

Abstract
The non-hydrolysing bacterial UDP-N-acetylglucosamine 2-epimerase (UDP-GlcNAc 2-epimerase) catalyses the conversion of UDP-GlcNAc into UDP-N-acetylmannosamine, an intermediate in the biosynthesis of several cell-surface polysaccharides. This enzyme is allosterically regulated by its substrate UDP-GlcNAc. The structure of the ternary complex between the Bacillus anthracis UDP-GlcNAc 2-epimerase, its substrate UDP-GlcNAc and the reaction intermediate UDP, showed direct interactions between UDP and its substrate, and between the complex and highly conserved enzyme residues, identifying the allosteric site of the enzyme. The binding of UDP-GlcNAc is associated with conformational changes in the active site of the enzyme. Kinetic data and mutagenesis of the highly conserved UDP-GlcNAc-interacting residues confirm their importance in the substrate binding and catalysis of the enzyme. This constitutes the first example to our knowledge, of an enzymatic allosteric activation by direct interaction between the substrate and the allosteric activator.
Introduction
The non-hydrolysing bacterial UDP-GlcNAc 2-epimerases catalyse the reversible conversion of UDP-N-acetylglucosamine (UDP-GlcNAc) into UDP-N-acetylmannosamine (UDP-ManNAc) (Kawamura et al, 1978, 1979). The latter is an intermediate in the biosynthesis of bacterial cell-surface polysaccharides and the enterobacterial common antigen (ECA). The ECA is a surface-associated glycolipid common to all members of the Enterobacteriacea family (Kuhn et al, 1988). In species such as Staphyloccocus aureus and Bacillus anthracis, the importance of the UDP-GlcNAc 2-epimerase in the biosynthesis of polysaccharides is highlighted by the presence of two functionally redundant copies of this enzyme (Kiser et al, 1999; Read et al, 2003). Studies support a mechanism in which the reaction proceeds through an initial anti-elimination of UDP to generate an intermediate 2-acetoamidoglucal, followed by the subsequent syn-addition of UDP to yield the product UDP-ManNAc (Tanner, 2002; Fig 1). This bacterial enzyme is related to the bifunctional mammalian UDP-GlcNAc 2-epimerase/ManNAc kinase, a hydrolysing enzyme that converts UDP-GlcNAc into UDP and ManNAc, and phosphorylates the latter into ManNAc 6-phosphate (Hinderlich et al, 1997; Stasche et al, 1997). The mammalian enzyme catalyses the rate-limiting step in sialic acid biosynthesis and is a crucial regulator of cell-surface sialylation in humans (Keppler et al, 1999).
Figure 1.

UDP-N-acetylglucosamine 2-epimerase-catalysed reaction. Reactions catalysed by the (A) bacterial and (B) mammalian enzymes.
The structure of the UDP-GlcNAc 2-epimerase in Escherichia coli shows structural homology to glycosyl transferases, such as T4 phage β-glucosyltransferase, and glycogen phosphorylase (Campbell et al, 2000). Although the enzyme was crystallized in the presence of UDP-GlcNAc, only UDP—an intermediate in the reaction—was observed in the structure bound at a site that has been established to be the catalytic site on the basis of mutagenesis studies (Campbell et al, 2000; Samuel & Tanner, 2004).
A unique characteristic of the bacterial UDP-GlcNAc 2-epimerases is their allosteric regulation by the substrate UDP-GlcNAc, which acts as an activator. In the absence of this activator, virtually no UDP-ManNAc is epimerized in the reverse reaction (Kawamura et al, 1979; Morgan et al, 1997; Samuel & Tanner, 2004); however, when trace amounts of UDP-GlcNAc are added, the reaction proceeds towards equilibrium. This indicates that UDP-GlcNAc is required for the enzyme to acquire a catalytically competent conformation. The structural studies of the E. coli epimerase discussed above showed that each subunit of the dimer of the enzyme adopted a slightly different conformation owing to a 10° interdomain rotation that was proposed to be a part of the allosteric regulatory mechanism. However, it remained unclear how UDP-GlcNAc could trigger these changes as it was absent from the structure (Campbell et al, 2000).
