

Abstract
Spatial working memory and the ability of a cholinesterase inhibitor to enhance memory were assessed at 4, 10, and 16 months of ages in control and Ts65Dn mice, a partial trisomy model of Down syndrome, with possibly significant relationships to Alzheimer’s Disease as well. In addition, ACh release during memory testing was measured in samples collected from the hippocampus using in vivo microdialysis at 4, 10, and 22–25 months of age. When tested on a four-arm spontaneous alternation task, the Ts65Dn mice exhibited impaired memory scores at both 4 and 10 months. At 16 months, control performance had declined toward that of the Ts65Dn mice and the difference in scores across genotypes was not significant. Physostigmine (50 μg/kg) fully reversed memory deficits in the Ts65Dn mice in the 4-month-old group but not in older mice. Ts65Dn and control mice exhibited comparable baseline levels of ACh release at all ages tested; these levels did not decline significantly across age in either genotype. ACh release increased significantly during alternation testing only in the young Ts65Dn and control mice. However, the increase in ACh release during alternation testing was significantly greater in control than Ts65Dn mice at this age. The controls exhibited a significant age-related decline in the testing-related increase in ACh release. With only a small increase during testing in young Ts65Dn mice, the age-related decline in responsiveness of ACh release to testing was not significant in these mice. Overall, these results suggest that diminished responsiveness of ACh release in the hippocampus to behavioral testing may contribute memory impairments in Ts65Dn mice.
Keywords: Down syndrome, Ts65Dn mice, acetylcholine, spontaneous alternation, physostigmine, microdialysis, hippocampus, aging and memory
1. Introduction
Down syndrome, a genetic form of mental retardation, is characterized by a trisomy of chromosome 21. While the degree of cognitive dysfunction varies with the individual, the disorder includes impairments of spatial, verbal, and memory abilities (e.g. Marcell and Armstrong, 1982; Nadel, 2003, Wishart, 1995). The trisome includes the gene for amyloid precursor protein (Glenner, 1988; Korenberg et al., 1989; Tanzi, 1989), perhaps related to the appearance in individuals with Down syndrome of many neuropathological traits that occur in Alzheimer’s Disease. For example, Down syndrome is accompanied by the appearance at about 40 years of age of neuritic plaques, neurofibrillary tangles, and loss of basal forebrain cholinergic neurons (Casanova et al., 1985; Holtzman et al., 1996), pathologies evident also in brain of individuals with Alzheimer’s Disease.
In recent years, several segmental trisomy mice have been developed as a model of Down syndrome (Davisson, Schmidt and Akeson, 1990; Galdzicki et al., 2001) and, with the homologies to Alzheimer’s disease, for that disorder as well. The Ts65Dn mouse has a partial trisomy for a segment of mouse chromosome 16 that is homologous to a portion of human chromosome 21, spanning from the gene for amyloid precursor protein to the myxovirus susceptibility gene (Davisson and Costa, 1999; Davisson et al., 2001; Reeves et al., 1995).
Ts65Dn mice exhibit learning and memory deficits on several tasks, comprehensively reviewed in Sérégaza et al. (2006). For example, impairments have been seen on spatial, operant conditioning and context discrimination tasks (Bimonte-Nelson et al., 2003; Demas et al., 1996, 1998; Escoriheula et al., 1995, 1998; Hunter, Bimonte and Granholm, 2003; Reeves et al., 1995; Seo and Isacson, 2005; Wenger et al., 2004), as well as on tasks involving sustained attention (Driscoll et al., 2004). However, learning and memory are not impaired in all tasks. Cued learning in the swim task is preserved in the Ts65Dn mice (Escorihuela et al., 1995; Sago et al., 2000), as is memory needed to discriminate very dissimilar contexts (Hyde and Crnic, 2001) and memory for inhibitory avoidance training (Coussons-Read and Crnic, 1996).
Neurobiological characterizations of the Ts65Dn mouse reveal decreases in cholinergic markers, including reductions in the number and size of cholinergic neurons in the medial septum, that appear after 4 – 6 months of age (Granholm, Sanders and Crnic, 2000; Holtzman et al., 1996). These reductions are analogous to the loss of forebrain cholinergic neurons seen in both Down syndrome (Casanova et al., 1985; Head et al., 2001; Mann and Esiri, 1989) and Alzheimer’s Disease (Candy et al., 1986; Coyle, Price and DeLong, 1983; Vogels et al., 1990; Whitehouse et al., 1985).
The early emergence of the decreases in cholinergic markers suggests that learning and memory deficits evident in old age in control mice might appear at earlier ages in Ts65Dn mice. Most assessments of learning and memory in the Ts65Dn mice have been done at a single age and compared to control mice of the same age. In one experiment in which the importance of age was assessed, mice were trained at 4 and 6 months of age in a water-escape version of a radial arm maze (Hunter et al., 2003). The findings revealed that while Ts65Dn mice exhibited learning deficits at both ages compared to controls, the older Ts65Dn mice had more severe impairments than did the younger mice. These findings suggest that the onset of learning and memory deficits appear at about the same time as do the decreases in cholinergic size and number in the medial septum (Granholm, Sanders and Crnic, 2000). A later experiment examined learning and memory for context discrimination across a broad range of ages, from 3 to 14 months. At 4 months of age, both the Ts65Dn and control mice exhibited comparable learning. However, at 5 – 8 months and at 10–14 months of age, the Ts65Dn mice showed slower acquisition of the context discrimination task than did controls or 4-month-old Ts65Dn mice (Hyde and Crnic, 2001). These experiments support the view that the onset of cognitive and cholinergic impairments may be related and may occur during relatively early adulthood in Ts65Dn mice.
The present experiment explores this possible relationship further, assessing memory, the ability of a cholinesterase inhibitor to enhance memory, and release of acetylcholine (ACh) in the hippocampus in Ts65Dn mice across a range of ages. A spontaneous alternation task was used to assess spatial working memory. This task was selected for several reasons. First, spontaneous alternation testing requires no reward and therefore no food or water motivation, avoiding potential complications of design based on different motivation levels across groups (cf. Dember and Richman, 1989; Lalonde, 2002). Similarly, because the task involves relatively low stress, differences in stress responses to training are less likely to interfere with interpretations regarding the bases for possible differences in learning and memory, in contrast to tasks such as the swim task where hypothermia-related stress responses in Ts65Dn mice vs. controls produced apparent differences in learning and memory that were better attributed to genotypic differences in stress responses (Stasko and Costa, 2004). Second, on the basis of studies of age-related changes in learning and memory, spontaneous alternation tests are likely to provide opportunities to assess age-related changes and enhancement of memory in Ts65Dn mice. Spontaneous alternation scores decrease in aged rodents (McNay and Gold, 2001; Stone, Rudd and Gold, 1992) and age-related and other impairments can be reversed by pharmacological agents including cholinergic agonists (Gold, 2001, 2007; McNay and Gold, 2001; Sarter et al., 1988). In addition, there is recent evidence that Ts65Dn mice exhibit deficits on this task (Fernandez et al., 2007). Thus, spontaneous alternation tasks provide opportunities to assess age-related impairments and pharmacological reversals of those impairments in Ts65Dn mice. Third, there is clear association in rats of spontaneous alternation scores and release of ACh in the hippocampus. Release of ACh in the hippocampus increases during exploratory behavior (Giovannini et al., 2001) and alternation testing; during spontaneous alternation testing, the magnitude of release is related to alternation scores under many conditions (Ragozzino, Unick and Gold, 1996; Ragozzino et al., 1998; Pych et al., 2005). Fourth, as a test of working memory, each mouse can be tested on multiple occasions under different conditions permitting use of fewer mice.
The acetylcholinesterase inhibitor, physostigmine, was selected as the drug with which to enhance memory because of past findings indicating that the drug enhances memory in aging and other conditions (e.g., Rispoli et al., 2006; Flood et al., 1993; Normile and Altman, 1992; Riekkinen et al., 1991; Ohta et al., 1991; Castellano et al., 1989; Chang and Gold, 2004; Degroot and Parent, 2000; Ukai et al., 1995; Walker and Gold, 1992; Beracochea et al., 1992; Stone et al., 1991; Murray and Fibiger, 1986; Maurice et la., 1996; Dong et al., 2005; Popovi et al., 1997; Santucci et al., 1991). In addition, physostigmine was selected because it is an indirect agonist that requires at least some remaining release of ACh in order to exert its pharmacological actions. Therefore, the drug might match the measures of ACh release using microdialysis, as included in the present experiment, more clearly than would, for example, a muscarinic receptor agonist.
