrophosphate elaboration from various articular tissues. Arthritis Rheum. 1993;36:539–542. doi: 10.1002/art.1780360415. [DOI] [PubMed] [Google Scholar]
- Rachow JW, Ryan LM. Inorganic pyrophosphate metabolism in arthritis. Rheum Dis Clin North Am. 1988;14:289–302. [PubMed] [Google Scholar]
- Howell DS, Martel-Pelletier J, Pelletier JP, Morales S, Muniz O. NTP pyrophosphohydrolase in human chondrocalcinotic and osteoarthritic cartilage. II. Further studies on histologic and subcellular distribution. Arthritis Rheum. 1984;27:193–199. doi: 10.1002/art.1780270211. [DOI] [PubMed] [Google Scholar]
- Ryan LM, Wortmann RL, Karas B, Lynch MP, McCarty DJ. Pyrophosphohydrolase activity and inorganic pyrophosphate content of cultured human skin fibroblasts. Elevated levels in some patients with calcium pyrophosphate dihydrate deposition disease. J Clin Invest. 1986;77:1689–1693. doi: 10.1172/JCI112487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AM, Johnson MD, Kingsley DM. Role of the mouse ank gene in control of tissue calcification and arthritis. Science. 2000;289:265–270. doi: 10.1126/science.289.5477.265. [DOI] [PubMed] [Google Scholar]
- Sweet HO, Green MC. Progressive ankylosis, a new skeletal mutation in the mouse. J Hered. 1981;72:87–93. doi: 10.1093/oxfordjournals.jhered.a109459. [DOI] [PubMed] [Google Scholar]
- Rosenthal AK, Ryan LM. Probenecid inhibits transforming growth factor-beta 1 induced pyrophosphate elaboration by chondrocytes. J Rheumatol. 1994;21:896–900. [PubMed] [Google Scholar]
- Xu Y, Pritzker KP, Cruz TF. Characterization of chondrocyte alkaline phosphatase as a potential mediator in the dissolution of calcium pyrophosphate dihydrate crystals. J Rheumatol. 1994;21:912–919. [PubMed] [Google Scholar]
- Guerne PA, Blanco F, Kaelin A, Desgeorges A, Lotz M. Growth factor responsiveness of human articular chondrocytes in aging and development. Arthritis Rheum. 1995;38:960–968. doi: 10.1002/art.1780380712. [DOI] [PubMed] [Google Scholar]
- Rosen F, McCabe G, Quach J, Solan J, Terkeltaub R, Seegmiller JE, Lotz M. Differential effects of aging on human chondrocyte responses to transforming growth factor beta: increased pyrophosphate production and decreased cell proliferation. Arthritis Rheum. 1997;40:1275–1281. doi: 10.1002/1529-0131(199707)40:7<1275::AID-ART12>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Ryan LM, McCarty DJ. Calcium pyrophosphate crystal deposition disease, pseudogout, and articular chondrocalcinosis. In: Koopman W, editor. Arthritis and Allied Conditions: A Textbook of Rheumatology. 13. Baltimore: Williams and Wilkins; 1997. pp. 2103–2126. [Google Scholar]
- Hirose J, Ryan LM, Masuda I. Up-regulated expression of cartilage intermediate-layer protein and ANK in articular hyaline cartilage from patients with calcium pyrophosphate dihydrate crystal deposition disease. Arthritis Rheum. 2002;46:3218–3229. doi: 10.1002/art.10632. [DOI] [PubMed] [Google Scholar]
- Sohn P, Crowley M, Slattery E, Serra R. Developmental and TGF-beta-mediated regulation of Ank mRNA expression in cartilage and bone. Osteoarthritis Cartilage. 2002;10:482–490. doi: 10.1053/joca.2002.0810. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-beta signal transduction. J Cell Sci. 2001;114:4359–4369. doi: 10.1242/jcs.114.24.4359. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. 2005;118:3573–3584. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- Kuettner KE, Pauli BU, Gall G, Memoli VA, Schenk RK. Synthesis of cartilage matrix by mammalian chondrocytes in vitro. I. Isolation, culture characteristics, and morphology. J Cell Biol. 1982;93:743–750. doi: 10.1083/jcb.93.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, Richardson MA, Topper JN, Gimbrone MA, Jr, Wrana JL, et al. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell. 1997;89:1165–1173. doi: 10.1016/S0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Terkeltaub R, Rosenbach M, Fong F, Goding J. Causal link between nucleotide pyrophosphohydrolase overactivity and increased intracellular inorganic pyrophosphate generation demonstrated by transfection of cultured fibroblasts and osteoblasts with plasma cell membrane glycoprotein-1. Relevance to calcium pyrophosphate dihydrate deposition disease. Arthritis Rheum. 1994;37:934–941. doi: 10.1002/art.1780370624. [DOI] [PubMed] [Google Scholar]
- Johnson K, Terkeltaub R. Upregulated ank expression in osteoarthritis can promote both chondrocyte MMP-13 expression and calcification via chondrocyte extracellular PPi excess. Osteoarthritis Cartilage. 2004;12:321–335. doi: 10.1016/j.joca.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Punzi L, Oliviero F, Ramonda R. Transforming growth factor-beta levels in synovial fluid of osteoarthritis with or without calcium pyrophosphate dihydrate crystals. J Rheumatol. 2003;30:420. author reply 420–421. [PubMed] [Google Scholar]