The structure of the non-hydrolysing UDP-GlcNAc 2-epimerase from B. anthracis reported here shows a UDP molecule bound to the active site and an adjacently bound UDP-GlcNAc molecule. The UDP and UDP-GlcNAc are hydrogen bonded to each other and with a common arginine residue. This not only identifies the allosteric site of this enzyme, but also provides the first observation of direct interaction between a substrate molecule and an allosteric activator in an enzyme active site. Residues coordinating the UDP-GlcNAc are highly conserved in non-hydrolysing bacterial UDP-GlcNAc 2-epimerases but not in their hydrolysing mammalian counterparts, providing a target for the development of antibacterial agents.
Results And Discussion
Overall structure
The structure of the B. anthracis UDP-GlcNAc 2-epimerase was solved to 1.7 Å from crystals grown in the presence of UDP-GlcNAc (Table 1). It is very similar to the previously determined structure of the homologous E. coli UDP-GlcNAc 2-epimerase in complex with UDP (Fig 2A; Protein Data Bank (PDB) ID 1F6D; Campbell et al, 2000) or with UDP-GalNAc (1VGV; Badger et al, 2005) and the Bacillus subtilis enzyme without substrate (1O6C; Badger et al, 2005). Both the latter structures were solved as part of a structural genomics programme and were not characterized functionally. In 1VGV, UDP-GalNAc was seen bound to the same position as the UDP intermediate in both the B. anthracis and E. coli (1F6D) structures, but it is not a natural substrate of the enzyme, which is probably why the full molecule was captured in the active site. In contrast to the UDP-bound E. coli structure (1F6D), both chains of the B. anthracis epimerase dimer are in the same conformation. Secondary structure matching superimposition of equivalent Cα atoms of the dimer subunits yields an r.m.s.d. of 0.06 Å and 1.88 Å for the B. anthracis and E. coli (1F6D) enzymes, respectively. Comparison between the closed form of the E. coli enzyme (1F6D) and the B. anthracis enzyme yields an r.m.s.d. of 1.63 Å for 341 Cα atoms. From here onwards, unless otherwise stated, the closed form of the E. coli structure (1F6D) will be used for all structural comparisons.
Table 1.
Crystallographic statistics
| Data collection | |
| Wavelength (Å) | 1.08090 |
| Resolution range | 60–1.7 |
| Number of unique reflections | 106,456 |
| Average redundancy | 3.5 (3.4) |
| Completeness | 99.4 (99.0) |
| Rmerge (%) | 8.7 (34.6) |
| I/σ | 14.6 (3.5) |
| Wilson B-factor (Å2) | 16.1 |
| Refinement | |
| Number of reflections | |
| Work set | 101,096 |
| Test set | 5,360 |
| Rfactor (%) | 22.1 |
| Rfree (%) | 26.2 |
| R.m.s.d. for bond lengths (Å) | 0.018 |
| R.m.s.d. for bond angles (deg) | 1.76 |
| R.m.s.d. B factor for main chain atoms (Å2) | 1.67 |
| R.m.s.d. B factor for side chain atoms (Å2) | 3.35 |
| Average B-factor (Å2) | |
| Main chains | 14.8 |
| Side chains | 16.7 |
| Waters | 27.4 |
| UDP | 12.4 |
| UDP-GlcNAc | 10.6 |
| Values in parenthesis refer to the highest-resolution shells. UDP-GlcNAc, UDP-N-acetylglucosamine. | |
Figure 2.

Structure of UDP-GlcNAc 2-epimerase. (A) Stereo view of the superimposition of the structures of UDP-GlcNAc 2-epimerase from Bacillus anthracis (light blue) and Escherichia coli (magenta). UDP (yellow) and UDP-GlcNAc (green) from the B. anthracis structure are shown as sticks. (B) Fo−Fc simulated annealing omit electron density map contoured at 3σ for UDP-GlcNAc and UDP calculated as the final model minus these molecules. UDP-GlcNAc, UDP-N-acetylglucosamine.
Active site
Clear electron density for both UDP and UDP-GlcNAc molecules was observed in the active site of the B. anthracis enzyme (Fig 2B). UDP and 2-acetoamidoglucal are thermodynamically favoured intermediates of the epimerase-catalysed reaction and are released into solution on prolonged incubation of the enzyme with UDP-GlcNAc (Morgan et al, 1997; Samuel & Tanner, 2004). All of the interactions between UDP and the epimerase are conserved between the B. anthracis and the E. coli enzymes, and a similar presence of water molecules and lack of electron density was observed at the active site of 2-acetoamidoglucal. Although the residues responsible for proton abstraction/addition have not been identified unambiguously, mutations in the E. coli enzyme of ionizable residues found in the region where the glucosamine moiety of the substrate would be positioned identified Asp 95 and Glu 131 as candidates for the proton abstraction, and Glu 117 as being involved in the second reaction step. These residues are conserved in B. anthracis (as Asp 100, Glu 136 and Glu 122) and are in similar positions in the structure, making them strong candidates for catalytic residues.