The present experiments examined Ts65Dn mice and control 2N mice on: (1) Spontaneous alternation performance to assess spatial working memory; (2) efficacy of physostigmine, an indirect ACh agonist, to enhance spatial working memory; and (3) release of ACh in the hippocampus at baseline and in response to alternation testing.
2. Materials and Methods
2.1. Animals
44 Ts65Dn mice and 52 wild-type control male mice were purchased from Jackson Laboratory (Bar Harbor, ME). Mice arrived in the laboratory at approximately 2 months of age and were maintained in the laboratory until the conclusion of testing. The mice were housed individually in translucent cages, with food and water available ad libitum. The mice were maintained in a 12:12 hr light-dark cycle (lights on at 8:00 am) throughout the experiments.
The first segmental trisomy Ts65Dn mutation was developed by Davisson et al. (1990, 1993). The trisomy is maintained and the experimental mice are generated by repeated backcrosses of Ts65Dn females to C57BL/6JEi X C3H/HeSnJ F1 males. To confirm the roughly 20% trisomic Ts65Dn mutation, karyotyping is performed by Jackson Laboratory using fluorescent in situ hybridization. Non-mutation 2N mice were used as controls in this study.
Availability of mice for this experiment was very low; the data were collected over a 4-year period. Because of the low availability and high cost of the mice, as well as the higher attrition of Ts65Dn mice after middle age, mice were tested on repeated trials for spontaneous alternation performance and physostigmine enhancement of that performance. Approximately 6 months later, the same mice were prepared for microdialysis to measure ACh release at baseline and during spontaneous alternation testing. For the microdialysis studies, a new cohort was used for the youngest group.
All procedures were approved by the University of Illinois Institutional Animal Care and Use Committee and comply with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
2.2. Spontaneous alternation testing
Spontaneous alternation scores were obtained in a plus-shaped maze in mice at approximately 4, 10, and 16 months of age. The testing apparatus was a plus-shaped maze made of a black Plexiglas floor and clear Plexiglas walls with an open top. The maze had four identical arms (25.5 x 7.7 x 10.0 cm, L x W x H) extending from a central platform (7.7 x 7.7 cm). Mice were allowed to traverse the maze freely as in previous studies with rats (Chang and Gold, 2004; McNay and Gold, 2001; Ragozzino et al., 1996, 1998; Pych et al., 2005; Stefani and Gold, 1998, 2001). After being placed in the central platform, mice were permitted to move freely through the maze for 6 minutes. During that time, the numbers and sequences of entries were recorded. An alternation was defined as entry into four different arms on overlapping sets of five consecutive arm entries. The use of 4/5 arms as a measure is one we have used often in the past (Ragozzino et al., 1996), because it provides lower within-group variability than does use of a 4/4 criterion to define an alternation. With this procedure, the number of possible alternations is equal to the number of arm entries minus four. A percent alternation score was calculated as the ratio of alternations made/alternations possible x 100; using the 4/5 criterion to define an alternation, chance performance is 44%.
An ABAB design was employed to test the effects of physostigmine on spontaneous alternation performance of mice at 4 months, 10 months and 16 months of age. With this design, each mouse was tested four times. Testing sessions were separated by one week. Baseline sessions were administered on Weeks 1 and 3, and treatment sessions were administered on Weeks 2 and 4. Mice were placed on the maze and were allowed to locomote freely for 8 min. On Week 1, the mice were tested without injection or with only saline injection 10 min prior to the tests. On Week 2, the same mice received physostigmine hemisulfate (50 μg/kg, IP) 10 min prior to another spontaneous alternation test. The dose was selected on the basis of pilot data together with past results (Stone et al., 1991). The sequence was then repeated in Weeks 3 and 4. Because alternation scores did not differ on Week 1 vs. Week 3 (for each age, P>0.8), these groups were combined such that each mouse had a single control score. Similarly, because alternation scores did not differ on the two physostigmine treatment sessions on Weeks 2 and 4 (for each age, P>0.7), these scores were similarly combined.
2.4. Surgery
Guide cannulae for microdialysis probes were implanted unilaterally in the hippocampus. The mice were anesthetized with sodium pentobarbital (60 mg/kg, IP, plus supplements as needed). They were then placed in a stereotaxic apparatus with horizontal skull position using two acute (18°) ear bars. One plastic guide cannulae (CMA/7, Carnegie Medicin, Stockholm, Sweden) was lowered into the ventral hippocampus (coordinates: 3.2 mm posterior to bregma, 3.0 mm lateral and 1.2 mm ventral from the surface of the skull) (Franklin and Paxinos, 1997). Three anchoring screws (Plastics One Inc, Roanoke, VA) were implanted in the skull and the assemblage was anchored in place with dental cement. A dummy probe was placed in the guide cannulae until the start of microdialysis procedures.
2.5. Microdialysis Procedures
ACh release was measured in 4-, 10- and 22-month-old groups of mice before, during and after spontaneous alternation testing in the plus-maze. As noted above, the 10- and 22-month-old mice were those tested 6 months earlier on the spontaneous alternation task; a new group of 4-month-old mice was added for this microdialysis experiment. Microdialysis probes (CMA/7, 2 mm, Carnegie Medicin, Stockholm, Sweden) were inserted through the guide cannula into the hippocampus. During microdialysis, the probes were perfused continuously at a rate of 1.0 μl/min with artificial cerebrospinal fluid (aCSF, 128 mM NaCl, 2.5 mM KCl, 1.3 mM CaCl2, 2.1 mM MgCl2, 0.9 mM NaH2PO4, 2.0 mM Na2HPO4, 1.0 mM dextrose and adjusted to pH 7.4). The perfusate also contained 100 nM of the acetylcholinesterase inhibitor, neostigmine, included to enable the observation of changes in acetylcholine release in the hippocampus during behavioral testing (Chang, Savage and Gold, 2006). Temporal resolution for ACh samples was 6 min (6.0 μl/sample).
To equilibrate the dialysate with hippocampal extracellular fluid and to avoid temporary changes in extracellular neurotransmitter levels caused by acute tissue damage, the first hour of dialysate was discarded (Westerink and Timmerman, 1999). During this hour, the mice were kept in a holding cage with fresh bedding. Next, for ACh sampling, dialysate samples were collected every 6 min into small vials. The first four samples (each sample = 6 μl / 6 min), were collected as baseline samples while mice remained undisturbed in the holding cage. After the baseline sampling period, mice were placed in the maze for 6 min (one sample) for a spontaneous alternation test. An additional 6 samples were collected after the conclusion of behavioral testing. After the completion of microdialysis, all samples were stored at −80°C until assayed for ACh content.
2.6. ACh assay procedures
ACh content in each dialysate sample was assayed by high performance liquid chromatography in combination with electrochemical detection (Bioanalytical Systems Inc., West Lafayette, IN). The assay system included an ion-exchange microbore analytical column, a microbore ACh/Ch immobilized enzyme reactor (IMER) containing acetylcholinesterase and choline oxidase, an auxiliary electrode with radical flow electrochemical thin-layer cell and thin-layer gasket, a ‘wired’ enzyme electrode (a redox polymer film containing horseradish peroxidase coated on the surface of a 6mm glassy carbon working electrode), a DA-5 interface between detector and computer, controlling and analyzing software and a low-dispersion Rheodyne injection valve (model 9725i) with a 10 μl PEEK loop. Stable and relatively pulse-free flow was achieved with a Shimadzu LC-10ADvp pump. The potential held by the working electrode was 100 mV vs. a Ag/AgCl reference electrode. The mobile phase contained 50 mM Na2HPO4 (pH 8.5) and 0.005% ProClin™ 150 microbiocide. The flow rate was 140 μl/min. Injection volume in this experiment was 5.0 μl. The detection limit was ≤ fmol. The assay was completed in 12.5 min.
2.7. Histology
After behavioral procedures were completed, mice were deeply anesthetized with sodium pentobarbital and perfused with 30 ml sodium phosphate buffer (100 mM, pH 7.3), followed by 30 ml paraformaldehyde-picric acid solution (4% paraformaldehyde (w/v) and 15% saturated picric acid solution (v/v) in 100 mM phosphate buffer). The brains were fixed in the paraformaldehyde-picric acid solution for 48 hr after perfusion, and were then moved to a 25% sucrose solution for cryoprotection. Each brain was then sectioned at 50 μm using a Leica 1800 cryostat. To check probe placements, sections through the areas with cannulae tracks were mounted on slides, dried and stained with cresyl violet. Cannulae placements were evaluated using light microscopy.