- Lotz M, Rosen F, McCabe G, Quach J, Blanco F, Dudler J, Solan J, Goding J, Seegmiller JE, Terkeltaub R. Interleukin 1 beta suppresses transforming growth factor-induced inorganic pyrophosphate (PPi) production and expression of the PPi-generating enzyme PC-1 in human chondrocytes. Proc Natl Acad Sci USA. 1995;92:10364–10368. doi: 10.1073/pnas.92.22.10364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessle L, Johnson KA, Anderson HC, Narisawa S, Sali A, Goding JW, Terkeltaub R, Millan JL. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci USA. 2002;99:9445–9449. doi: 10.1073/pnas.142063399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, de Caestecker MP, Yamada Y. Transcriptional cross-talk between Smad, ERK1/2, and p38 mitogen-activated protein kinase pathways regulates transforming growth factor-beta-induced aggrecan gene expression in chondrogenic ATDC5 cells. J Biol Chem. 2001;276:14466–14473. doi: 10.1074/jbc.M005724200. [DOI] [PubMed] [Google Scholar]
- Qureshi HY, Sylvester J, El Mabrouk M, Zafarullah M. TGF-beta-induced expression of tissue inhibitor of metalloproteinases-3 gene in chondrocytes is mediated by extracellular signal-regulated kinase pathway and Sp1 transcription factor. J Cell Physiol. 2005;203:345–352. doi: 10.1002/jcp.20228. [DOI] [PubMed] [Google Scholar]
- Badger AM, Roshak AK, Cook MN, Newman-Tarr TM, Swift BA, Carlson K, Connor JR, Lee JC, Gowen M, Lark MW, et al. Differential effects of SB 24 a selective p38 mitogen-activated protein kinase inhibitor, on IL-1 treated bovine and human cartilage/chondrocyte cultures. Osteoarthritis Cartilage. 2002;8:434–443. doi: 10.1053/joca.1999.0319. [DOI] [PubMed] [Google Scholar]
- Poleni PE, Bianchi A, Etienne S, Koufany M, Sebillaud S, Netter P, Terlain B, Jouzeau JY. Agonists of peroxisome proliferators-activated receptors (PPAR) alpha, beta/delta or gamma reduce transforming growth factor (TGF)-beta-induced proteoglycans' production in chondrocytes. Osteoarthritis Cartilage. 2007;15:493–505. doi: 10.1016/j.joca.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Krejci P, Masri B, Fontaine V, Mekikian PB, Weis M, Prats H, Wilcox WR. Interaction of fibroblast growth factor and C-natriuretic peptide signaling in regulation of chondrocyte proliferation and extracellular matrix homeostasis. J Cell Sci. 2005;118:5089–5100. doi: 10.1242/jcs.02618. [DOI] [PubMed] [Google Scholar]
- Mulder KM, Morris SL. Activation of p21ras by transforming growth factor beta in epithelial cells. J Biol Chem. 1992;267:5029–5031. [PubMed] [Google Scholar]
- Reimann T, Hempel U, Krautwald S, Axmann A, Scheibe R, Seidel D, Wenzel KW. Transforming growth factor-beta1 induces activation of Ras, Raf-1, MEK and MAPK in rat hepatic stellate cells. FEBS Lett. 1997;403:57–60. doi: 10.1016/S0014-5793(97)00024-0. [DOI] [PubMed] [Google Scholar]
- Ryan LM, Kurup IV, Cheung HS. Transduction mechanisms of porcine chondrocyte inorganic pyrophosphate elaboration. Arthritis Rheum. 1999;42:555–560. doi: 10.1002/1529-0131(199904)42:3<555::AID-ANR21>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Rosado E, Schwartz Z, Sylvia VL, Dean DD, Boyan BD. Transforming growth factor-beta1 regulation of growth zone chondrocytes is mediated by multiple interacting pathways. Biochim Biophys Acta. 2002;1590:1–15. doi: 10.1016/S0167-4889(02)00194-5. [DOI] [PubMed] [Google Scholar]
- Li TF, O'Keefe RJ, Chen D. TGF-beta signaling in chondrocytes. Front Biosci. 2005;10:681–688. doi: 10.2741/1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S, DiBattista JA, Sun Y, Li WQ, Zafarullah M. Up-regulation of tissue inhibitor of metalloproteinases-3 gene expression by TGF-beta in articular chondrocytes is mediated by serine/threonine and tyrosine kinases. J Cell Biochem. 1998;70:517–527. doi: 10.1002/(SICI)1097-4644(19980915)70:4<517::AID-JCB8>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Jaken S. Protein kinase C isozymes and substrates. Curr Opin Cell Biol. 1996;8:168–173. doi: 10.1016/S0955-0674(96)80062-7. [DOI] [PubMed] [Google Scholar]
- Scharstuhl A, Diepens R, Lensen J, Vitters E, van Beuningen H, van der Kraan P, van den Berg W. Adenoviral overexpression of Smad-7 and Smad-6 differentially regulates TGF-beta-mediated chondrocyte proliferation and proteoglycan synthesis. Osteoarthritis Cartilage. 2003;11:773–782. doi: 10.1016/S1063-4584(03)00165-1. [DOI] [PubMed] [Google Scholar]
- Blom AB, van der Kraan PM, van den Berg WB. Cytokine targeting in osteoarthritis. Curr Drug Targets. 2007;8:283–292. doi: 10.2174/138945007779940179. [DOI] [PubMed] [Google Scholar]
- Goekjian PG, Jirousek MR. Protein kinase C in the treatment of disease: signal transduction pathways, inhibitors, and agents in development. Curr Med Chem. 1999;6:877–903. [PubMed] [Google Scholar]
- Hanauske AR, Oberschmidt O, Hanauske-Abel H, Lahn MM, Eismann U. Antitumor activity of enzastaurin ( LY317615.HCl) against human cancer cell lines and freshly explanted tumors investigated in vitro soft-agar cloning experiments. Invest New Drugs. 2007;25:205–210. doi: 10.1007/s10637-007-9038-7. [DOI] [PubMed] [Google Scholar]