

Summary
The co-crystal structure of Thermus aquaticus EF-Tu·GDPNP bound to yeast Phe-tRNAPhe reveals that EF-Tu interacts with the tRNA body primarily through contacts with the phosphodiester backbone. Twenty amino acids in the tRNA binding cleft of Thermus thermophilus EF-Tu were each mutated to structurally conservative alternatives and the affinities of the mutant proteins to yeast Phe-tRNAPhe determined. Eleven of the twenty mutations reduced the binding affinity from 4-fold to >100 fold, while the remaining ten had no effect. The thermodynamically important residues were spread over the entire tRNA binding interface, but were concentrated in the region which contacts the tRNA T-stem. Most of the data could be reconciled by considering the crystal structures of both free EF-Tu·GTP and the ternary complex and allowing for small (1.0 Å) movements in the amino acid side chains. Thus, despite the non-physiological crystallization conditions and crystal lattice interactions, the crystal structures reflect the biochemically relevant interaction in solution.
Keywords: indirect readout, tRNA-protein interaction, alanine scanning, protein mutagenesis
Introduction
EF-Tu is a bacterial G protein whose catalytic cycle is coupled to the binding and release of aminoacyl-tRNA (aa-tRNA) during protein synthesis. The GTP bound form of the protein binds all elongator aa-tRNA with similar affinities and the resulting ternary complex binds the ribosomal A/T site in a multistep mechanism 1; 2; 3; 4; 5. Upon correct matching of the codon with the anticodon, GTP hydrolysis occurs and the aa-tRNA is released from EF-Tu·GDP and enters the ribosomal A site 6. Thus, binding of aa-tRNA to EF-Tu and its subsequent release is central to the mechanism of protein synthesis. The 405 amino acids of T. aquaticus EF-Tu are folded into an N-terminal GTP binding domain and two domains adopting β-barrel motifs. The co-crystal structures of T. aquaticus EF-Tu·GDPNP complexed with either yeast Phe-tRNAPhe or E. coli Cys-tRNACys reveal that the protein interacts with both the esterified amino acid and the upper part of the extended helix formed by the A and T stems of the tRNA tertiary structure 7; 8. An analysis of the binding of a series of E. coli tRNAs misacylated with valine to T. thermophilus EF-Tu·GTP has revealed that different tRNA bodies bind the protein with very different affinities 9. These affinities are arranged in a way that offsets the variable contribution of the esterified amino acid such that correctly aminoacylated elongator tRNAs bind EF-Tu uniformly 10. Thus, EF-Tu shows considerable specificity for different tRNA bodies 5; 9.
This work focuses on mutagenizing the tRNA binding surface of EF-Tu with the goal of understanding how the tight binding of aminoacyl-tRNAs is achieved. The X-ray co-crystal structure of yeast Phe-tRNAPhe complexed with T. aquaticus EF-Tu indicates that the side chains or main chain functional groups of at least 15 amino acids could potentially participate in hydrogen bonds or ionic interactions with eight 2′ hydroxyl groups, eight phosphates and a single base on the tRNA. Thus any specificity of EF-Tu for tRNAs would appear to primarily be the result of “indirect readout” of the A and T-stem sequences where subtle differences in the position of the RNA phosphodiester dictate the strength of the interaction between the tRNA body and the protein. However, since very high ionic strengths were used for crystallization, the crystal structure may not accurately reflect the interactions made between EF-Tu and the tRNA body in solution at lower ionic strengths. Thus, it remains possible that the co-crystal structure reflects a binding intermediate and not the final complex that participates in ribosome binding. This is suggested by fluorescent studies that have shown that both the aa-tRNA and EF-Tu undergo rearrangement upon complex formation which is inconsistent with the crystal structures11; 12; 13; 14. An analysis of the binding properties of mutant proteins will provide insight on whether the molecular interactions seen in the co-crystal structure actually reflects thermodynamically relevant interactions of the EF-Tu·tRNA complex in solution.
Similar experiments with other protein-protein and nucleic acid-protein complexes often show that not all of the amino acid side chains that appear to be involved in a contact in a co-crystal structure actually contribute substantially to the net free energy of binding 15; 16; 17; 18. Instead, a limited number of mutagenically sensitive residues define “hot spots” in the binding interface 18. Such thermodynamic “hot spots” are candidates for contributing to the specificity of EF-Tu for different tRNA bodies.
Results
Site Directed Mutagenesis Of T. thermophilus EF-Tu
The crystal structure of T. aquaticus EF-Tu complexed with GDPNP and yeast Phe-tRNAPhe shows that all three domains of the protein interact with the A and T-stems of the tRNA tertiary structure. A total of 20 residues of EF-Tu which appear to interact with the tRNA backbone were chosen for mutagenesis. These included all the amino acids with side chains capable of participating in hydrogen bonds or electrostatic interactions that are within 3.8 Å of the tRNA in the co-crystal structure as well as Arg389 which is more distal, but could potentially reorient to intereact with the tRNA. Not mutated were the amino acids that formed the binding pocket for the amino acid esterified to the end of tRNA. The mutated residues can conveniently be considered to be located in four adjacent regions of the tRNAPhe body which extend from the 3′-terminal adenosine to the closing base pair of the T-loop (Figure 1).
Figure 1.
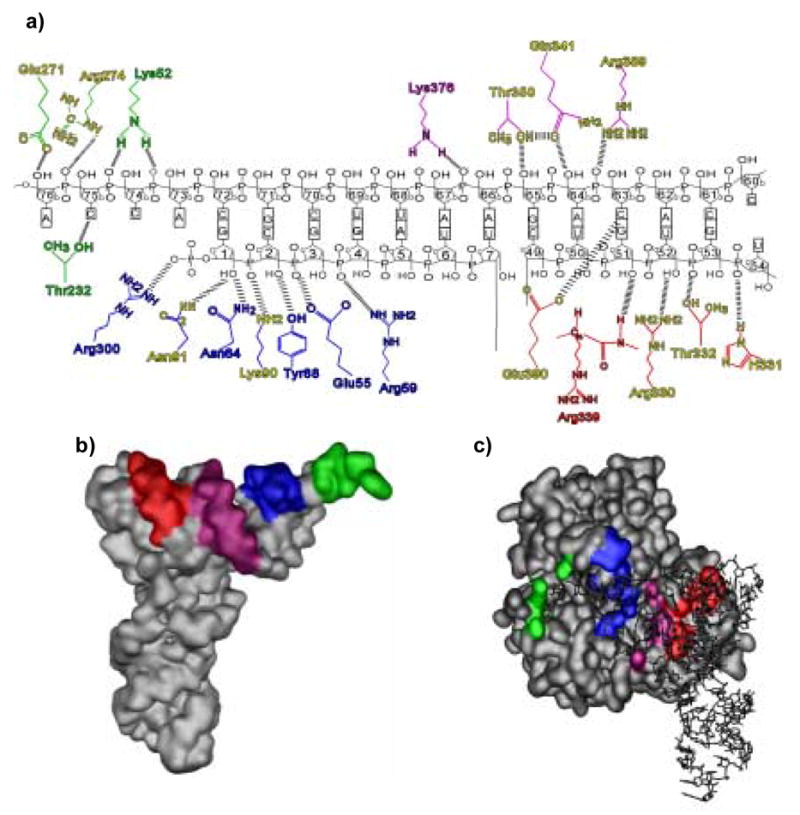
Four regions of EF-Tu interaction with yeast Phe-tRNAPhe. (a) Schematic of T. aquaticus EF-Tu amino acids contacting yeast Phe-tRNAPhe backbone. The hashed lines represent possible interactions between the amino acid and the tRNA up to 7Å. Region 1 contacts the 3′ end (green). Region 2 contacts the 5′ end (blue). Region 3 contacts the junction of the acceptor and T-stems (magenta). Region 4 contacts the T-stem (red). Residues which exhibit a ΔΔG0 >0.5 kcal/mol are highlighted in yellow.(b) Surface representation of yeast Phe-tRNAPhe with regions contacted by EF-Tu highlighted. (c) Surface representation of T. aquaticus EF-Tu with yeast Phe-tRNAPhe contact regions highlighted with the same color code as in (a).