2.8. Data analyses
Results for baseline behavior and chemistry experiments were analyzed by ANOVA for genotype, age and genotype by age interactions, followed by planned comparison t-tests by condition within age and genotype. Behavior results were also analyzed using one-sample t-tests vs. chance performance and chemistry results were also similarly analyzed for within-rat increases in ACh release during training. Analyses of physostigmine effects on alternation scores were similarly performed with repeated measure (saline x drug) ANOVAs.
3. Results
3.1. Spontaneous alternation scores
BASELINE PERFORMANCE
Ts65Dn mice exhibited alternation scores in the plus-maze that did not differ from chance at any age (Ps > 0.2). Control mice exhibited alternation scores above chance in both the 4- and 10-month-old groups (Ps < 0.05), but not in the 16-month-old group (P > 0.2). A trend toward a decrease in alternation scores by age was seen in the control mice [F(2,28) = 2.60, P < 0.1] but not the Ts65Dn mice [F(2,25) = 0.02, P > 0.9], the latter having near-chance scores at all ages. Ts65Dn mice had scores significantly lower than those of the same-age controls at both 4 and 10 months of age (P <0.05 and P < 0.01, respectively), but not at 16 months of age (P > 0.1) when the performance of controls had declined toward chance (Figure 1). Total number of arm entries during the 6-min test did not differ significantly by age or by strain.
Figure 1.
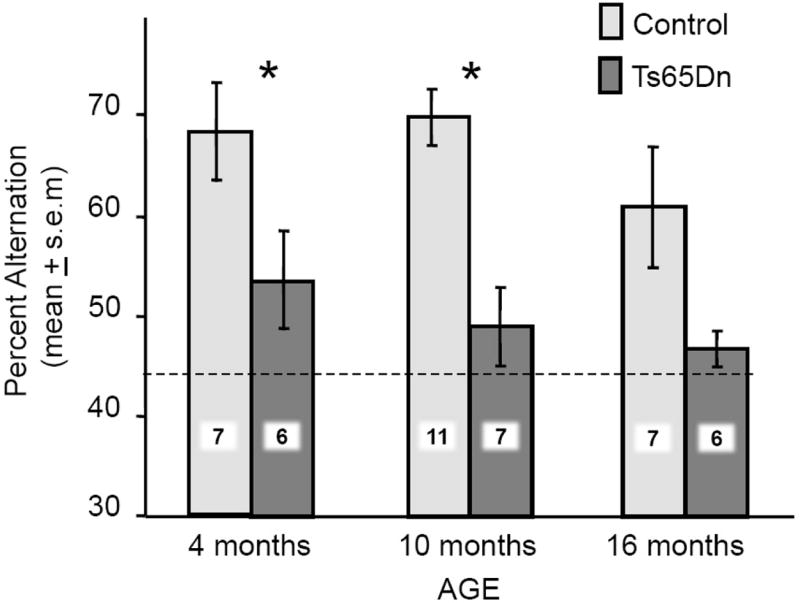
Spontaneous alternation scores across age in Ts65Dn and control mice. The dashed horizontal line shows chance performance. Note that the scores of Ts65Dn mice were significantly lower than those of same-age controls at 3–4 and 9–10 month of ages (*P< 0.05 and **P < 0.01 respectively). There was no difference at the oldest (16–17 month) age tested when controls also had relatively low scores. The numbers superimposed on bars indicate numbers of mice / group.
PHYSOSTIGMINE ENHANCEMENT
The effects of physostigmine on spontaneous alternation scores in the plus-shaped maze are presented in Figure 2 for the Ts65Dn and control mice; for clarity, the baseline (saline) scores for both genotypes are repeated from Figure 1. The main result was that physostigmine reversed deficits in alternation scores in young Ts65Dn mice, but the ability of physostigmine to enhance memory decreased with age. Physostigmine did not significantly modify alternation scores in 10- or 16-month-old Ts65Dn mice. A repeated (saline, physostigmine) measure ANOVA revealed significant effects of both age [F(2,38)=4.39, P<0.02] and genotype [F(1,38)=16.27, P<0.001]. The effect of age is evident in the scores of the Ts65Dn mice. In the 4-month-old Ts65Dn mice, but not older ages, physostigmine significantly enhanced alternation scores compared to those observed under the saline condition (P<0.001). The alternation scores attained by the young Ts65Dn mice under the physostigmine condition resulted in scores comparable to those of the control saline-treated mice (P>0.7). The interaction of genotype by drug condition was also statistically significant [F(1,31)=9.86, P<0.01], reflecting enhancement of alternation scores after physostigmine treatment in Ts65Dn mice but not controls.
Figure 2.
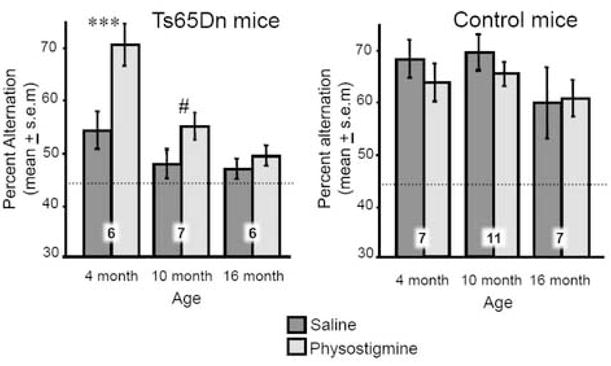
Effects of physostigmine on spontaneous alternation scores in Ts65Dn and control mice. The dashed horizontal line shows chance performance. The numbers superimposed on bars indicate numbers of mice / group. (Left.) Note that physostigmine treatment fully reversed the deficit in alternation performance in 3–4 month-old Ts65Dn mice (***P<0.001) but not at older ages. (Right). Physostigmine did not significantly enhance memory in the control mice at any age.
3.2. ACh release in the hippocampus before and during alternation testing
Only data from mice with correct cannulae placements, as in Figure 3, were included in this experiment. Basal values of ACh release in the hippocampus are shown in Figure 4. Ts65Dn and control mice had comparable baseline levels at all ages [F(2,31)=0.14, P>0.9]. Baseline release in Ts65Dn and control mice decreased slightly with age, but the declines were not statistically significant (Ps>0.2 for both genotypes).
Figure 3.
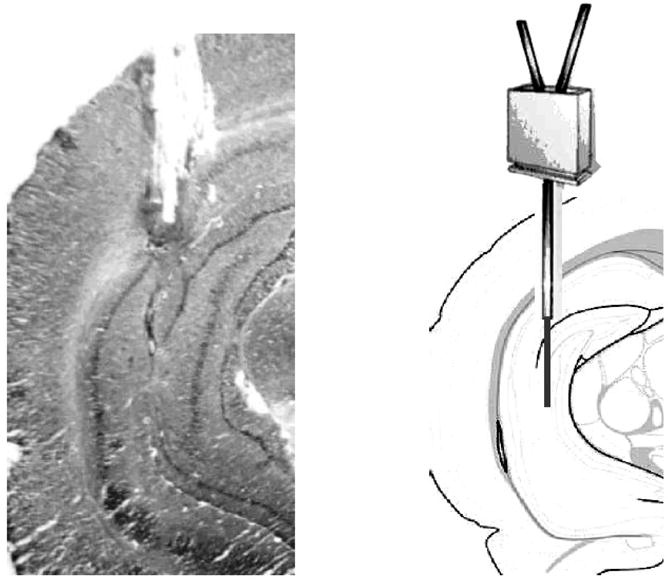
(Left.) Example of an acceptable cannula placement in which the active portion of the probe was positioned entirely within the hippocampus. (Right.) Schematic of a probe scaled to size overlaying a brain atlas section adapted from Franklin and Paxinos (1997).
Figure 4.
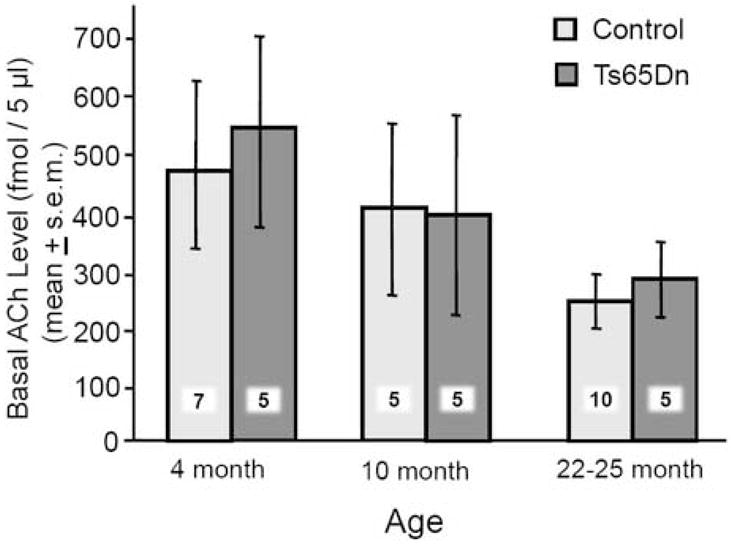
Baseline levels of ACh in the hippocampus of Ts65Dn and control mice at three ages. All values were calibrated for recovery. Note that the two sets of mice exhibited comparable baseline levels of ACh release at all ages. Both groups showed a slight, non-significant, decline in baseline ACh levels with age. The numbers superimposed on bars indicate numbers of mice / group.