This is the first time that UDP-GlcNAc has been observed in the active site of UDP-GlcNAc 2-epimerase. The molecule lies in an extended pocket lined by several hydrophilic side chains and forms hydrogen bonds to the side chains of residues Gln 43, Gln 46, Gln 70, His 44, His 242, Arg 210 and Glu 136 (Fig 3). It also makes hydrogen bonds to the main chain of the enzyme and to water molecules. UDP-GlcNAc makes two hydrogen bonds to the α- and β-phosphates of the adjacent UDP molecule, helping to hold it in place for the second step of the epimerization reaction.
Figure 3.

Interactions between the enzyme and UDP-GlcNAc. Stereo view of the hydrogen bond interactions between UDP-GlcNAc (pink), UDP (green) and Bacillus anthracis epimerase residues (light blue). Water molecules are shown as red spheres and hydrogen bonds as dotted lines. UDP-GlcNAc, UDP-N-acetylglucosamine.
Sequence alignments of UDP-GlcNAc 2-epimerases from several bacterial species show that all of the residues that interact with UDP-GlcNAc through their side chains are highly conserved in the non-hydrolysing enzymes, but are variant in the hydrolysing enzymes (supplementary information online). Therefore, this UDP-GlcNAc-binding site is conserved and probably unique to the non-hydrolysing bacterial UDP-GlcNAc 2-epimerases.
Although the crystals of the E. coli enzyme (1F6D) were also grown in the presence of UDP-GlcNAc and belong to the same space group with related unit cell dimensions, they were obtained at pH 5.25 in which the enzyme is only marginally active (Kawamura et al, 1979). The B. anthracis epimerase crystals were grown in a pH range of 8.2–8.8, which is the optimal pH range for the enzyme. Thus, it might be that UDP-GlcNAc binds optimally to the enzyme only at a higher pH, which might help to explain how we trapped the enzyme in its UDP-GlcNAc-bound form.
Conformational changes induced by UDP-GlcNAc
Comparison between our UDP-GlcNAc 2-epimerase structure and that of the E. coli enzyme in complex with UDP (1F6D; Campbell et al, 2000) shows that binding of the substrate UDP-GlcNAc triggers major but localized conformational changes that occur mostly in three loops (His 209–Gly 215, Ile 65–Leu 72 and Val 241–Pro 245; Fig 4) containing the residues that interact with the pyrophosphate and glucosamine moieties of the UDP-GlcNAc.
Figure 4.

Conformational changes triggered by UDP-GlcNAc. The Bacillus anthracis UDP-GlcNAc 2-epimerase is shown superimposed with the closed form of the Escherichia coli enzyme (Protein Data Bank ID 1F6D; chain B). The largest conformational changes are shown in blue and pink for the B. anthracis and E. coli enzymes, respectively. The side chains of Arg 210 and His 242 are shown for both enzymes (asterisks indicate E. coli), and UDP-GlcNAc in pink. UDP-GlcNAc, UDP-N-acetylglucosamine.
The flipping of the His 209–Gly 215 loop (His 213–Gly 219 in E. coli) is the most striking conformational change, and leads to shifts of 6.5 Å and 12 Å in the Cα atom and guanidium group positions of residue Arg 210 (Arg 214). This is in contrast to a shift of only 0.5 Å for the Cα atom of His 209 (His 213), which establishes hydrogen bonds with the β-phosphate of UDP in both the B. anthracis and the E. coli enzyme structures. This loop flip brings the side chain of Arg 210 into position to make hydrogen bonds to the β-phosphate of UDP, a hydroxyl group of UDP-GlcNAc, the carboxyl group of Glu 136 (Glu 131 in E. coli) and a water molecule (Fig 3). Thus, Arg 210 has an important role in anchoring both UDP and UDP-GlcNAc, and, as its guanidium is likely to be fully protonated at the optimum pH of the enzyme (between 7 and 9), it probably stabilizes the leaving UDP in the anti-elimination reaction.