Region 1 contacts the backbone of the conserved 3′ terminal CCA of the tRNA and includes Lys52 in domain I and Thr232, Arg274 and Glu271 in domain II (Figure 2a). Region 2 contacts the backbone of residues 1, 2 and 3 at the 5′ terminus of the tRNA and contains Glu55, Arg59, Asn64, Tyr88, Lys90 and Asn91 in domain I and Arg300 in domain II (Figure 2b). Region 3 contacts nucleotides 64–67 at the junction of the A and T-stems and is comprised of Gln341, Thr350, Lys376 and Arg389 in domain III (Figure 2c). Finally, region 4 contacts nucleotides 51–54 in the T-stem and includes Arg330, His331, Thr332, Arg339 and Glu390 in domain III (Figure 2d). Previous experiments in which single deoxynucleotides were substituted into a variant of yeast Phe-tRNAPhe and tested for EF-Tu binding have established that contacts in all four regions contribute to the total binding energy between the aminoacyl-tRNA and EF-Tu 19. With the exception of Lys52, Asn64, Thr232 and Lys376 all of the mutated amino acids were greater than 80% identical among 188 complete bacterial EF-Tu sequences in the UniProt database and most were greater than 90% identical 20. The high percentage of identity of the mutagenized residues suggests that these positions are important for initial aa-tRNA binding or a later step in the EF-Tu pathway. Mutations were made in T. thermophilus EF-Tu which differs from the T. aquaticus EF-Tu used for co-crystal structures at 7 surface positions which are all distant from the tRNA binding interface and thus unlikely to affect binding. Isosterically conservative mutations were made to minimize the impact on the local structure of EF-Tu. Amino acids which do not branch at the β-carbon were replaced by alanine, while amino acids which do branch at the side chain β-carbon were replaced by valine. Tyr88 was replaced with phenylalanine. For ease of purification, an N-terminal His6 tag was added and the presence of a downstream TEV protease site permitted in vitro cleavage to restore the native N-terminus of each mutant protein. Each of the mutant proteins was found to express well and were purified to near homogeneity.
Figure 2.
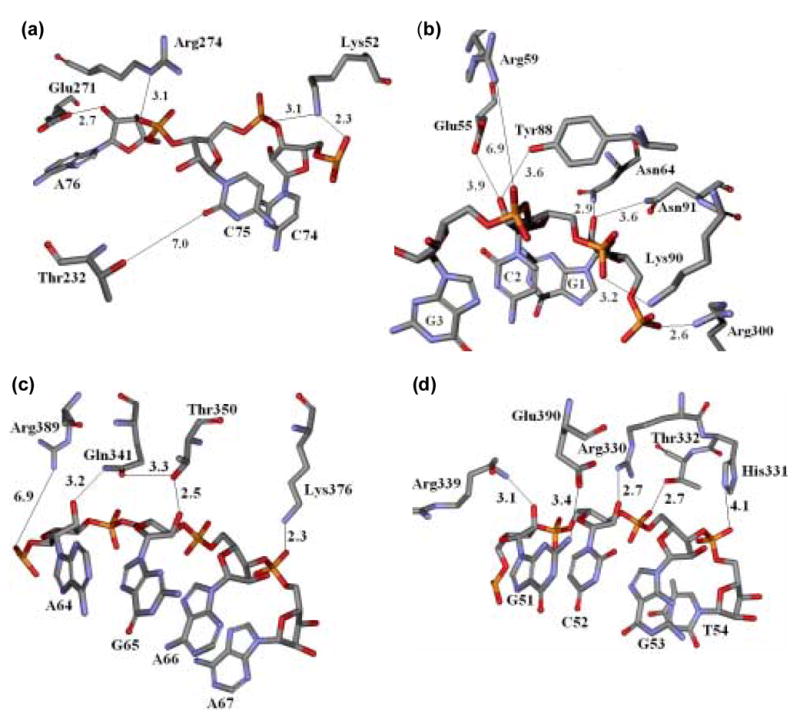
Atomic details of the interactions of the four regions of T. aquaticus EF-Tu with the acceptor and T-stems of yeast Phe-tRNAPhe from the co-crystal structure. The solid black lines represent possible contacting groups of EF-Tu and the tRNA within 7Å. (a) Region 1 interacting with the 3′ end of yeast Phe-tRNAPhe. (b) Region 2 interacting with the 5′ end of yeast Phe-tRNAPhe. (c) Region 3 interacting with the junction of the acceptor and T-stem of yeast Phe-tRNAPhe. (d) Region 4 interacting with the T-stem of yeast Phe-tRNAPhe.
Although all the mutations were made on amino acids that lie on the surface of EF-Tu, it was important to test whether each point mutation could effect the global folding of the protein. To do this, the stoichiometry of GTP binding of a set of mutant protein was measured relative to the native protein. Since the nucleotide binding pocket of EF-Tu is located in domain 1 on the face opposite the aminoacyl-tRNA binding cleft, an assay for GTP binding is a reasonable way to assess whether any of the EF-Tu mutations unexpectedly interfere with the global folding of the protein. The binding of GTP to a set of EF-Tu mutants was measured utilizing a non-enzymatic nucleotide exchange assay in which the EF-Tu·GDP bound form was converted to EF-Tu·GTP in the presence of excess [3H]GTP. All of the tested EF-Tu mutants bound with similar molar ratio of EF-Tu to GTP as the wild type protein, suggesting that none of the tested mutations impaired the overall folding (Table 1).
Effects of T. thermophilus EF-Tu mutations of yeast Phe-tRNAPhe binding
| Mutation | Percent Conserved* | tRNA Contact | % GTP Binding** | KD (nM)measured | koff (min−1) | KD (nM)calculated† | ΔΔG0 (kcal/mol)‡ |
|---|---|---|---|---|---|---|---|
| Native | (100) | 13.7 (±4) | 0.10 (±.03) | 15 (± 7) | |||
| Region 1 | |||||||
| Lys52Ala | 24 | C74 OP1 | 120 (± 43) | 27 (±18) | 0.14 (±0.07) | 20 (±13) | 0.18 |
| Thr232Ala | 46 | C75 O2 | 0.08 (±0.02) | 12 (±3.2) | −0.10 | ||
| Glu271Ala | 100 | A76 2′OH | 75 (±23) | >1.8 | >300 | >1.5 | |
| Arg274Ala | 81 | A76 OP2 | 74 (±23) | 0.33 (±0.17) | 72 (±37) | 0.85 | |
| Region 2 | |||||||
| Glu55Ala | 98 | C2 2′OH | 0.06 (±0.01) | 8.6 (±2) | −0.29 | ||
| Arg59Ala | 100 | G3 OP1 | 0.09 (±0.02) | 13 (±6) | −0.08 | ||
| Asn64Ala | 23 | G1 2′OH | 127 (±19) | 21 (±15) | 0.07 (±0.03) | 10 (±6) | −0.19 |
| Tyr88Phe | 100 | C2 2′OH | 151 (±23) | 13 (±10) | 0.01 (±0.07) | 11 (±5) | −0.16 |
| Lys90Ala | 100 | G1 OP3 | 162 (±44) | 25 (± 9) | 0.23 (±0.08) | 35 (±18) | 0.50 |
| Asn91Ala | 100 | G1 2′OH | 41(±10) | 0.51 (±0.06) | 78 (±30) | 0.91 | |
| Arg300Ala | 100 | G1 OP1 | 90 (±18) | 0.15 (±0.5) | 23 (±11) | 0.24 | |
| Region 3 | |||||||
| Gln341Ala | 100 | A64 2′OH | 102 (±33) | 126 (±57) | 0.64 (±0.07) | 98 (±24) | 1.0 |
| Thr350Val | 99 | G65 2′OH | 129 (±44) | 0.72 (±0.11) | 103 (±28) | 1.1 | |
| Lys376Ala | 12 | A67 OP2 | 84 (± 4) | 0.12 (±0.02) | 18 (±6) | 0.12 | |
| Arg389Ala | 100 | A65 OP2 | 104 (±37) | >1.8 | >300 | >1.5 | |
| Region 4 | |||||||
| Arg330Ala | 99 | U52 2′OH | 84 (±17) | >1.8 | >300 | >1.5 | |
| His331Val | 99 | U54 OP2 | 152 (±9) | 92 (±16) | 0.41 (±0.05) | 62 (±24) | 0.79 |
| Thr332Val | 89 | G53 OP1 | 84 (±52) | 0.62 (±0.07) | 94 (±36) | 1.0 | |
| Arg339Ala | 95 | G51 2′OH | 82 (±20) | 0.12 (±0.03) | 18 (±5) | 0.12 | |
| Glu390Ala | 100 | G51 N2 | n.d. | 0.57 (±0.2) | 86 (±30) | 1.0 | |
Percent conservation of amino acid among 189 bacterial EF-Tu sequences aligned with ClustalW.