The changes in ACh release during spontaneous alternation testing are shown in Figure 5. In these dialysis samples, the percent increase in ACh release was significantly above baseline in both control and Ts65Dn mice only in the 4-month-old groups (Ps < 0.01, one-sample t-tests). The magnitude of the percent increase in release of ACh during alternation testing declined with age in both control and Ts65Dn mice. The overall declines across age for percent increase in release of ACh during spontaneous alternation testing were statistically significant [F(2,31)=13.31, P<0.001]; the age-related decline was significant within the control mice [F(2,19)=7.85, P<0.01] and there was a similar trend within the Ts65Dn mice [F(2,12)=3.23, P<0.07]. Of particular interest, testing-related increases in the release of ACh were higher in the control mice than in Ts65Dn mice at 4-month of age (P <0.05), but not at older ages (Ps > 0.5), resulting in a significant interaction of genotype by age [F(2,31)=3.51, P<0.05].
Figure 5.
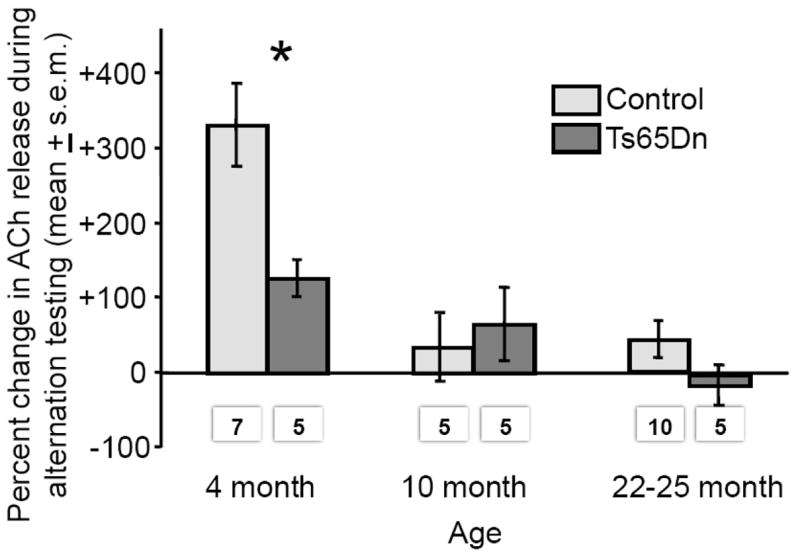
Percent change in ACh release in the hippocampus during spontaneous alternation testing. Note that ACh release increased significantly during testing only in 4-month-old controls and Ts65Dn mice. The magnitude of that increase declined with age in both genotypes. Ts65Dn mice had lower percent increase in ACh release than did same-age controls at 4 months (*Ps<0.05) but not at older ages. The numbers below bars indicate numbers of mice / group.
4. Discussion
The results of these experiments extend in several respects the findings about learning deficits and cholinergic dysfunctions in Ts65Dn mice. The main findings are: 1) Relative to controls, the Ts65Dn mice have impaired spatial working memory at 4 and 10 months of age. 2) Physostigmine ameliorates the impairment in alternation scores in the Ts65Dn mice, but only at the youngest age tested. 2) Increases in ACh release in the hippocampus in response to alternation testing are muted in young Ts65Dn mice compared to controls.
4.1. Spontaneous Alternation
Ts65Dn mice failed to exceed chance performance at any age tested, from 4–16 months of age. Therefore, the present findings do not demonstrate age-related impairments in these mice, although it is quite possible that memory in the spontaneous alternation task would be intact at ages younger than 4 months. This possibility is supported by the findings of several other experiments in which spontaneous alternation performance deteriorated with age in normal rats and mice and in models of Alzheimer’s Disease (Corcoran et al., 2002; Lalonde et al., 2003; Lamberty and Gower, 1990; McNay and Gold, 2001; Miller et al., 1999; Stone et al., 1992; Willig et al., 1987; Zornetzer et al, 1982).
The findings of memory deficits in the plus-maze in 3–4 month-old Ts65Dn mice are near the young ages of the onset of impairments reported before. For example, deficits in context discrimination learning were seen at 3 months, not at 4 months, but again at 5 months and older (Hyde and Crnic, 2001). On a test of working and reference memory in a water-escape radial-arm maze task, learning was impaired in both 4- and 6-month-old Ts65Dn mice (Hunter, Bimonte and Granholm, 2003). In most other studies, learning and memory deficits in Ts65Dn mice were found at 6 months or older (Demas et al., 1996; Escorihuela et al., 1995, 1998; Holtzman et al., 1996; Reeves et al., 1995), although deficits in Ts65Dn mice on some versions of swim tasks are evident as early as 4 months of age (Seo and Isacson, 2005). Thus, the present findings are generally consistent with many reports of memory impairments in this partial trisomy model of Down syndrome, most examining mice at 6 months and older (cf. Sérégaza et al., 2006). With multiple task-dependent neurobiological mechanisms involved in learning and memory, different tasks are likely to show different functional sensitivities and therefore different ages of onset of impairments.
The control mice exhibited alternation scores above chance at 4 and 10 months, but not at 16 months. However, even at 16 months, the values had not fully reached chance levels, leading to a non-significant trend for development of impairments with aging. A clear demonstration of age-related deficits in this task likely requires tests at older ages. In Fischer 344 rats, deficits on a similar task were evident at 24 months of age (McNay and Gold, 2001).
4.2. Physostigmine enhancement of memory
As a cholinesterase inhibitor, physostigmine augmentation of cholinergic functions relies on at least partial integrity of acetylcholine neurons. Results obtained here show that physostigmine enhanced alternation scores of Ts65Dn mice in young mice. Of particular interest, the effectiveness of physostigmine in enhancing memory decreased as the age of the Ts65Dn mice increased. On the basis of these findings alone, it would appear that ACh functions in the hippocampus had deteriorated at older ages to an extent below which enhancement of residual functions were inadequate to support memory. The results obtained with measures of ACh release during behavioral were consistent with this view. At older ages, alternation testing did not result in increases in release of ACh. An important future experiment to address the significance of the age-related decline in the efficacy of physostigmine in enhancing memory in Ts65Dn mice will be to obtain parallel measures of ACh release in the presence and absence of physostigmine during spontaneous alternation testing.
Physostigmine did not enhance memory in controls at any age group. The failure to see enhancement of spontaneous alternation performance may reflect the high scores evident in each age group, from 4–16 months. Across multiple studies (e.g. Pych et al., 2005; Ragozzino, Unick and Gold, 1996, 1998; Stefani and Gold, 2001, Stone, Rudd and Gold, 1992), alternation scores appear to be maximal at values approximately half-way between chance performance (here 44%) and a simple right/left response bias (100%), i.e. at 72%. At the ages tested in the present experiment, control performance under the saline condition did not differ significantly from this theoretical maximum score. Physostigmine might be more likely to enhance memory after an age-related deficit in alternation scores emerges, presumably at older ages in the control mice. This view is supported by evidence from other studies showing that acetylcholinesterase inhibitors enhance memory in aged rats and mice (e.g., Rispoli et al., 2006; Flood et al., 1993; Normile and Altman, 1992; Riekkinen et al., 1991), and that such drugs can be effective in memory-impaired aged rats but not in young rats (Ohta et al., 1991; Castellano et al., 1989). Similarly, acetylcholinesterase inhibitors are particularly effective in enhancing memory in rats and mice with compromised cholinergic functions and with other memory-impairing pharmacological manipulations (Chang and Gold, 2004; Degroot and Parent, 2000; Ukai et al., 1995; Walker and Gold, 1992; Beracochea et al., 1992; Stone et al., 1991; Murray and Fibiger, 1986). Acetylcholinesterase inhibitors are also effective enhancers of memory in several rodent models of Alzheimer’s Disease (Maurice et al., 1996; Dong et al., 2005; Popovi et al., 1997; Santucci et al., 1991). Thus, based on the results seen in the Ts65Dn mice, physostigmine might be effective in older control mice only if the cholinergic system has sufficient remaining function. This interpretation is consistent with many findings indicating that physostigmine and other similar acetylcholinesterase inhibitors enhance memory in spontaneous alternation tasks in rats and mice that are senescent or that have compromised cholinergic functions. An alternative possibility is that the 50 mg/kg dose of physostigmine that enhanced memory in Ts65Dn mice is not optimal for enhancement in controls.
4.3. Acetylcholine release
Spontaneous alternation tasks as used in present study have previously been found to elicit ACh release in the hippocampus of rats in a manner related to memory performance on this task (Chang and Gold, 2004; Ragozzino and Gold, 1995; Ragozzino et al., 1996, 1998; Pych et al., 2005). The microdialysis results presented here suggest that the onset of dysfunctions in septohippocampal cholinergic functions, i.e. ACh release during maze testing but not at baseline, occurs prior to 3–4 months in Ts65Dn mice. Although baseline measures were comparable between Ts65Dn and control mice, the responsiveness to the challenge posed by behavioral testing revealed significant decrements in the Ts65Dn mice, potentially contributing to the impairments in working memory seen at the youngest ages tested in the present experiments. It is also possible that ACh release increased more at the young ages because the maze was more novel for the young mice that were naive with respect to alternation testing than for the older mice, which had been tested 6 months earlier. This is an explanation for which additional tests are needed. Nonetheless, it is clear that the young control mice exhibit greater increases in release during alternation testing than do the young Ts65Dn mice.
The Ts65Dn mice retained the capacity to increase release of ACh in the hippocampus at four months. However, even at four months of age, the youngest age tested here,Ts65Dn mice had responses of ACh release to behavioral testing that were significantly reduced relatively to the controls. Together with the findings that physostigmine enhanced memory in the Ts65Dn mice only in the 4-month-old group, the findings suggest that residual cholinergic functions can support memory at that age. Prior findings indicate that some markers of ACh function in Ts65Dn mice are intact at young ages but emerge in adulthood, generally evident after 6 months of age (Granholm, Sanders and Crnic, 2000). The development of cholinergic dysfunctions in Ts65Dn mice parallels a similar situation in humans with Down syndrome, in whom the basal forebrain cholinergic system appears normal at birth but degenerates in early adulthood (Casanova et al., 1985; Head et al., 2001; Yates et al., 1983). The cholinergic neurons that project to the hippocampus and provide the source of the ACh release measured in the present experiment are located in the medial septum. Therefore, it is particularly relevant to note findings using choline acetyltransferase immunocytochemistry showing that the cell size of cholinergic neurons in the medial septum declines after 4 months of age (Granholm et al., 2002). Likely related to these results, immunostaining for the NGF receptor, trkA, a supportive factor for ACh functions, also declines between 4 and 6 months of aged (Granholm, Sanders and Crnic, 2000; Holtzman et al., 1996).
4.4 General conclusions
The parallel age-related declines in memory and behaviorally-stimulated ACh release in Ts65Dn mice are compatible with past evidence using other tasks and other measures of ACh function (cf. Sérégaza et al., 2006). In particular, previous studies have noted a decrease at early ages in immunostaining for choline acetyltransferase-positive (ChAT+) neurons in the medial septum in Ts65Dn mice (Granholm, Sanders and Crnic, 2000; Holtzman et al., 1996). Decreases in ChAT+ neurons appear as early as 6 months of age in these mice, with a decline during aging that is distinctly earlier in Ts65Dn mice than in controls. It is interesting that in the present experiment, the baseline levels of ACh release are comparable at all ages tested. However, the responsiveness of ACh release to behavioral testing procedures is reduced in Ts65Dn mice compared to controls even at 3–4 months of age. Similarly, physostigmine enhances memory in the 3–4 month-old but not older Ts65Dn mice. These findings suggest that the decline in ACh functions noted by anatomical measures are evident in terms of release only under conditions when the system is challenged by conditions of behavior or drug administration. According to this view, the function of the residual ChAT+ neurons is sufficient to maintain baseline levels of release but is not sufficient to meet the demands of responding to spontaneous alternation testing or to physostigmine treatment.
The interest in the parallel changes in ACh and memory is derived from extensive evidence that ACh regulates memory formation, neural plasticity, and the interaction of multiple memory systems in selection of strategies for learning (cf. Gold, 2004), as well as attention mechanisms that might be important for spatial working memory (cf. Sarter, Gehring and Kozak, 2006). The present findings suggest that there may be linkage between cholinergic functions and cognitive impairments in the Ts65Dn mice, particularly evident with the use of comparisons across ages. The ability of physostigmine to enhance memory in the Ts65Dn mice clearly decreases with age. Also, the magnitude of the increase in ACh release in response to training declines with age. However, comparable levels of ACh release are seen in the middle-aged control mice that have good memory scores and in the middle-aged Ts65Dn mice that have poor memory scores. Thus, the relationship between ACh release and performance is imperfect and may well reflect other consequences of age such as additive dysfunctions and compensatory changes within cholinergic systems. In addition, the imperfect association of ACh markers with behavior may also reflect independent consequences of age in the partial trisomy mice, for example in other neurotransmitters (Fernandez et al., 2007) or neurobiological processes. These other processes include impaired long-term potentiation (Costa and Grybko, 2005; Fernandez et al., 2007; Kleschevnikov et al., 2004; Siarey et al., 1997), enhanced long-term depression (Siarey et al., 1999), impaired expression of brain-derived neurotrophic factor (Seo and Isacson, 2005), altered signaling pathways (Siarey et al., 2006), and altered neuroanatomical organization of the hippocampus (Belichenko et al., 2004; Hanson et al., 2007; Kurt et al., 2004; Lorenzi and Reeves, 2006).
Most studies of brain functions in Ts65Dn mice, including the present experiments, have focused on measures of the hippocampus in Ts65Dn mice. However, it is certainly the case that alterations in other brain areas are important to the neural and cognitive impairments in these mice. In this regard, changes in brain-derived neurotrophic functions are evident in frontal cortex (Bimonte-Nelson et al., 2003) as well as hippocampus. Also, as in humans with Down syndrome (cf. Shapiro, 2001), the volume of the cerebellum of Ts65Dn mice is reduced, with decreased numbers of both granule and Purkinje cells perhaps resulting from dysfunctions of cell signaling mechanisms during development (Olson et al., 2004; Roper et al., 2006). Thus, understanding of the neural and cognitive phenotypic differences between partial trisomy mice and diploid mice will require assessments of many variables across age.
In summary, the current study reveals a deficit in spatial working memory in young and middle aged Ts65Dn mice, a model of Down syndrome. Comparisons of the release of ACh in the hippocampus of these mice and controls at a wide range of ages revealed comparable levels of release at baseline but smaller increases in the Ts65Dn mice in response to behavioral testing. In addition, the ability of an indirect acetylcholine agonist to reverse memory impairments in Ts65Dn mice declined with age. Because baseline release of ACh in the hippocampus of Ts65Dn mice declined with age but was comparable to the levels seen in control mice throughout the lifespan, these findings suggest that the memory deficits in young to middle-aged Ts65Dn mice reflect in part disruption of the ability of forebrain cholinergic mechanisms to respond when needed.