The Ile 65–Leu 72 (Ile 60–Leu 67) loop was partly disordered in the E. coli epimerase enzyme structures in which residues 66–69 (61–64) were not seen (Campbell et al, 2000). In the B. anthracis structure, it makes four hydrogen bonds from Arg 69 and Gln 70 to the ribose and α-phosphate of UDP-GlcNAc (Fig 3).
The conformational changes that occurs on the Val 241–Pro 245 (Val 245–Pro 249) loop are reminiscent of those in the His 209–Gly 215 (His 213–Gly 219) loop, shifting the Cα and Nɛ2 atoms of residue His 242 (His 246) by approximately 3 Å and 9 Å, respectively, between both structures. These movements bring His 242 into hydrogen bonding position to both the β-phosphate and a hydroxyl of the UDP-GlcNAc molecule.
Importance of UDP-GlcNAc-interacting residues
Bacterial UDP-GlcNAc 2-epimerases are tightly allosterically regulated by the substrate UDP-GlcNAc as, in the absence of this epimer, the rate of epimerization is extremely slow. This and the strict conservation of the UDP-GlcNAc-interacting residues indicate that the conformational changes discussed above are crucial for converting the enzyme to an active state. To investigate this, we generated point mutants with alanine substitutions of UDP-GlcNAc-interacting residues. All mutants, except His44Ala, were soluble and behaved as wild-type enzyme during purification. As the His44Ala mutation completely abolished the solubility of the protein, a histidine to glutamine change was made that overcame the problem of solubility. Kinetic data for the wild-type and mutant enzymes are summarized in Table 2.
Table 2.
Kinetic data (±s.e.m., n=3) for the epimerization of UDP-GlcNAc by wild-type and mutant Bacillus anthracis UDP-GlcNAc 2-epimerase
| Enzyme | kcat (s−1) | KM (mM) | kcat/KM (s−1 mM−1) | n |
|---|---|---|---|---|
| Wild type | 7.9±0.3 | 2.2±0.2 | 3.6 | 1.4±0.1 |
| Arg210Ala | 0.021±0.001 | 20.9±2.2 | 0.001 | 1.15±0.07 |
| His242Ala | 0.22±0.10 | 10.4±1.0 | 0.021 | 1.47±0.15 |
| His44Gln | 4.0±0.2 | 7.3±0.8 | 0.55 | 1.21±0.08 |
| Gln70Ala | 2.06±0.07 | 11.1±0.8 | 0.19 | 1.24±0.06 |
| Gln43Ala | 1.20±0.07 | 17.2±1.6 | 0.070 | 1.77±0.23 |
| UDP-GlcNAc, UDP-N-acetylglucosamine. | ||||
The KM for the wild-type enzyme (2.2 mM) is two- to threefold higher than those reported for the E. coli (0.6 mM) and Bacillus cereus enzymes (1.1 mM), although the kcat is similar to E. coli (7.9 versus 7.1 s−1), whereas the Hill coefficient is slightly lower than both (1.4 versus 1.8 s−1; Kawamura et al, 1978, 1979; Morgan et al, 1997; Samuel & Tanner, 2004). A possible explanation is that B. anthracis contains two redundant and identical copies of the UDP-GlcNAc 2-epimerase gene in contrast to the single copy present in E. coli. Thus, it might still achieve comparable UDP-GlcNAc turnover rates to E. coli by using higher amounts of UDP-GlcNAc 2-epimerase.