Molar ratio of GTP bound relative to wild type EF-Tu.
KD = koff/kon (kon = 6.6×106 M−1 min−1)
ΔΔG0 = −RT ln (KDwt/KDmut)
n.d. = no data
koff & KD determined at pH 7.0, 20 mM MgCl2, 0.5 M NH4Cl
Phe-tRNAPhe Binding To T. thermophilus EF-Tu Mutants
The equilibrium dissociation constants of wild type EF-Tu and six mutants; Lys52Ala, Asn64Ala, Tyr88Phe, Lys90Ala, His331Val, and Gln341Ala were measured directly utilizing a modified RNase protection assay 1; 5. Apparent KD measurements were made by monitoring the fraction of yeast Phe-tRNAPhe that was protected from rapid RNase A digestion as function of EF-Tu·GTP concentration. The use of high specific activity [3′-32P] Phe-tRNAPhe made it possible to keep the aa-tRNA concentration low enough to be sub-saturating throughout the assay (Figure 3a). The apparent KD values varied by no more than two fold in four independent measurements. In order to convert the experimental apparent KD value to actual KD values, it was necessary to determine the fraction of each mutant protein that was capable of binding aa-tRNA by titrating a known concentration of [32P] Phe-tRNAPhe with varying amounts of protein with concentrations well above the KD. The number of moles of protein required to bind all the Phe-tRNAPhe divided by the moles of input protein defined the fraction of the active protein. To accomplish this, Phe-tRNAPhe was added to a final concentration of 300 nM to varying concentrations of EF-Tu·GTP ranging from 10 nM to 6 μM and the fraction of the Phe-tRNAPhe that was protected from RNase digestion was determined (Figure 3b). The ratio of the concentration of the added Phe-tRNAPhe over the concentration of EF-Tu required for complete RNase protection was used to determine the fraction of active EF-Tu. For wild type EF-Tu and the six mutants tested, the fraction of the active protein was found to vary between 10 and 15%. This low activity of aa-tRNA binding to EF-Tu has long been observed but rarely discussed 21; 22; 23. While it is possible that the protein inactivates during purification, perhaps due to aggregation 28, it is more likely that the low activity is the result of post-translational modification of the over-expressed protein. An unknown E. coli protein kinase is known to phosphorylate Thr382 and abolish aa-tRNA binding24; 25; 26. It is also possible that the methylation of Lys56 may modify the aa-tRNA binding activity of the protein26; 27.
Figure 3.
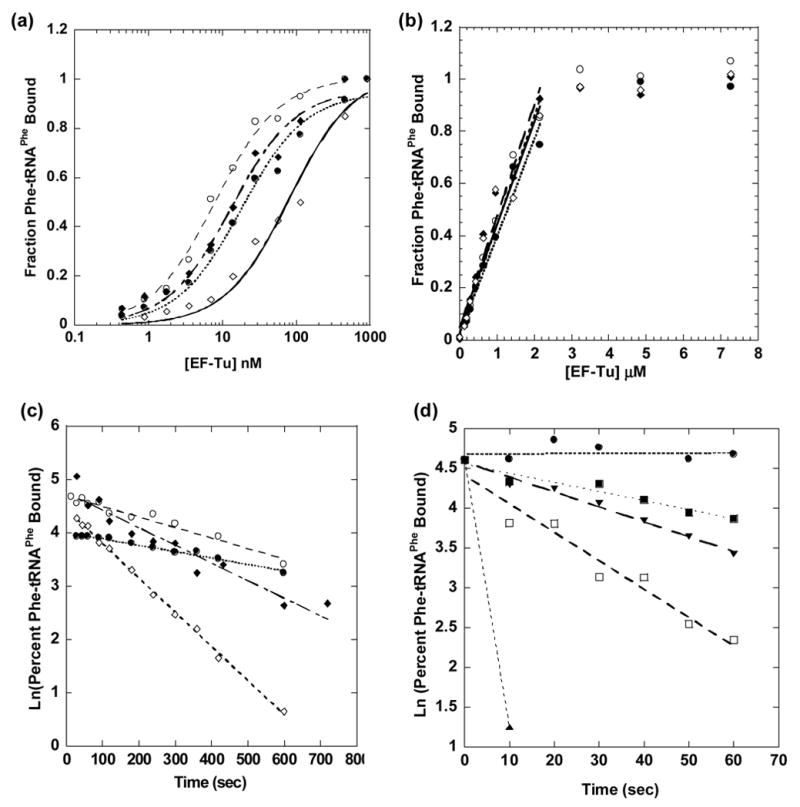
Binding of T. thermophilus EF-Tu mutants to yeast Phe-tRNAPhe in buffer B at 00C. (a) Representative equilibrium binding curves fit to a single binding isotherm (●) wild type EF-Tu, KD=15.6 nM, (◆) K52A, KD= 34 nM, (○) N64A, KD= 33.8 nM, (◇) H331V, KD= 93.6 nM. (b) Representative active site titration curves with 300 nM of Phe-tRNAPhe with the initial slopes for wild type and the same three mutations are fit to a linear equation and the intercept at 1.0 corresponds to 13, 6.3, 8.2, and 7.7 percent active EF-Tu, respectively. (c) Dissociation rate curves fit to single exponential for (●) wild type EF-Tu, koff = 0.0016 s−1, (◆) K52A, koff = 0.0024 s−1, (○) N64A, koff = 0.0021s−1 (◇) H331V, koff = .0068 s−1. (d) Dissociation rate curves of weak binding EF-Tu under tight binding conditions (50 mM NH4Cl, 1 M NH4SO4) fit to single exponential for (●) wild-type EF-Tu koff = .0002 s−1, (▼) E271A, koff = 0.019 s−1, (□) R330A, koff = 0.036 s−1, (■) R389A, koff =0.011 s−1, (▲) No EF-Tu, rate = 0.35 s−1.
In order to more efficiently assay the ability of yeast Phe-tRNAPhe to bind with the EF-Tu mutants, a second version of the RNase protection assay that measures the dissociation rate was employed 5. When measuring the dissociation rate it is not necessary to know the tRNA binding activity of the individual mutant proteins thereby eliminating the need for active site titrations which are required when measuring the equilibrium dissociation constant. The dissociation rate was determined by measuring the fraction of [3H] Phe-tRNAPhe protected as a function of time after the addition of pancreatic RNase A (Figure 3c). The addition of RNase A perturbs the equilibrium of the system by rapidly degrading the free aa-tRNA and preventing re-association with EF-Tu. By plotting the natural log of the fraction of tRNA protected versus time, the dissociation rate was calculated. For the fourteen mutants in which the dissociation rate could be measured, the values for quadruplicate experiments typically varied by less than 20%, but occasionally varied by as much as 50%.
Three of the mutations, Glu271Ala, Arg330Ala and Arg389Ala, were completely defective in yeast Phe-tRNAPhe binding at the conditions tested. This may be due to the loss of a contact which is crucial for aa-tRNA binding or the mutation may have resulted in a significant structural change in the tRNA binding cleft. To examine this further, the dissociation rate of yeast [3H] Phe-tRNAPhe from the three mutant proteins was measured under conditions where EF-Tu binding was considerably tighter due to the reduction of the NH4Cl concentration from 0.5 M to 50 mM and the addition of NH4SO4 to 1 M 29; 30. Under these tight binding conditions, detectable RNase protection was observed for all three mutations, although the dissocation rates were very fast. This suggests that the mutations did not grossly distort the tRNA binding cleft of EF-Tu, but instead disrupted a critically important contact.