Footnotes
Now at Department of Animal Sciences, University of Illinois, Urbana, Illinois 61801
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Belichenko PV, Masliah E, Kleschevnikov AM, Villar AJ, Epstein CJ, Salehi A, Mobley WC. Synaptic structural abnormalities in the Ts65Dn mouse model of Down Syndrome. Journal of Comparative Neurology. 2004;480:281–298. doi: 10.1002/cne.20337. [DOI] [PubMed] [Google Scholar]
- Beracochea D, Micheau J, Jaffard R. Memory deficits following chronic alcohol consumption in mice: relationships with hippocampal and cortical cholinergic activities. Pharmacology, Biochemistry & Behavior. 1992;42:749–753. doi: 10.1016/0091-3057(92)90024-a. [DOI] [PubMed] [Google Scholar]
- Bimonte-Nelson HA, Hunter CL, Nelson ME, Granholm AC. Frontal cortex BDNF levels correlate with working memory in an animal model of Down syndrome. Behavioural Brain Research. 2003;139:47–57. doi: 10.1016/s0166-4328(02)00082-7. [DOI] [PubMed] [Google Scholar]
- Candy JM, Perry EK, Perry RH, Court JA, Oakley AE, Edwardson JA. The current status of the cortical cholinergic system in Alzheimer’s disease and Parkinson’s disease. Progress in Brain Research. 1998;70:105–132. doi: 10.1016/s0079-6123(08)64300-9. [DOI] [PubMed] [Google Scholar]
- Casanova MF, Walker LC, Whitehouse PJ, Price DL. Abnormalities of the nucleus basalis in Down’s syndrome. Annals of Neurology. 1985;18:310–313. doi: 10.1002/ana.410180306. [DOI] [PubMed] [Google Scholar]
- Castellano C, Petkov VV, Kehayov R, Panuscheva N. Age-determined changes in the effects of a newly synthesized cholinesterase inhibitor on exploratory behavior, memory and striatal cholinesterase activity in rats. Acta Physiologica et Pharmacologica Bulgarica. 1989;15:3–9. [PubMed] [Google Scholar]
- Chang Q, Gold PE. Impaired and spared cholinergic functions in the hippocampus after lesions of the medial septum/vertical limb of the diagonal band with 192 IgG-saporin. Hippocampus. 2004;14:170–179. doi: 10.1002/hipo.10160. [DOI] [PubMed] [Google Scholar]
- Chang Q, Savage LM, Gold PE. Microdialysis measures of functional increases in ACh release in the hippocampus with and without inclusion of acetylcholinesterase inhibitors in the perfusate. Journal of Neurochemistry. 2006;97:697–706. doi: 10.1111/j.1471-4159.2006.03765.x. [DOI] [PubMed] [Google Scholar]
- Corcoran KA, Lu Y, Turner RS, Maren S. Overexpression of hAPPswe impairs rewarded alternation and contextual fear conditioning in a transgenic mouse model of Alzheimer’s disease. Learning & Memory. 2002;9:243–252. doi: 10.1101/lm.51002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa AC, Grybko MJ. Deficits in hippocampal CA1 LTP induced by TBS but not HFS in the Ts65Dn mouse: a model of Down syndrome. Neuroscience Letters. 2005;382:317–322. doi: 10.1016/j.neulet.2005.03.031. [DOI] [PubMed] [Google Scholar]
- Coussons-Read ME, Crnic LS. Behavioral assessment of the Ts65Dn mouse, a model for Down syndrome: altered behavior in the elevated plus maze and open field. Behavioral Genetics. 1996;26:7–13. doi: 10.1007/BF02361154. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Price DL, DeLong MR. Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science. 1983;219:1184–1190. doi: 10.1126/science.6338589. [DOI] [PubMed] [Google Scholar]
- Davisson MT, Bechtel LJ, Akeson EC, Fortna A, Slavov D, Gardiner K. Evolutionary breakpoints on human chromosome 21. Genomics. 2001;78:99–106. doi: 10.1006/geno.2001.6639. [DOI] [PubMed] [Google Scholar]
- Davisson MT, Costa AC. Mouse models of human genetic neurological disease. In: Popko B, editor. Mouse models of Down syndrome. New York: Plenum; 1999. pp. 297–327. [Google Scholar]
- Davisson MT, Schmidt C, Akeson EC. Segmental trisomy of murine chromosome 16: a new model system for studying Down syndrome. Progress in Clinical and Biological Research. 1990;360:263–280. [PubMed] [Google Scholar]
- Davisson MT, Schmidt C, Reeves RH, Irving NG, Akeson EC, Harris BS, Bronson RT. Segmental trisomy as a mouse model for Down syndrome. Progress in Clinical and Biological Research. 1993;384:117–133. [PubMed] [Google Scholar]
- Degroot A, Parent MB. Increasing acetylcholine levels in the hippocampus or entorhinal cortex reverses the impairing effects of septal GABA receptor activation on spontaneous alternation. Learning & Memory. 2000;7:293–302. doi: 10.1101/lm.32200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demas GE, Nelson RJ, Krueger BK, Yarowsky PJ. Spatial memory deficits in segmental trisomic Ts65Dn mice. Behavioural Brain Research. 1996;82:85–92. doi: 10.1016/s0166-4328(97)81111-4. [DOI] [PubMed] [Google Scholar]
- Demas GE, Nelson RJ, Krueger BK, Yarowsky PJ. Impaired spatial working and reference memory in segmental trisomy (Ts65Dn) mice. Behavioural Brain Research. 1998;90:199–201. doi: 10.1016/s0166-4328(97)00116-2. [DOI] [PubMed] [Google Scholar]
- Dember WN, Richman CL. Spontaneous Alternation Behavior. New York: Springer-Verlag; 1989. [Google Scholar]
- Dong H, Csernansky CA, Martin MV, Bertchume A, Vallera D, Csernansky JG. Acetylcholinesterase inhibitors ameliorate behavioral deficits in the Tg2576 mouse model of Alzheimer’s disease. Psychopharmacology. 2005;181:145–152. doi: 10.1007/s00213-005-2230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll LL, Carroll JC, Moon J, Crnic LS, Levitsky DA, Strupp BJ. Impaired sustained attention and error-induced stereotypy in the aged Ts65Dn mouse: a mouse model of Down syndrome and Alzheimer’s disease. Behavioral Neuroscience. 2004;118:1196–1205. doi: 10.1037/0735-7044.118.6.1196. [DOI] [PubMed] [Google Scholar]
- Escorihuela RM, Fernandez-Teruel A, Vallina IF, Baamonde C, Lumbreras MA, Dierssen M, Tobena A, Florez J. A behavioral assessment of Ts65Dn mice: a putative Down syndrome model. Neuroscience Letters. 1995;199:143–146. doi: 10.1016/0304-3940(95)12052-6. [DOI] [PubMed] [Google Scholar]
- Escorihuela RM, Vallina IF, Martinez-Cue C, Baamonde C, Dierssen M, Tobena A, Florez J, Fernandez-Teruel A. Impaired short- and long-term memory in Ts65Dn mice, a model for Down syndrome. Neuroscience Letters. 1998;247:171–174. doi: 10.1016/s0304-3940(98)00317-6. [DOI] [PubMed] [Google Scholar]
- Fernandez F, Morishita W, Zuniga E, Nguyen J, Blank M, Malenka RC, Garner CC. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nature Neuroscience. 2007;10:411–413. doi: 10.1038/nn1860. [DOI] [PubMed] [Google Scholar]
- Flood JF, Morley JE, La Reginna M. Age-related changes in the pharmacological improvement of retention in senescence accelerated mouse (SAM) Neurobiology of Aging. 1993;14:159–166. doi: 10.1016/0197-4580(93)90092-p. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. San Diego, CA: Academic Press; 1997. [Google Scholar]
- Galdzicki Z, Siarey R, Pearce R, Stoll J, Rapoport SI. On the cause of mental retardation in Down syndrome: extrapolation from full and segmental trisomy 16 mouse models. Brain Research - Brain Research Reviews. 2001;35:115–145. doi: 10.1016/s0926-6410(00)00074-4. [DOI] [PubMed] [Google Scholar]
- Giovannini MG, Rakovska A, Benton RS, Pazzagli M, Bianchi L, Pepeu G. Effects of novelty and habituation on acetylcholine, GABA, and glutamate release from the frontal cortex and hippocampus of freely moving rats. Neuroscience. 2001;106:43–53. doi: 10.1016/s0306-4522(01)00266-4. [DOI] [PubMed] [Google Scholar]
- Glenner GG. Alzheimer’s disease: its proteins and genes. Cell. 1988;52:307–308. doi: 10.1016/s0092-8674(88)80021-7. [DOI] [PubMed] [Google Scholar]
- Gold PE. Drug enhancement of memory in aged rodents and humans. In: Carroll ME, Overmier JB, editors. Animal research and human health: Advancing human welfare through behavioral science. Washington, DC: American Psychological Association.; 2001. pp. 293–304. [Google Scholar]