The Arg210Ala mutant shows a 10-fold increase in KM and a 400-fold decrease in kcat, indicating that this residue has an important role both in stabilizing the incoming UDP-GlcNAc that enters the active and allosteric sites, and the resulting UDP produced in the anti-elimination reaction. As Arg 210 is hydrogen bonded to a UDP-GlcNAc hydroxyl that interacts with UDP, in addition to its direct hydrogen bond to one β-phosphate of UDP, it essentially has a role in stabilizing the two β-phosphate oxygen atoms of the latter. The lower Hill coefficient of the Arg210Ala mutant indicates that this residue is also directly involved in the allosteric regulation of the enzyme, as the cooperativity between the allosteric and the catalytic sites is significantly reduced, probably through the disruption of interactions in the His 209–Gly 215 loop. Notably, Arg 210 also interacts with Glu 136 (Glu 131) through two hydrogen bonds (one direct and one water-mediated), and at the optimal pH range of the epimerase reaction (pH 7–9), these side chains are likely to be ionized with opposite charges. The mutation Glu131Gln in the E. coli epimerase led to a 10,000-fold decrease in kcat (Samuel & Tanner, 2004), and the effect of the disruption of this interaction on Glu 136 in the Arg210Ala mutant might explain its large decrease in kcat. Interestingly, Arg 210 is conserved only in the non-hydrolysing bacterial UDP-GlcNAc 2-epimerases, whereas Glu 136 (Glu 131) is conserved in all UDP-GlcNAc 2-epimerases, indicating that the crucial role of Arg 210 and the conformational changes associated with it are unique to the bacterial non-hydrolysing UDP-GlcNAc 2-epimerases.
The His242Ala mutant also shows a moderate increase in KM (fivefold) and a pronounced decrease in kcat (36-fold). This residue makes hydrogen bonds to the β-phosphate and to a hydroxyl group of UDP-GlcNAc that interacts with the neighbouring UDP molecule. Thus, it is likely that it has a role in both the binding of UDP-GlcNAc at the allosteric site and the stabilization of the UDP intermediate. Interestingly, its unchanged Hill coefficient indicates that the cooperativity is preserved, with minimal effect on communication between the two sites. Although interactions with the UDP-GlcNAc and UDP are disrupted (as shown by the KM and kcat), the conformational regulation remains similar to wild type.
The mutants Gln70Ala, Gln43Ala and His44Gln have moderate decreases (two- to sevenfold) in kcat and increases in KM (three- to eightfold). All of these residues form hydrogen bonds with the axial oxygens of the α-phosphate of the UDP-GlcNAc. His 44 also forms hydrogen bonds with one of the axial oxygens of the β-phosphate, and Gln 43 with the carbonyl of the acetyl moiety of UDP-GlcNAc. Thus, it seems that interactions between the epimerase and the α-phosphate of UDP-GlcNAc help in positioning the latter for optimal interaction with the UDP in the catalytic site. The weakened α-phosphate interactions owing to the loss of a few hydrogen bonds in these mutants probably lead to a lower affinity of the allosteric site for UDP-GlcNAc and also as a consequence for the bound UDP. The Hill coefficients for Gln70Ala and His44Gln have also decreased, showing less cooperativity between the catalytic and allosteric site, which is probably due to disruptions of the hydrogen-bonding network in the Ile 65–Leu 72 loop. Paradoxically, Gln43Ala shows an increase in cooperativity between sites. The reason for this is unclear from the structure, because this mutation would lead to loss of hydrogen bonds to both UDP-GlcNAc and Thr 102. Perhaps replacing a larger glutamine residue with a smaller alanine residue reduces the barrier to loop movement, increasing cooperativity when a UDP-GlcNAc binds (although a higher KM means that this happens at higher substrate concentrations). However, it should be noted that the uncertainty range for the measured n value is double that of the others, making confident conclusions difficult.
The relatively larger changes in the KM, kcat and Hill coefficient of the Arg210Ala mutant compared with those of the other mutants confirm that this residue is crucial to the conformational changes that lead to the UDP-GlcNAc-induced activation of the B. anthracis UDP-GlcNAc 2-epimerase, and to the stabilization of the interaction between UDP-GlcNAc molecules in the active and allosteric site.
Binding of UDP-GlcNAc obstructs the active site
The binding of UDP-GlcNAc to the allosteric site of the enzyme not only triggers closure of the active site but also obstructs its access to the solvent, thus preventing the products of the anti-elimination reaction from diffusing away before they can react again in the syn-addition. Consequently, following every catalytic cycle, the active site must open for product release and substrate uptake. These enzymes release the intermediates UDP and 2-acetoamidoglucal into solution at a rate of about 1/400 that of the UDP-GlcAc epimerization (Morgan et al, 1997; Tanner, 2002), and, as these are thermodynamically more stable than either the substrate or the product of the reaction, the enzyme must kinetically trap them to avoid their release into solution.