Although the dissociation rates are convenient to compare the tRNA binding properties of a set of EF-Tu mutants, they cannot provide thermodynamically useful quantities such as KD and ΔG0. From the experimentally determined KD and koff values of wild type and six mutant EF-Tu, their kon values were calculated to be 6.6×106 +/− 2.4×106 M−1min−1, which is in agreement with published association rates 5; 31. Since the association rate was determined to be constant for the wild type and six mutant EF-Tu, it was assumed that the remainder of the mutant had similar kon values. Thus the KD values were calculated directly from the measured dissociation rate by dividing by an association rate of 6.6×106 M−1min−1. Subsequently, the free energy of binding, ΔG0, was calculated as ΔG0= −RT ln(KD). The change in the free energy of binding resulting from the mutation,ΔΔG0, was defined as ΔΔG0 = −RT ln(KDwt/KDmut).
As summarized in Table 1, the results of the binding experiments showed three categories of effects: ten EF-Tu mutants bound Phe-tRNAPhe similar (−0.3 to 0.3 kcal/mol) to the wild type protein, seven mutants showed ΔΔG0 values between 0.5 and 1.1 kcal/mol and three mutants showed binding which was too weak to be quantified (ΔΔG0 > 1.5 kcal/mol).
Analysis of Individual Mutants
Of the four mutations made in region 1, two had no effect on binding (Thr232 and Lys52) and two did (Arg274 and Glu271). The side chain of Thr232 is 7.0 Å from the nearest tRNA atom, well outside of hydrogen bond distance. Thus, it is not surprising that no change in binding energy was observed for the Thr232Ala mutation. However, the lack of an effect for the Lys52Ala mutation is not easy to understand. In the co-crystal structure, the ε-amino group of Lys52 lies 2.3 and 3.1 Å from phosphate 74 and phosphate 75 respectively (Figure 2a), suggesting that an electrostatic interaction forms that would be expected to contribute to the binding energy. Since Lys52 does not appear to interact with anything in the free EF-Tu·GDPNP crystal structure it is unlikely that any ion pairs are broken upon tRNA binding 32. Thus it is unclear why no binding energy is lost when the Lys52Ala mutation is made.
The replacement of Arg274 with alanine in region 1, resulted in a moderate 0.6 kcal/mol reduction in binding energy compared to the wild-type protein. Since the guanidinium group of Arg274 is 3.1 Å from a non-bridging oxygen of phosphate 76. The observed loss in binding energy is consistent with the disruption of an electrostatic interaction between protein and the tRNA (Figure 2a). The Glu271Ala mutation reduces binding by greater than 1.5 kcal/mol. The corresponding glutamine to alanine mutation in E. coli EF-Tu decreases the binding to yeast Phe-tRNAPhe by a comparable amount 33. Since the carboxylate of Glu271 is within 2.7 Å of the 2′ hydroxyl of ribose 76, the weaker binding could simply be due to the loss of a hydrogen bond (Figure 2a). However, a derivative of yeast Phe-tRNAPhe containing a 2′ deoxy modification of ribose 76 which should also prevent the formation of the hydrogen bond was found to bind EF-Tu with same affinity as the unmodified tRNA 19. Thus, either the 2′ deoxy aa-tRNA has some alternate means to maintain the binding free energy or a different reason for the weaker binding of the Glu271Ala mutation must exist. For example, Pedersen et al. have suggested that the cavity created by the mutation in the E. coli ternary complex may permit the 3′ terminus of the tRNA to be more flexible, reducing the net binding free energy 33. Alternatively, the carboxylate group of Glu271 may actually derive its binding energy by contacting the nearby the nearby (3.4 Å) N7 of A76 instead of the 2′ hydroxyl group. Finally, it is possible that removing Glu271 allows the esterified amino acid to freely migrate between the 2′ and 3′ hydroxyl groups thereby some how weakening its contribution to binding.
Mutation of two of the seven residues within region 2 (Lys90 and Asn91) had a clear effect upon Phe-tRNAPhe binding, while mutation of the remaining five (Glu55, Arg59, Asn64, Tyr88 and Arg300) had no effect. The 0.5 kcal/mol loss in the free energy of binding for the Lys90Ala mutation is likely to be due to the disruption of the ion pair formed between the ε-amino group and a non bridging oxygen of phosphate 2 (Figure 2b). A similar conclusion was reached for the corresponding Lys89Ala mutation in E. coli EF-Tu suggesting that this highly conserved lysine may play a similar role in aa-tRNA binding for all bacterial species34. Upon mutation of the Asn91 to alanine a 0.9 kcal/mol loss in binding free energy was observed. The corresponding mutation Asn90Ala in E. coli EF-Tu showed a greater than 1.7 kcal/mol decrease in the free energy when binding yeast Phe-tRNAPhe 34. In the co-crystal structure, the nearest approach of the Asn91 side chain to the tRNA is 3.6 Å away from the 2′ hydroxyl group of ribose 1 which is too far for a hydrogen bond. However, when ribose 1 of yeast Phe-tRNAPhe was substituted with a deoxyribose, a comparable decrease in the binding free energy was observed 19. Considering their proximity, it seems likely that the two essential groups interact with each other in solution and contribute to the overall free energy of binding.
The absence of any effect on aa-tRNA binding for the Glu55Ala, Arg59Ala, and Tyr88Phe mutations is not unexpected since the side chains are all greater that 3.6Å away from the nearest tRNA functional group (Figure 2b). However, reconciling the absence of thermodynamic effects for the two remaining mutations in region 2 (Arg300Ala and Asn64Ala) with the co-crystal structure is more difficult. Despite the fact that guanidinium group of Arg300 is only 2.6 Å away from the negatively charged 5′ phosphate of the tRNA (Figure 2b) the Arg300Ala mutation has no effect on binding. The absence of a thermodynamic effect associated with this putative salt bridge is confirmed by the fact that the removal of the 5′ phosphate from the tRNA also has no effect on binding 19. It is possible that this could be explained by proposing that the guanidinium of Arg300 is slightly misplaced in the structural model and instead of interacting with phosphate 1, it interacts with the nearby (3.4 Å) carboxylate of Asp348. This would account for the biochemical data and does not represent a large rearrangement at the resolution of the model. The Asn64Ala mutation shows no effect on binding despite its proximity (2.9 Å) to the essential 2′ hydroxyl group of ribose 1. Asn64 is too far (4.6 Å) from any other amino acid in the free EF-Tu·GDPNP structure to make it unlikely that a compensatory interaction could explain this result. As discussed above, it is probably Asn91 that makes the critical contact with ribose 1.
Of the four residues in region 3, three (Arg389, Gln341 and Thr350) show substantial thermodynamic effects when mutated. Surprisingly, the only mutation that has little or no effect on binding, Lys376Ala, is hard to reconcile with the crystal structure since the side chain amino group is 2.3Å from a non-bridging oxygen of phosphate 67 (Figure 2c). Lys376 does not appear to contact any amino acids in the EF-Tu·GDPNP structure suggesting that a compensatory intra-molecular interaction is not responsible for the apparent lack of effect on binding upon its mutation to alanine.
Replacement of Arg389 with alanine resulted in a complete loss of the binding affinity of aa-tRNA to EF-Tu, but the effect is probably not directly related to an EF-Tu-tRNA interaction. The nearest approach of the guanidinium group is 6.9 Å from a non-bridging oxygen of phosphate 64 (Figure 2c), which is far for an ion pair. Instead, the observed decrease in binding energy may be a consequence of disrupting the ion pair that forms between the guanidinium group of Arg389 and the nearby (2.6 Å) carboxylate group of Glu118. Since Glu118 is part of the adjacent domain I, it is likely that this interaction is critical for the maintenance of the proper alignment of the two domains which form part of the tRNA binding cleft. Since the pair is present in both the free EF-Tu·GTP and the ternary complex, it is not directly involved in binding the tRNA body.
Within region 3, the Gln341Ala and Thr350Val mutations resulted in moderate decreases in binding energy to Phe-tRNAPhe with ΔΔG0 values of 1.0 and 1.1 kcal/mol, respectively. According to the X-ray co-crystal structure, Thr350 is sufficiently close (2.5 Å) to form a hydrogen bond with the 2′ hydroxyl group of ribose 65 while Gln341 is rather far (3.2 Å) from the hydroxyl group of ribose 64 (Figure 2c). However, substitution of ribose 64, but not ribose 65, with a deoxynucleotide reduces the binding affinity of T. thermophilus EF-Tu 19. To further investigate this situation, the Gln341Ala/Thr350Val double mutant was prepared and found to have a ΔΔG0 =1.2 kcal/mol which is similar to either of the single mutants, suggesting that the two amino acids only form a single thermodynamically relevant contact with the tRNA. The most reasonable way to reconcile the biochemical data with the structure is to propose that a thermodynamically relevant hydrogen bond forms between Gln341 and ribose 64, accounting for the ΔΔG0 when either partner is deleted. The role of Thr350 is to help position Gln341 to make this critical contact properly, presumably through the intramolecular hydrogen bond between the hydroxyl group of Thr350 and the carbonyl group of Gln341 shown in Figure 2c. Upon replacement of Thr350 with valine, this interaction is lost and Gln341 may not be correctly positioned to interact with ribose 64. This explanation implies that the positioning of the two side chains in solution may need to be a little different than shown in the co-crystal structure such that Gln341 would be closer to ribose 64 and Thr350 somewhat further from ribose 65.