- Gold PE. Coordination of multiple memory systems. Neurobiology of Learning and Memory. 2004;82:230–242. doi: 10.1016/j.nlm.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Gold PE. Memory enhancers. In: Byrne J, editor. Learning and Memory: a Comprehensive Reference. Elsevier Science; Oxford: 2007. In press. [Google Scholar]
- Granholm AC, Ford KA, Hyde LA, Bimonte HA, Hunter CL, Nelson M, Albeck D, Sanders LA, Mufson EJ, Crnic LS. Estrogen restores cognition and cholinergic phenotype in an animal model of Down syndrome. Physiology & Behavior. 2002;77:371–385. doi: 10.1016/s0031-9384(02)00884-3. [DOI] [PubMed] [Google Scholar]
- Granholm AC, Sanders LA, Crnic LS. Loss of cholinergic phenotype in basal forebrain coincides with cognitive decline in a mouse model of Down’s syndrome. Experimental Neurology. 2000;161:647–663. doi: 10.1006/exnr.1999.7289. [DOI] [PubMed] [Google Scholar]
- Hanson JE, Blank M, Valenzuela RA, Garner CC, Madison DV. The functional nature of synaptic circuitry is altered in area CA3 of the hippocampus in a mouse model of Down’s syndrome. Journal of Physiology. 2007;579(Part 1):53–67. doi: 10.1113/jphysiol.2006.114868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head E, Azizeh BY, Lott IT, Tenner AJ, Cotman CW, Cribbs DH. Complement association with neurons and beta-amyloid deposition in the brains of aged individuals with Down Syndrome. Neurobiology of Disease. 2001;8:252–265. doi: 10.1006/nbdi.2000.0380. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Santucci D, Kilbridge J, Chua-Couzens J, Fontana DJ, Daniels SE, Johnson RM, Chen K, Sun Y, Carlson E, Alleva E, Epstein CJ, Mobley WC. Developmental abnormalities and age-related neurodegeneration in a mouse model of Down syndrome. Proceedings of the National Academy of Sciences USA. 1996;93:13333–13338. doi: 10.1073/pnas.93.23.13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter CL, Bimonte HA, Granholm AC. Behavioral comparison of 4 and 6 month-old Ts65Dn mice: age-related impairments in working and reference memory. Behavioural Brain Research. 2003;138:121–131. doi: 10.1016/s0166-4328(02)00275-9. [DOI] [PubMed] [Google Scholar]
- Hyde LA, Crnic LS. Age-related deficits in context discrimination learning in Ts65Dn mice that model Down syndrome and Alzheimer’s disease. Behavioral Neuroscience. 2001;115:1239–1246. [PubMed] [Google Scholar]
- Kleschevnikov AM, Belichenko PV, Villar AJ, Epstein CJ, Malenka RC, Mobley WC. Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. Journal of Neuroscience. 2004;24:8153–8160. doi: 10.1523/JNEUROSCI.1766-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korenberg JR, Pulst SM, Neve RL, West R. The Alzheimer amyloid precursor protein maps to human chromosome 21 bands q21.105-q21.05. Genomics. 1989;5:124–127. doi: 10.1016/0888-7543(89)90095-5. [DOI] [PubMed] [Google Scholar]
- Kurt MA, Kafa MI, Dierssen M, Davies DC. Deficits of neuronal density in CA1 and synaptic density in the dentate gyrus, CA3 and CA1, in a mouse model of Down syndrome. Brain Research. 2004;1022:101–109. doi: 10.1016/j.brainres.2004.06.075. [DOI] [PubMed] [Google Scholar]
- LaLonde R. The neurobiological basis of spontaneous alternation. Neuroscience and Biobehavioral Reviews. 2002;26:91–104. doi: 10.1016/s0149-7634(01)00041-0. [DOI] [PubMed] [Google Scholar]
- Lalonde R, Lewis TL, Strazielle C, Kim H, Fukuchi K. Transgenic mice expressing the betaAPP695SWE mutation: effects on exploratory activity, anxiety, and motor coordination. Brain Research. 2003;977:38–45. doi: 10.1016/s0006-8993(03)02694-5. [DOI] [PubMed] [Google Scholar]
- Lamberty Y, Gower AJ. Age-related changes in spontaneous behavior and learning in NMRI mice from maturity to middle age. Physiology & Behavior. 1990;47:1137–1144. doi: 10.1016/0031-9384(90)90364-a. [DOI] [PubMed] [Google Scholar]
- Lorenzi HA, Reeves RH. Hippocampal hypocellularity in the Ts65Dn mouse originates early in development. Brain Research. 2006;1104:153–159. doi: 10.1016/j.brainres.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Mann DM, Esiri MM. The pattern of acquisition of plaques and tangles in the brains of patients under 50 years of age with Down’s syndrome. Journal of Neurological Science. 1989;89:169–179. doi: 10.1016/0022-510x(89)90019-1. [DOI] [PubMed] [Google Scholar]
- Marcell MM, Armstrong V. Auditory and visual sequential memory of Down syndrome and nonretarded children. American Journal of Mental Deficiencies. 1982;87:86–95. [PubMed] [Google Scholar]
- Maurice T, Lockhard BP, Privat A. Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Research. 1996;706:181–193. doi: 10.1016/0006-8993(95)01032-7. [DOI] [PubMed] [Google Scholar]
- McNay EC, Gold PE. Age-related differences in hippocampal extracellular fluid glucose concentration during behavioral testing and following systemic glucose administration. Journal of Gerontology Series A - Biological Sciences and Medical Sciences. 2001;56:B66–B71. doi: 10.1093/gerona/56.2.b66. [DOI] [PubMed] [Google Scholar]
- Miller MM, Hyder SM, Assayag R, Panarella SR, Tousignant P, Franklin KB. Estrogen modulates spontaneous alternation and the cholinergic phenotype in the basal forebrain. Neuroscience. 1999;91:1143–1153. doi: 10.1016/s0306-4522(98)00690-3. [DOI] [PubMed] [Google Scholar]
- Murray CL, Fibiger HC. Pilocarpine and physostigmine attenuate spatial memory impairments produced by lesions of the nucleus basalis magnocellularis. Behavioral Neuroscience. 1986;100:23–32. doi: 10.1037//0735-7044.100.1.23. [DOI] [PubMed] [Google Scholar]
- Nadel L. Down’s syndrome: a genetic disorder in biobehavioral perspective. Genes Brain and Behavior. 2003;2:156–166. doi: 10.1034/j.1601-183x.2003.00026.x. [DOI] [PubMed] [Google Scholar]
- Normile HJ, Altman HJ. Effects of combined acetylcholinesterase inhibition and serotonergic receptor blockade on age-associated memory impairments in rats. Neurobiology of Aging. 1992;13:735–740. doi: 10.1016/0197-4580(92)90097-h. [DOI] [PubMed] [Google Scholar]
- Ohta H, Ni XH, Matsumoto K, Watanabe H. Working memory deficit in aged rats in delayed nonmatching to position task and effect of physostigmine on performance of young and aged rats. Japanese Journal of Pharmacology. 1991;56:303–309. doi: 10.1254/jjp.56.303. [DOI] [PubMed] [Google Scholar]
- Olson LE, Roper RJ, Baxter LL, Carlson EJ, Epstein CJ, Reeves RH. Down syndrome mouse models Ts65Dn, Ts1Cje, and Ms1Cje/Ts65Dn exhibit variable severity of cerebellar phenotypes. Developmental Dynamics. 2004;230:581–589. doi: 10.1002/dvdy.20079. [DOI] [PubMed] [Google Scholar]
- Popovi M, Popovi N, Jovanova-Nesi K, Bokonji D, Dobri S, Kosti VS, Rosi N. Effect of physostigmine and verapamil on active avoidance in an experimerntal model of Alzheimer’s disease. International Journal of Neuroscience. 1997;90:87–97. doi: 10.3109/00207459709000628. [DOI] [PubMed] [Google Scholar]
- Pych JC, Chang Q, Colon-Rivera C, Gold PE. Acetylcholine release in hippocampus and striatum during training on a rewarded spontaneous alternation task. Neurobiology of Learning and Memory. 2005;84:93–101. doi: 10.1016/j.nlm.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Ragozzino ME, Gold PE. Glucose injections into the medial septum reverse the effects of intraseptal morphine infusions on hippocampal acetylcholine output and memory. Neuroscience. 1995;68:981–988. doi: 10.1016/0306-4522(95)00204-v. [DOI] [PubMed] [Google Scholar]