Gene knockout has impaired growth
To examine the significance of the enzyme to B. anthracis, a deletion of one of the two copies of the epimerase gene was constructed. The resulting BA5509 mutant was grown alongside the wild-type strain and showed a significant growth defect (Fig 5). We have been unable to obtain a double mutant strain, indicating that such a mutant is non-viable; however, even the deletion of a single copy has marked and adverse effects on the growth of the organism. Our findings, taken with the essential nature of UDP-GlcNAc 2-epimerase activity in both Listeria monocytogenes (Dubail et al, 2006) and B. subtilis (Soldo et al, 2002), confirm the importance of both the epimerase activity for growth and its viability as an antibiotic target.
Figure 5.

Growth curve of BA5509 mutant and wild-type strains. Each point represents the average of at least three independent experiments. Error bars are not shown, as they are smaller than the symbols used to mark the data points. OD, optical density.
In summary, the structure reported indicates that non-hydrolysing bacterial UDP-GlcNAc 2-epimerases use a new allosteric regulatory mechanism that involves direct interaction between one substrate molecule in the active site and another in the allosteric site. This regulatory mechanism helps the enzyme to hold its substrates and stable intermediates in the active site until the completion of a two-step elimination/addition reaction. The conservation of the allosteric site residues in the non-hydrolysing UDP-GlcNAc 2-epimerases indicates that this mechanism is used exclusively by this class of bacterial enzymes, thus providing a selective way of targeting them, particularly in the case of B. anthracis, for the development of antibacterial agents.
Methods
Protein expression and purification. Both UDP-GlcNAc 2-epimerase and UDP-ManNAc dehydrogenase enzymes were expressed and purified as hexahistidine fusion proteins, and purified to homogeneity by affinity, anion and gel filtration chromatography (supplementary information online).
Crystallization and structural determination. Crystals of the B. anthracis UDP-GlcNAc 2-epimerase were grown in hanging drops containing an equal volume of protein (5 mg/ml in 50 mM Tris–HCl (pH 8.0), 5 mM UDP-GlcNAc (Sigma, St Louis, MO, USA), 200 mM NaCl and 2 mM dithiothreitol) and well solution (22–26% PEG 4,000–6,000, 100 mM Tris–HCl (pH 8.2–8.8) and 0.2 M Li2SO4) and were cryoprotected by mother liquor supplemented with 30% glycerol. Data were collected at the National Synchrotron Light Source (NSLS) beamline X29, and the structure solved by molecular replacement using the structure of the UDP-GlcNAc 2-epimerase without substrate from B. subtilis (PDB ID 1O6C) as a search model (Badger et al, 2005). The structure was refined with Refmac5 (Murshudov et al, 1997), using noncrystallographic symmetry restraints (supplementary information online). The final model contains all 750 protein residues (375 from each chain including 4 vector-encoded residues), 2 UDPs, 2 UDP-GlcNAcs and 577 water molecules. The Ramachandran plot shows 91.6% of residues in the most favoured regions, and 8.1% and 0.3% in the generously and additionally allowed regions, respectively. Figs 2, 3 and 4 were generated using PyMol (DeLano, 2002).
Enzyme kinetics. The kinetic parameters of the B. anthracis UDP-GlcNAc 2-epimerase for the epimerization of UDP-GlcNAc were obtained by a previously described coupled assay with UDP-ManNAc dehydrogenase (Morgan et al, 1997; supplementary information online). Initial reaction velocities were fit to the Hill equation using Prism 4 (GraphPad Software Inc., San Diego, CA, USA): 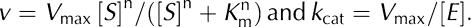
Mutant strain construction. The shuttle plasmid pASD2 was used to disrupt gene BA5509 in B. anthracis ΔSterne using a previously described method (Bozue et al, 2005). Growth of BA5509 and wild-type strains in BHI media was then monitored in a microplate assay at 24°C (supplementary information online).
Supplementary information is available at EMBO reports online (http://www.emboreports.org)
Supplementary Material
supplementary information
Acknowledgments
We thank W. Shi for access to Brookhaven beamline X29 and B. Manjasetty for assistance with data collection. L.M.V. was supported by a postdoctoral fellowship from the Svenska Sällskapet för Medicinsk Forskning. S.S.B. was partly supported by National Research Service Award Training grant GM066699. This work was funded in part by research funds to C.E.S. from the Rockefeller University and by U.S. Public Health Service grant AI056510 to C.E.S. and V.A.F. Coordinates and structure factors have been deposited in the Protein Data Bank under accession code 3BEO. Crystallographic work was performed by L.M.V., biochemical work was performed by S.S.B. and L.M.V., and microbiology by R.S.