Of the five amino acids in region 4, one (Arg339) has no effect on binding when mutated while the remaining four (Arg330, His331, Thr332, and Glu390) do. As expected, the mutation of Arg339 to alanine does not alter binding since it is the main chain amide group of this residue which appears to make a hydrogen bond with the essential 2′ hydroxyl group of ribose 51 (Figure 2d) and mutating the side chain would not be expected to disrupt the contact. The charged guanidinium group of the Arg339 side chain is also too far (>8.0 Å) from any of the phosphate groups in the tRNA to contribute to the binding affinity.
Mutation of the four remaining amino acids in region 4, all reduce the free energy of binding and all make a clear interaction in the co-crystal structure. First, the Arg330Ala mutation results in a >1.5 kcal/mol decrease in the free energy of binding, presumably due to the loss of a contact with the 2′hydroxyl group of ribose 52. In agreement with this, the replacement of ribose 52 with a deoxyribose results in a similar, large decrease in the free energy of binding 19. Second, the mutation of His331 to valine reduces the free energy of binding by 0.8 kcal/mol, presumably due to the loss of a hydrogen bond with the non-bridging O2 of the nearby phosphate 54 (Figure 2d). However, at the 2.7 Å resolution of the co-crystal structure it is not possible to determine the rotational orientation of the imidazole ring of H331, so either the τ or π amino group may be involved in the interactions. Thirdly, the replacement of Thr332 with valine gave a ΔΔG0 of 1 kcal/mol which presumably reflects the loss of the hydroxyl bond between the hydroxyl group of Thr332 and a non-bridging oxygen of phosphate 53 (Figure 2d). Finally, when Glu390 was mutated to alanine a ΔΔG0 of 1.0 kcal/mol was observed. Although the carboxylate group of Glu390 rather far (3.4 Å) from the amino group of G51 to make a hydrogen bond (Figure 2d), in the complex of T. aquaticus EF-Tu with Cys-tRNACys Glu390 makes a clear hydrogen bond with the amino group of G63 (which pairs with C51). Thus it is reasonable to suggest that Glu390 also makes a productive hydrogen bond with G51 of tRNAPhe in solution.
Combining the data from the EF-Tu mutagenesis experiments presented here and the previous yeast Phe-tRNAPhe modification experiments, it is apparent that thermodynamically relevant contacts are made throughout the protein-RNA interface. The contacts involve the side chains of nine (or possibly ten) amino acid and a main chain of another which interact with five 2′ hydroxyl groups (at residues 1, 51, 52, 64, and 67) and 4 phosphate groups (at residues 1, 53, 54 and 76) and a single base (the minor groove amino group of the 51–63 base pair). This includes the amino acids in region 1 (Arg274 and Glu271), two or possibly three in region 2 (Lys90, Asn91 and possibly Arg300), one in region 3 (Gln341) and five in region 4 (Arg330, His331, Thr332, Glu390 and the main chain of Arg339). Although the ΔΔG0 values obtained from the EF-Tu mutagenesis experiments may not directly reflect the binding energy associated with each contact, it is apparent from the number of thermodynamically relevant contacts that the majority of the total free energy of binding between EF-Tu and yeast Phe-tRNAPhe is derived from contacts with the T-stem. This suggests that this part of the interface should also contribute to the specificity of different tRNAs.
Discussion
The overall goal of this work was to use site directed mutagenesis of T. thermophilus EF-Tu to better understand which parts of the extensive interface with Phe-tRNAPhe contributes to the overall binding affinity. Conservative point mutations were made in all of the amino acids that could potentially contact the tRNA as suggested by the co-crystal structure. The binding affinity of each of the proteins was compared to the wild type protein to calculate a ΔΔG0. While this straightforward approach has been used with several other protein-nucleic acid complexes 35, 15 as well as protein-protein complexes36; 37, interpretation of the data is often complicated by several issues. First, when interpreting how a mutant protein interacts with the tRNA, it is important to consider the structures of both the free protein and bound complex. This is especially important if the mutated amino acid undergoes a structural change upon tRNA binding since the mutation could exert its thermodynamic effect in the free form, in the complex, or in both forms. A second complicating factor is the precise amino acid change that is made. By only mutating surface residues and always introducing smaller uncharged side chains, the likelihood of disrupting the overall structure of the protein is minimized. However, it is possible that the mutation could have unanticipated structural effects by modifying the solvation. Finally, it is also important to acknowledge that when designing and interpreting experiments based on X-ray crystal structures, attention is focused on the enthalpic component of the free energy and that the dynamics and solvation effects associated with the entropic component of ΔG0 are hard to assess.
Considering the above caveats, it is notable that most of the biochemical data can be reconciled with the available structural data. Of the 20 point mutations in the tRNA binding cleft that were tested, the aa-tRNA binding properties of 11 of them could have been correctly predicted by simply inspecting the structural model of the ternary complex. Mutations in four amino acids that were quite far from the tRNA showed no effect on binding and therefore can be considered controls. Mutations of seven amino acids that either did or did not reduce the aa-tRNA binding affinity could be explained by either disrupting (or not disrupting) the hydrogen bond network between the protein and the tRNA present in the structural model. For example, five of these amino acids make direct hydrogen bonds with the tRNA and bind less well when they are mutated.
There are nine mutations whose tRNA binding properties would have been more difficult to predict from the structural model. For five (Glu271Ala, Asn91Ala, Gln341Ala, Thr350Val and Glu390Ala) the mutation causes a clear effect on tRNA binding, but the reason is not immediately clear since the amino acids do not contact the tRNA in the X-ray structural model of the ternary complex. For most of these, the data was reconciled with the structure by proposing that the amino acid side chain moves by 1 Å or less such that it can form a hydrogen bond with a tRNA functional group. In several of these cases, the proposed hydrogen bond was either supported by the biochemical data identifying essential tRNA functional groups or because it was present in the structure of EF-Tu complexed with Cys-tRNACys. Adjustments of this magnitude are not unreasonable considering that the structural model was built from an X-ray data set with 2.7 Å resolution. The four remaining mutations (Lys52Ala, Arg300Ala, Asn64Ala, and Lys376Ala) were found to have no effect on binding despite the fact that a clear contact between EF-Tu and tRNA is observed in the structural model. In each case, the absence of an effect could not be explained by an obvious compensating interaction in the EF-Tu·GDPNP structure, although it remains possible that the hydration or dynamics of these residues in the free form minimizes thermodynamic impact. It is interesting that in contrast with most of the other amino acids in the tRNA binding interface, three of the amino acids in this group are not phylogentically conserved among bacteria, supporting the idea that they do not contribute to affinity.
The reasonable correlation between the results of the protein mutagenesis data presented here and other biochemical data clearly suggests that the X-ray co-crystal structure determined in 3 M NH4SO4, 7 mM MgCl2 and 20 mM Tris HCl (pH 7.6) is fairly consistent with the complex formed in solution 0.5 M NH4Cl, 20 mM MgCl2, and 50 mM Hepes (pH 7.0). Thus, neither the formation of the crystal lattice nor the crystallization conditions appreciably affect the interactions between the aa-tRNA and EF-Tu observed in solution. This also implies that the crystal structure reflects the biochemically relevant ternary complex and not a binding intermediate.
It is common in protein-nucleic acid interfaces that the majority of the binding free energy between the two interacting molecules is concentrated into relatively small, localized clusters18; 38. The mutagenesis data presented here for the EF-Tu tRNA interaction indicates that while the binding free energy encompasses the entire protein-nucleic acid interface, the most important residues are clustered in region 3 at the intersection of the acceptor and T-stems and in region 4 at the end of the T-stem. Smaller effects are seen in regions 1 and 2 which interact with the 3′ and 5′ ends of the tRNA. One can propose several reasons why the energetics of the interaction may have evolved in this way. First, there is some evidence that those aa-tRNAs which are products of class 1 aminoacyl tRNA synthetases bind EF-Tu by a “hand off” mechanism involving an intermediate where both proteins are bound to tRNA simultaneously39. In such a mechanism, a tRNA would initially bind EF-Tu through contacts in regions 3 and 4, while its 3′ end was bound within in the active site of the synthetase. After aminoacylation, the aminoacylated 3′ end of the tRNA would transfer to regions 1 and 2 of EF-Tu and the synthetase would dissociate. While the crystal structures of most class 1 aminoacyl tRNA synthetases bound to their cognate tRNAs are compatible with the simultaneous binding of EF-Tu to the T stem, the formation of such a complex would be unlikely unless EF-Tu bound tightly to this part of the tRNA.
A second rationale why a substantial fraction of the binding free energy involves contacts in the T-stem of tRNA is that this region may contain the majority of the specificity determinants. In the initial experiments establishing the specificity of EF-Tu to different tRNA bodies9, it was noted that the tighter binding tRNAs contained a preponderance of AU or UA base pairs in the 49–65 and 7–66 positions which contact EF-Tu in regions 3 and 4. Since yeast tRNAPhe used in these experiments lies in the middle of the hierarchy of tRNA binding affinities, this raises the possibility that ΔΔG0 values for several of the mutations will be quite different for other tRNAs.
Finally, the role that EF-Tu plays in the mechanism of decoding on the ribosome may also have contributed to the evolution of the energetics of its interaction with tRNA. Cryoelectron microscopy studies suggest that when the ternary complex binds its cognate codon on 70S ribosomes, it also makes extensive interactions with the large ribosomal subunit and the structures of both the protein and tRNA parts of the complex are subtly altered 40; 41. Upon hydrolysis of GTP, the interdomain stability of EF-Tu is weakened and the aa-tRNA is released from EF-Tu and transfered to the A site in a process termed accommodation 42. It seems likely that the energetics of the EF-Tu – tRNA interaction will be altered upon ribosome binding and change again after GTP hydrolysis. Indeed, it is possible that the tight binding of EF-Tu to the T-stem may be a critical feature of the translational mechanism or may be necessary to maintain translational accuracy. Thus, it will be interesting to assay the translational performance of the mutants in the tRNA binding interface delivered in this work.
Materials & Methods
EF-Tu Mutagensis & Purification
The initial plasmid containing the T. thermophilus tuf1 gene was provided by Anidya Banerjee and Marvin Mackinen (University of Chicago). A sequence coding for (His)6-TEV cleavable linker was appended to the 5′ end of the EF-Tu gene by PCR and cloned into a pET-3a vector between NdeI and BamHI sites. The N-terminal amino acid sequence of the resulting protein was MH6GNKYFQA so that cleavage by TEV after the glutamine resulted in the native N-terminal alanine residue. Individual EF-Tu mutants were generated using Quick Change primer mediated, site directed mutagenesis (Stratagene). The sequence of each EF-Tu variant was confirmed by DNA sequencing.
Wild type and mutant EF-Tu purification was accomplished by growing transformed BL21(DE3) pLysS cells to an OD600 of 0.6 at 37°C. The cells were then induced by the addition of 1 mM IPTG and grown at 37°C for 4 hours. The cells were then harvested by centrifugation and stored frozen at −20°C. Thawed cells were lysed by sonication in buffer A (50 mM HEPES (pH 7.5), 10 mM MgCl2, 5% glycerol, 10mM imidazole, 20 μM ABSF, 5 mM β-mercaptoethanol, 100 μM GDP). The cell lysate was cleared by centrifugation at 10,000 g for 30 minutes followed by filtration through a 0.22 micron cellulose acetate filter. The resulting filtrate was loaded onto a 1 mL HiTrap Ni2+ chelating column (Amersham). The column was washed with 10 mL of wash buffer (50 mM HEPES (pH 7.5), 10 mM MgCl2, 5% glycerol, 10 mM imidazole, 1 M NaCl, 0.5% Tween-20, 5 mM β-mercaptoethanol, 100 μM GDP). EF-Tu was then eluted from the column with a 20 mL gradient of 10 mM to 1 M imidazole in buffer A. Elution fractions were run on SDS-PAGE to determine the presence of EF-Tu. Fractions containing EF-Tu were pooled and DTT was added to a final concentration of 10 mM. Purified His6-TEV protease43 was a generous gift from Robert Batey (University of Colorado) and was added to the pooled EF-Tu fractions to a final concentration of 1μM. TEV digestion was allowed to proceed at 10°C for 15 hours. The EF-Tu/TEV mixture was then dialyzed against buffer A to remove DTT. To remove any uncleaved His6-EF-Tu and His6-TEV protease, the dialysate was mixed with Ni2+ agarose at 4°C for 20 minutes followed by filtration. The purified fraction of EF-Tu which did not bind to the Ni2+ agarose was then stored at a concentration >15 uM at −20°C in buffer A containing 50% glycerol. The purity of the final EF-Tu was determined to be >90% by SDS-PAGE. The concentration of each EF-Tu was determined by the Bradford assay44.
Preparation of RNA
Purified yeast tRNAPhe was purchased from Sigma-Aldrich. The [3′ 32P] labeling of the tRNA was performed as previously described45 using His6 tagged tRNA-terminal nucleotidyl transferase (provided by C. McHenry, University of Colorado Health Science Center) with the following modification. After the addition of pyrophosphatase the reaction was incubated at 37 °C for 10 minutes as opposed to 30 seconds.
The plasmid containing the yeast PheRS gene was a generous gift from Inchan Kwon and David Tirrell (Cal Tech). Protein purification was accomplished by growing BL21(DE3) pLysS cells to an OD600 of 0.6 at 37°C. Cells were induced with 1 mM IPTG and grown at 37°C for an additional 4 hours. Cells were harvested by centrifugation and stored frozen at −20 °C. Cell pellet was resuspended in lysis buffer (50 mM NaH2PO4 (pH 8.0), 300 mM NaCl, 10 mM imidazole). Cells were lysed by the addition of lysozyme to a final concentration of 1.0 mg/ml followed by sonication. The resulting cell lysate was cleared by centrifugation at 10,000 g for 30 minutes at 4°C. Cleared cell lysate was loaded onto a charged 1mL Ni2+ chelating column and washed with 8 column volumes of wash buffer (50 mM NaH2PO4 (pH 8.0), 300 mM NaCl, 20 mM imidazole). The column was then eluted with a 10 mM to 250 mM gradient of imidazole in lysis buffer. The purified yeast PheRS was then stored at −20°C in lysis buffer containing 50% glycerol. The purity of the final PheRS was greater than 90% as determined by SDS-PAGE. The concentration of PheRS was determined by the Bradford assay using BSA as a standard.
The aminoacylation of [3′-32P] labeled yeast tRNAPhe with phenylalanine was performed with 1μM [3′-32P] tRNAPhe, 4 mM ATP, 30 mM KCl, 15 mM MgCl2, 5 mM DTT, 30 mM HEPES (pH 7.0), 250 μM phenylalanine, 0.025 units/ul yeast inorganic pyrophosphatase and 0.2 μM of yeast PheRS. The extent of aminoacylation was determined to be > 60% using the a P1 nuclease assay45. The aminoacylation of unlabeled yeast tRNAPhe with [3H]phenylalanine was performed as in Dale et al. 10, except the final tRNAPhe concentration was 5 μM rather than 2 to 3 μM.
EF-Tu Assays
Since EF-Tu is stored in the GDP bound form, it was necessary to activate the protein before formation of ternary complex. To accomplish this, 0.5μM EF-Tu was incubated for 3 hours in 50 mM HEPES (pH 7.0), 20 mM MgCl2, 0.5 M NH4Cl, 5 mM DTT, 20 μM GTP, 3 mM phosphoenolpyruvate and 50 μg/ml pyruvate kinase (Buffer B).
The determination of the relative stoichiometry of GTP binding of each mutant EF-Tu was accomplished by using a GDP binding assay modified from that described by Miller and Weissbach46. 200 μL of 1μM EF-Tu in buffer B containing 2 mM [3H] GTP was incubated at 37 °C for 2 hours. Any contaminating GDP was converted to GTP by the addition of 3 mM phosphoenolpyruvate and 50 ug/ml pyruvate kinase. The [8-3H] GTP activated EF-Tu was diluted in 2 mL of 10 mM Tris, 10 mM NH4Cl, 10 mM MgCl2 (buffer C) at 0°C and then filtered through nitrocellulose filters that had been presoaked in buffer C. The samples were then washed three times with 3 mL of buffer C at 0°C. The filters were then dissolved in 10 mL of scintillation fluid and counted. The GTP binding of each mutant EF-Tu was measured at least three times and is reported as the percent GTP bound compared to wild-type EF-Tu (Table 1).
The dissociation rate constant of each EF-Tu·GTP·Phe-tRNAPhe complex was determined with a ribonuclease protection assay adapted to a 96 well format 19. Ternary complex was formed by incubation of 0.5 μM EF-Tu·GTP with <0.1 μM yeast [3H]Phe-tRNAPhe at 0°C in 125 μL buffer B for 20 minutes. The reaction was initiated by the addition of 13 μL of 1 mg/mL RNase A. 10 μL aliquots were removed at various time points and quenched were in 110 μL of 10% trichloroacetic acid containing 0.1 mg/mL unfractionated yeast tRNA. The resulting quenched time points were then filtered through nitrocellulose, washed and counted on a phosphorimager as previously described 10. The dissociation rate constants were determined as the negative slope of the ln(cpm) versus time plot. The dissociation rate constant of each mutant EF-Tu with yeast Phe-tRNAPhe was measured at least three times, and the mean value and standard deviation of each koff was calculated. Control experiments in which no EF-Tu was present, the addition of RNase A typically resulted in the digestion of >95% of the Phe-tRNAPhe within 15 seconds. In order to calculate the KD of each EF-Tu mutant with yeast Phe-tRNAPhe from the experimentally determined koff, the association rate constant of 6.6×106 M−1min−1 was used.
Equilibrium dissociation constants for the selected EF-Tu’s with yeast Phe-tRNAPhe were determined as previously described 10. 20 μL of twelve different EF-Tu concentrations (0.5 nM – 1 μM) in buffer B were each mixed with 0.4 μL of 100 nM [3′-32P] yeast Phe-tRNAPhe and incubated at 0°C for 20 minutes. After equilibration, 10 μL of 1 mg/mL RNaseA was added and quenched 20 seconds later with the addition of 110 μL of 10% trichloroacetic acid containing 0.1 mg/ml unfractionated yeast tRNA. To correct for any aa-tRNA that may remain after the 20 second treatment with RNase, a no EF-Tu control was performed and the counts remaining after RNAase digestion was substracted from the experimental data. The equilibrium dissociation data for each of the EF-Tu variants was normalized to maximum fraction of aa-tRNA bound. The resulting KD data was plotted as fraction of aa-tRNA protected versus EF-Tu concentration.
Abbreviations
- EF-Tu
Elongation Factor Tu
- GDPNP
Guanosine 5′-[β, γ-imido]triphosphate
- aa-tRNA
aminoacyl-tRNA
- TEV
Tobacco Etch Virus
- PheRS
Phenylalanyl-tRNA Synthetase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Louie A, Ribeiro NS, Reid BR, Jurnak F. Relative affinities of all Escherichia coli aminoacyl-tRNAs for elongation factor Tu-GTP. J Biol Chem. 1984;259:5010–6. [PubMed] [Google Scholar]
- 2.Ott G, Schiesswohl M, Kiesewetter S, Forster C, Arnold L, Erdmann VA, Sprinzl M. Ternary complexes of Escherichia coli aminoacyl-tRNAs with the elongation factor Tu and GTP: thermodynamic and structural studies. Biochim Biophys Acta. 1990;1050:222–5. doi: 10.1016/0167-4781(90)90170-7. [DOI] [PubMed] [Google Scholar]
- 3.Abrahamson JK, Laue TM, Miller DL, Johnson AE. Direct determination of the association constant between elongation factor Tu X GTP and aminoacyl-tRNA using fluorescence. Biochemistry. 1985;24:692–700. doi: 10.1021/bi00324a023. [DOI] [PubMed] [Google Scholar]
- 4.Janiak F, Dell VA, Abrahamson JK, Watson BS, Miller DL, Johnson AE. Fluorescence characterization of the interaction of various transfer RNA species with elongation factor Tu.GTP: evidence for a new functional role for elongation factor Tu in protein biosynthesis. Biochemistry. 1990;29:4268–77. doi: 10.1021/bi00470a002. [DOI] [PubMed] [Google Scholar]
- 5.LaRiviere FJ, Wolfson AD, Uhlenbeck OC. Uniform binding of aminoacyl-tRNAs to elongation factor Tu by thermodynamic compensation. Science. 2001;294:165–8. doi: 10.1126/science.1064242. [DOI] [PubMed] [Google Scholar]
- 6.Rodnina MV, Fricke R, Kuhn L, Wintermeyer W. Codon-dependent conformational change of elongation factor Tu preceding GTP hydrolysis on the ribosome. Embo J. 1995;14:2613–9. doi: 10.1002/j.1460-2075.1995.tb07259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nissen P, Kjeldgaard M, Thirup S, Polekhina G, Reshetnikova L, Clark BF, Nyborg J. Crystal structure of the ternary complex of Phe-tRNAPhe, EF-Tu, and a GTP analog. Science. 1995;270:1464–72. doi: 10.1126/science.270.5241.1464. [DOI] [PubMed] [Google Scholar]
- 8.Nissen P, Thirup S, Kjeldgaard M, Nyborg J. The crystal structure of Cys-tRNACys-EF-Tu-GDPNP reveals general and specific features in the ternary complex and in tRNA. Structure. 1999;7:143–56. doi: 10.1016/s0969-2126(99)80021-5. [DOI] [PubMed] [Google Scholar]
- 9.Asahara H, Uhlenbeck OC. The tRNA specificity of Thermus thermophilus EF-Tu. Proc Natl Acad Sci U S A. 2002;99:3499–504. doi: 10.1073/pnas.052028599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dale T, Sanderson LE, Uhlenbeck OC. The affinity of elongation factor Tu for an aminoacyl-tRNA is modulated by the esterified amino acid. Biochemistry. 2004;43:6159–66. doi: 10.1021/bi036290o. [DOI] [PubMed] [Google Scholar]
- 11.Adkins HJ, Miller DL, Johnson AE. Changes in aminoacyl transfer ribonucleic acid conformation upon association with elongation factor Tu-guanosine 5′-triphosphate. fluorescence studies of ternary complex conformation and topology. Biochemistry. 1983;22:1208–17. doi: 10.1021/bi00274a034. [DOI] [PubMed] [Google Scholar]
- 12.Jonak J, Anborgh PH, Parmeggiani A. Histidine-118 of elongation factor Tu: its role in aminoacyl-tRNA binding and regulation of the GTPase activity. FEBS Lett. 1994;343:94–8. doi: 10.1016/0014-5793(94)80614-4. [DOI] [PubMed] [Google Scholar]
- 13.Haruki M, Matsumoto R, Hara-Yokoyama M, Miyazawa T, Yokoyama S. Conformational changes of aminoacyl-tRNA and uncharged tRNA upon complex formation with polypeptide chain elongation factor Tu. FEBS Lett. 1990;263:361–4. doi: 10.1016/0014-5793(90)81414-j. [DOI] [PubMed] [Google Scholar]
- 14.Watson BS, Hazlett TL, Eccleston JF, Davis C, Jameson DM, Johnson AE. Macromolecular arrangement in the aminoacyl-tRNA.elongation factor Tu.GTP ternary complex. A fluorescence energy transfer study. Biochemistry. 1995;34:7904–12. doi: 10.1021/bi00024a015. [DOI] [PubMed] [Google Scholar]
- 15.Hobson D, Uhlenbeck OC. Alanine scanning of MS2 coat protein reveals protein-phosphate contacts involved in thermodynamic hot spots. J Mol Biol. 2006;356:613–24. doi: 10.1016/j.jmb.2005.11.046. [DOI] [PubMed] [Google Scholar]
- 16.Nuss JM, Whitaker PB, Air GM. Identification of critical contact residues in the NC41 epitope of a subtype N9 influenza virus neuraminidase. Proteins. 1993;15:121–32. doi: 10.1002/prot.340150204. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Z, Palzkill T. Dissecting the protein-protein interface between beta-lactamase inhibitory protein and class A beta-lactamases. J Biol Chem. 2004;279:42860–6. doi: 10.1074/jbc.M406157200. [DOI] [PubMed] [Google Scholar]
- 18.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267:383–6. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 19.Pleiss JA, Uhlenbeck OC. Identification of thermodynamically relevant interactions between EF-Tu and backbone elements of tRNA. J Mol Biol. 2001;308:895–905. doi: 10.1006/jmbi.2001.4612. [DOI] [PubMed] [Google Scholar]
- 20.Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N, Yeh LS. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2005;33:D154–9. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asahara H, Uhlenbeck OC. Predicting the binding affinities of misacylated tRNAs for Thermus thermophilus EF-Tu.GTP. Biochemistry. 2005;44:11254–61. doi: 10.1021/bi050204y. [DOI] [PubMed] [Google Scholar]
- 22.Hunter SE, Spremulli LL. Mutagenesis of glutamine 290 in Escherichia coli and mitochondrial elongation factor Tu affects interactions with mitochondrial aminoacyl-tRNAs and GTPase activity. Biochemistry. 2004;43:6917–27. doi: 10.1021/bi036068j. [DOI] [PubMed] [Google Scholar]
- 23.Louie A, Jurnak F. Kinetic studies of Escherichia coli elongation factor Tu-guanosine 5′-triphosphate-aminoacyl-tRNA complexes. Biochemistry. 1985;24:6433–9. doi: 10.1021/bi00344a019. [DOI] [PubMed] [Google Scholar]
- 24.Alexander C, Bilgin N, Lindschau C, Mesters JR, Kraal B, Hilgenfeld R, Erdmann VA, Lippmann C. Phosphorylation of elongation factor Tu prevents ternary complex formation. J Biol Chem. 1995;270:14541–7. doi: 10.1074/jbc.270.24.14541. [DOI] [PubMed] [Google Scholar]
- 25.Lippmann C, Lindschau C, Vijgenboom E, Schroder W, Bosch L, Erdmann VA. Prokaryotic elongation factor Tu is phosphorylated in vivo. J Biol Chem. 1993;268:601–7. [PubMed] [Google Scholar]
- 26.Kraal B, Lippmann C, Kleanthous C. Translational regulation by modifications of the elongation factor Tu. Folia Microbiol (Praha) 1999;44:131–41. doi: 10.1007/BF02816232. [DOI] [PubMed] [Google Scholar]
- 27.Wilkins MR, Gasteiger E, Gooley AA, Herbert BR, Molloy MP, Binz PA, Ou K, Sanchez JC, Bairoch A, Williams KL, Hochstrasser DF. High-throughput mass spectrometric discovery of protein post-translational modifications. J Mol Biol. 1999;289:645–57. doi: 10.1006/jmbi.1999.2794. [DOI] [PubMed] [Google Scholar]
- 28.Krab IM, te Biesebeke R, Bernardi A, Parmeggiani A. Elongation factor Ts can act as a steric chaperone by increasing the solubility of nucleotide binding-impaired elongation factor-Tu. Biochemistry. 2001;40:8531–5. doi: 10.1021/bi0104930. [DOI] [PubMed] [Google Scholar]
- 29.Antonsson B, Leberman R. Stabilization of the ternary complex EF-Tu.GTP.valyl-tRNAval by ammonium salts. Biochimie. 1982;64:1035–40. doi: 10.1016/s0300-9084(82)80384-2. [DOI] [PubMed] [Google Scholar]
- 30.Delaria K, Guillen M, Louie A, Jurnak F. Stabilization of the Escherichia coli elongation factor Tu-GTP-aminoacyl-tRNA complex. Arch Biochem Biophys. 1991;286:207–11. doi: 10.1016/0003-9861(91)90029-i. [DOI] [PubMed] [Google Scholar]
- 31.Nazarenko IA, Harrington KM, Uhlenbeck OC. Many of the conserved nucleotides of tRNA(Phe) are not essential for ternary complex formation and peptide elongation. Embo J. 1994;13:2464–71. doi: 10.1002/j.1460-2075.1994.tb06531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kjeldgaard M, Nissen P, Thirup S, Nyborg J. The crystal structure of elongation factor EF-Tu from Thermus aquaticus in the GTP conformation. Structure. 1993;1:35–50. doi: 10.1016/0969-2126(93)90007-4. [DOI] [PubMed] [Google Scholar]
- 33.Pedersen GN, Rattenborg T, Knudsen CR, Clark BF. The role of Glu259 in Escherichia coli elongation factor Tu in ternary complex formation. Protein Eng. 1998;11:101–8. doi: 10.1093/protein/11.2.101. [DOI] [PubMed] [Google Scholar]
- 34.Wiborg O, Andersen C, Knudsen CR, Clark BF, Nyborg J. Mapping Escherichia coli elongation factor Tu residues involved in binding of aminoacyl-tRNA. J Biol Chem. 1996;271:20406–11. doi: 10.1074/jbc.271.34.20406. [DOI] [PubMed] [Google Scholar]
- 35.Kassavetis GA, Driscoll R, Geiduschek EP. Mapping the Principal Interaction Site of the Brf1 and Bdp1 Subunits of Saccharomyces cerevisiae TFIIIB. J Biol Chem. 2006;281:14321–9. doi: 10.1074/jbc.M601702200. [DOI] [PubMed] [Google Scholar]
- 36.Dall’Acqua W, Goldman ER, Eisenstein E, Mariuzza RA. A mutational analysis of the binding of two different proteins to the same antibody. Biochemistry. 1996;35:9667–76. doi: 10.1021/bi960819i. [DOI] [PubMed] [Google Scholar]
- 37.Tsiang M, Jain AK, Dunn KE, Rojas ME, Leung LL, Gibbs CS. Functional mapping of the surface residues of human thrombin. J Biol Chem. 1995;270:16854–63. doi: 10.1074/jbc.270.28.16854. [DOI] [PubMed] [Google Scholar]
- 38.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 39.Zhang CM, Perona JJ, Ryu K, Francklyn C, Hou YM. Distinct kinetic mechanisms of the two classes of Aminoacyl-tRNA synthetases. J Mol Biol. 2006;361:300–11. doi: 10.1016/j.jmb.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 40.Stark H, Rodnina MV, Wieden HJ, Zemlin F, Wintermeyer W, van Heel M. Ribosome interactions of aminoacyl-tRNA and elongation factor Tu in the codon-recognition complex. Nat Struct Biol. 2002;9:849–54. doi: 10.1038/nsb859. [DOI] [PubMed] [Google Scholar]
- 41.Valle M, Zavialov A, Li W, Stagg SM, Sengupta J, Nielsen RC, Nissen P, Harvey SC, Ehrenberg M, Frank J. Incorporation of aminoacyl-tRNA into the ribosome as seen by cryo-electron microscopy. Nat Struct Biol. 2003;10:899–906. doi: 10.1038/nsb1003. [DOI] [PubMed] [Google Scholar]
- 42.Pape T, Wintermeyer W, Rodnina MV. Complete kinetic mechanism of elongation factor Tu-dependent binding of aminoacyl-tRNA to the A site of the E. coli ribosome. Embo J. 1998;17:7490–7. doi: 10.1093/emboj/17.24.7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lucast LJ, Batey RT, Doudna JA. Large-scale purification of a stable form of recombinant tobacco etch virus protease. Biotechniques. 2001;30:544–6. 548–550. doi: 10.2144/01303st06. passim. [DOI] [PubMed] [Google Scholar]
- 44.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 45.Wolfson AD, Uhlenbeck OC. Modulation of tRNAAla identity by inorganic pyrophosphatase. Proc Natl Acad Sci U S A. 2002;99:5965–70. doi: 10.1073/pnas.092152799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller DL, Weissbach H. Elongation factor Tu and the aminoacyl-tRNA-EFTu-GTP complex. Methods Enzymol. 1974;30:219–32. doi: 10.1016/0076-6879(74)30024-9. [DOI] [PubMed] [Google Scholar]