- Ragozzino ME, Pal SN, Unick K, Stefani MR, Gold PE. Modulation of hippocampal acetylcholine release and spontaneous alternation scores by intrahippocampal glucose injections. Journal of Neuroscience. 1998;18:1595–1601. doi: 10.1523/JNEUROSCI.18-04-01595.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragozzino ME, Unick KE, Gold PE. Hippocampal acetylcholine release during memory testing in rats: augmentation by glucose. Proceedings of the National Academy of Sciences USA. 1996;93:4693–4698. doi: 10.1073/pnas.93.10.4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, Schmidt C, Bronson RT, Davisson MT. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nature Genetics. 1995;11:177–184. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- Riekkinen P, Jr, Riekkinen Ml, Lahtinen H, Sirvio J, Valjakka A, Riekkinen P. Tetrahydroaminoacridine improves passive avoidance retention defects induced by aging and medial septal lesion but not by fimbria-fornix lesion. Brain Research Bulletin. 1991;27:587–594. doi: 10.1016/0361-9230(91)90031-e. [DOI] [PubMed] [Google Scholar]
- Rispoli V, Marra R, Costa N, Scipione L, Rotiroti D, De Vita D, Liveratore F, Carelli V. Choline pivaloyl ester strengthened the benefit effects of Tacrine and Galantamine on electroencephalographic and cognitive performances in nucleus basalis magnocellularis-lesioned and aged rats. Pharmacology, Biochemistry & Behavior. 2006;84:453–467. doi: 10.1016/j.pbb.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Roper RJ, Baxter LL, Saran NG, Klinedinst DK, Beachy PA, Reeves RH. Defective cerebellar response to mitogenic Hedgehog signaling in Down syndrome mice. Proceedings of the National Academy of Sciences USA. 2006;103:1452–1456. doi: 10.1073/pnas.0510750103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sago H, Carlson EJ, Smith DJ, Rubin EM, Crnic LS, Huang TT, Epstein CJ. Genetic dissection of region associated with behavioral abnormalities in mouse models for Down syndrome. Pediatric Research. 2000;48:606–613. doi: 10.1203/00006450-200011000-00009. [DOI] [PubMed] [Google Scholar]
- Santucci AC, Haroutunian V, Davis KL. Pharmacological alleviation of combined cholinergic/noradrenergic lesion-induced memory deficits in rats. Clinical Neuropharmacology, 14, Suppl. 1991;1:S1–S8. doi: 10.1097/00002826-199114001-00002. [DOI] [PubMed] [Google Scholar]
- Sarter M, Bodewitz G, Stephens DN. Attenuation of scopolamine-induced impairment of spontaneous alteration behaviour by antagonist but not inverse agonist and agonist beta-carbolines. Psychopharmacology (Berlin) 1988;94:491–495. doi: 10.1007/BF00212843. [DOI] [PubMed] [Google Scholar]
- Sarter M, Gehring WJ, Kozak R. More attention must be paid: the neurobiology of attentional effort. Brain Research - Brain Research Reviews. 2006;51:145–160. doi: 10.1016/j.brainresrev.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Seo H, Isacson O. Abnormal APP, cholinergic and cognitive function in Ts65Dn Down’s model mice. Experimental Neurology. 2005;193:469–480. doi: 10.1016/j.expneurol.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Sérégaza Z, Roubertoux PL, Jamon M, Soumireu-Mourat B. Mouse models of cognitive disorders in trisomy 21: a review. Behavior Genetics. 2006;36:387–404. doi: 10.1007/s10519-006-9056-9. [DOI] [PubMed] [Google Scholar]
- Shapiro BL. Developmental instability of the cerebellum and its relevance to Down syndrome. Journal of Neural Transmission Supplementum. 2001;61:11–34. doi: 10.1007/978-3-7091-6262-0_2. [DOI] [PubMed] [Google Scholar]
- Siarey RJ, Carlson EJ, Epstein CJ, Balbo A, Rapoport SI, Galdzicki Z. Increased synaptic depression in the Ts65Dn mouse, a model for mental retardation in Down syndrome. Neuropharmacology. 1999;38:1917–1920. doi: 10.1016/s0028-3908(99)00083-0. [DOI] [PubMed] [Google Scholar]
- Siarey RJ, Kline-Burgess A, Cho M, Balbo A, Best TK, Harashima C, Klann E, Galdzicki Z. Altered signaling pathways underlying abnormal hippocampal synaptic plasticity in the Ts65Dn mouse model of Down syndrome. Journal of Neurochemistry. 2006;98:1266–1277. doi: 10.1111/j.1471-4159.2006.03971.x. [DOI] [PubMed] [Google Scholar]
- Siarey RJ, Stoll J, Rapoport SI, Galdzicki Z. Altered long-term potentiation in the young and old Ts65Dn mouse, a model for Down Syndrome. Neuropharmacology. 1997;36:1549–1554. doi: 10.1016/s0028-3908(97)00157-3. [DOI] [PubMed] [Google Scholar]
- Stasko MR, Costa AC. Experimental parameters affecting the Morris water maze performance of a mouse model of Down syndrome. Behavioural Brain Research. 2004;154:1–17. doi: 10.1016/j.bbr.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Stefani MR, Gold PE. Intra-septal injections of glucose and glibenclamide attenuate galanin-induced spontaneous alternation performance deficits in the rat. Brain Research. 1998;813:50–56. doi: 10.1016/s0006-8993(98)00876-2. [DOI] [PubMed] [Google Scholar]
- Stefani MR, Gold PE. Intrahippocampal infusions of K-ATP channel modulators influence spontaneous alternation performance: relationships to acetylcholine release in the hippocampus. Journal of Neuroscience. 2001;21:609–614. doi: 10.1523/JNEUROSCI.21-02-00609.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone WS, Rudd RJ, Gold PE. Glucose attenuation of deficits in spontaneous alternation behavior and augmentation of relative brain 2-deoxyglucose uptake in old and scopolamine-treated mice. Psychobiology. 1992;20:270–279. [Google Scholar]
- Stone WS, Walser B, Gold SD, Gold PE. Scopolamine- and morphine-induced impairments of spontaneous alternation performance in mice: reversal with glucose and with cholinergic and adrenergic agonists. Behavioral Neuroscience. 1991;105:264–271. doi: 10.1037//0735-7044.105.2.264. [DOI] [PubMed] [Google Scholar]
- Tanzi RE. Molecular genetics of Alzheimer’s disease and the amyloid beta peptide precursor gene. Annals of Medicine. 1989;21:91–94. doi: 10.3109/07853898909149191. [DOI] [PubMed] [Google Scholar]
- Ukai M, Shinkai N, Kameyama T. Cholinergic receptor agonists inhibit pirenzepine-induced dysfunction of spontaneous alternation performance in the mouse. General Pharmacology. 1995;26:1529–1532. doi: 10.1016/0306-3623(95)00038-0. [DOI] [PubMed] [Google Scholar]
- Vogels OJM, Broere CAJ, Ter Laak HJ, Ten Donkelaar HJ, Nieuwenhuys R, Schulte BPM. Cell loss and shrinkage in the nucleus basalis Meynert complex in Alzheimer’s disease. Neurobiology of Aging. 1990;11:3–13. doi: 10.1016/0197-4580(90)90056-6. [DOI] [PubMed] [Google Scholar]
- Walker DL, Gold PE. Impairment of spontaneous alternation performance by an NMDA antagonist: attenuation with non-NMDA treatments. Behavioral & Neural Biology. 1992;58:69–71. doi: 10.1016/0163-1047(92)90952-z. [DOI] [PubMed] [Google Scholar]
- Wenger GR, Schmidt C, Davisson MT. Operant conditioning in the Ts65Dn mouse: learning. Behavioral Genetics. 2004;34:105–119. doi: 10.1023/B:BEGE.0000009480.79586.ee. [DOI] [PubMed] [Google Scholar]
- Westerink BHC, Timmerman W. Do neurotransmitters sampled by brain microdialysis reflect functional release? Analytica Chimica Acta. 1999;379:263–274. [Google Scholar]
- Whitehouse PJ, Struble RG, Hedreen JC, Clark AW, Price DL. Alzheimer’s disease and related dementias: selective involvement of specific neuronal systems. CRC Critical Reviews in Clinical Neurobiology. 1985;1:319–339. [PubMed] [Google Scholar]
- Willig F, Palacios A, Monmaur P, M’Harzi M, Laurent J, Delacour J. Short-term memory, exploration and locomotor activity in aged rats. Neurobiology of Aging. 1987;8:393–402. doi: 10.1016/0197-4580(87)90033-9. [DOI] [PubMed] [Google Scholar]
- Wishart JG. Cognitive abilities in children with Down syndrome: developmental instability and motivational deficits. Progress in Clinical and Biological Research. 1995;393:57–91. [PubMed] [Google Scholar]
- Yates CM, Simpson J, Gordon A, Maloney AF, Allison Y, Ritchie IM, Urquhart A. Catecholamines and cholinergic enzymes in pre-senile and senile Alzheimer-type dementia and Down’s syndrome. Brain Research. 1983;280:119–126. doi: 10.1016/0006-8993(83)91179-4. [DOI] [PubMed] [Google Scholar]
- Zornetzer SF, Thompson R, Rogers J. Rapid forgetting in aged rats. Behavioral and Neural Biology. 1982;36:49–60. doi: 10.1016/s0163-1047(82)90234-5. [DOI] [PubMed] [Google Scholar]