References
- Badger J et al. (2005) Structural analysis of a set of proteins resulting from a bacterial genomics project. Proteins 60: 787–796 [DOI] [PubMed] [Google Scholar]
- Bozue JA, Parthasarathy N, Phillips LR, Cote CK, Fellows PF, Mendelson I, Shafferman A, Friedlander AM (2005) Construction of a rhamnose mutation in Bacillus anthracis affects adherence to macrophages but not virulence in guinea pigs. Microb Pathog 38: 1–12 [DOI] [PubMed] [Google Scholar]
- Campbell RE, Mosimann SC, Tanner ME, Strynadka NC (2000) The structure of UDP-N-acetylglucosamine 2-epimerase reveals homology to phosphoglycosyl transferases. Biochemistry 39: 14993–15001 [DOI] [PubMed] [Google Scholar]
- DeLano WL (2002) The PyMOL molecular graphics system, on world wide web http://wwwpymolorg
- Dubail I, Bigot A, Lazarevic V, Soldo B, Euphrasie D, Dupuis M, Charbit A (2006) Identification of an essential gene of Listeria monocytogenes involved in teichoic acid biogenesis. J Bacteriol 188: 6580–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinderlich S, Stasche R, Zeitler R, Reutter W (1997) A bifunctional enzyme catalyzes the first two steps in N-acetylneuraminic acid biosynthesis of rat liver. Purification and characterization of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase. J Biol Chem 272: 24313–24318 [DOI] [PubMed] [Google Scholar]
- Kawamura T, Kimura M, Yamamori S, Ito E (1978) Enzymatic formation of uridine diphosphate N-acetyl-D-mannosamine. J Biol Chem 253: 3595–3601 [PubMed] [Google Scholar]
- Kawamura T, Ishimoto N, Ito E (1979) Enzymatic synthesis of uridine diphosphate N-acetyl-D-mannosaminuronic acid. J Biol Chem 254: 8457–8465 [PubMed] [Google Scholar]
- Keppler OT, Hinderlich S, Langner J, Schwartz-Albiez R, Reutter W, Pawlita M (1999) UDP-GlcNAc 2-epimerase: a regulator of cell surface sialylation. Science 284: 1372–1376 [DOI] [PubMed] [Google Scholar]
- Kiser KB, Bhasin N, Deng L, Lee JC (1999) Staphylococcus aureus cap5P encodes a UDP-N-acetylglucosamine 2-epimerase with functional redundancy. J Bacteriol 181: 4818–4824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn HM, Meier-Dieter U, Mayer H (1988) ECA, the enterobacterial common antigen. FEMS Microbiol Rev 4: 195–222 [DOI] [PubMed] [Google Scholar]
- Morgan PM, Sala RF, Tanner ME (1997) Eliminations in the reactions catalyzed by UDP-N-acetylglucosamine 2-epimerase. J Am Chem Soc 119: 10269–10277 [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Read TD et al. (2003) The genome sequence of Bacillus anthracis Ames and comparison to closely related bacteria. Nature 423: 81–86 [DOI] [PubMed] [Google Scholar]
- Samuel J, Tanner ME (2004) Active site mutants of the ‘non-hydrolyzing' UDP-N-acetylglucosamine 2-epimerase from Escherichia coli. Biochim Biophys Acta 1700: 85–91 [DOI] [PubMed] [Google Scholar]
- Soldo B, Lazarevic V, Pooley HM, Karamata D (2002) Characterization of a Bacillus subtilis thermosensitive teichoic acid-deficient mutant: gene mnaA (yvyH) encodes the UDP-N-acetylglucosamine 2-epimerase. J Bacteriol 184: 4316–4320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasche R, Hinderlich S, Weise C, Effertz K, Lucka L, Moormann P, Reutter W (1997) A bifunctional enzyme catalyzes the first two steps in N-acetylneuraminic acid biosynthesis of rat liver. Molecular cloning and functional expression of UDP-N-acetyl-glucosamine 2-epimerase/N-acetylmannosamine kinase. J Biol Chem 272: 24319–24324 [DOI] [PubMed] [Google Scholar]
- Tanner ME (2002) Understanding nature's strategies for enzyme-catalyzed racemization and epimerization. Acc Chem Res 35: 237–246 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary information