

Abstract
The gastrin/cholecystokinin-2 (CCK-2) receptor has been identified as a possible target for peptide receptor radionuclide imaging and therapy. Several radiolabeled peptides binding to this receptor have been explored in animal models and clinical trials but either low tumor uptake or high renal retention has been found. The aim of this study was to identify a peptide with improved tumor-to-kidney pharmacodynamics when compared with current candidates.
Methods:
A small peptide-chelator library of 34 compounds based on the C-terminal sequences of CCK-8 or minigastrin was constructed. The peptides were radiolabeled with 111In with high labeling efficiency (>90%), as determined by high-performance liquid chromatographic analysis. The labeled peptides were screened by assessing tumor and kidney uptake in pancreatic xenograft nude mouse models, including AR42J. An extensive biodistribution analysis was performed on the lead candidate from the library.
Results:
Minigastrin analogs containing a pentaglutamate sequence showed the highest tumor uptake but very high renal retention. CCK analogs showed the lowest tumor and renal uptake. Deletion of the pentaglutamate sequence in the gastrin analogs lowered the tumor uptake by a factor of 3 but decreased the kidney uptake by a factor of 20. Insertion of histidine residues in the sequence reduced kidney uptake by a further factor of almost 2-fold. In AR42J tumor-bearing mice, the peptide with the sequence DOTA-HHEAYGWMDF-NH2 (DOTA is tetraazacyclododecane tetraacetic acid) showed the highest tumor-to-kidney ratio of all peptides studied, with saturable uptake in target organs and low uptake by nontarget tissues other than the kidney.
Conclusion:
This peptide is a worthwhile candidate for clinical studies to determine whether it is suitable for use in peptide receptor–targeted radionuclide therapy.
The gastrin/cholecystokinin-2 (CCK-2) receptor has been identified as a possible target for peptide receptor radionuclide imaging and therapy. Levels of expression of the receptor are increased in several tumor types, including medullary thyroid cancer (1), neuroendocrine tumors (2), small cell lung cancer, and others (3).
CCK-2 receptor–targeted peptides originate from 2 main families. Human gastrin is a 34-amino-acid peptide that also exists in several C-terminal truncated forms, including minigastrin, a 13-residue peptide with the sequence LEEEEEAYGWMDF-NH2. Full-length CCK is also a 34-residue peptide but again exists in several shorter bioactive forms, including CCK-8, which has the structure DYMGWMDF-NH2.
The ability of both gastrin- and CCK-derived peptides to target the gastrin receptor in vivo has been studied by Behr et al. (4,5), who found that the labeled minigastrin analogs showed the greatest uptake in receptor-positive tissues but were also associated with extremely high uptake in the kidney, whereas CCK-8 exhibited low kidney uptake and tumor uptake. Recently published data suggest that renal uptake of gastrin analogs is mediated through a receptor that recognizes the pentaglutamic acid sequence (6). Behr explored the clinical utility of radiolabeled minigastrin in patients (7,8) and, for these studies, used the novel bifunctional agent DTPA-d-Glu (DTPA is diethylenetriaminepentaacetic acid), which showed improved kinetic stability when compared with conventional monofunctional DTPA (9). They were able to show a sensitivity of 91% in 75 patients with medullary thyroid cancer imaged with 111In-DTPA d-Glu-minigastrin. Eight patients were also treated with 90Y-labeled minigastrin and, although some evidence of efficacy was seen, hematologic and renal toxicities were observed.
Reubi et al. explored the potential of a series of radiolabeled CCK-8 peptides. They identified a modified analog of CCK-8, dYNleGWNleDF-NH2 with high affinity and selectivity for the CCK-2 receptor (10). When labeled with 111In using tetraazacyclododecane tetraacetic acid (DOTA) as a bifunctional chelating agent, the radioligand showed high specific internalization rates in the receptor-positive cell line AR42J, and biodistribution studies in tumor-bearing rats showed a modest but specific uptake into receptor-positive tissues (11). In 7 patients with medullary thyroid cancer imaged with the 111In-DTPA analog, some lesions were visualized but there was relatively low uptake both at these sites and in the receptor-positive stomach.
Aloj et al. evaluated the potential of 111In-DTPA-Glu-Gly-CCK-8 using models based on cells transfected with the CCK-2 receptor. Rapid internalization of the radioligand by cells was observed in vitro and uptake was seen in tumor-bearing nude mice, although levels of kidney uptake were greater than those seen in tumor (12).
The potential of 99mTc-labeled analogs for imaging gastrin receptors has also been recognized. von Guggenberg et al. have showed very high tumor uptake of 99mTc-HYNIC-minigastrin complexes (HYNIC is hydrazinonicotinamide) in AR42J-bearing nude mice (13), and Nock et al. have recently described the characterization of a series of tetraamine-conjugated gastrin analogs in the same animal model (14).
The aim of our research presented in this article was to identify a radioligand that combined the relatively high tumor uptake shown by peptides belonging to the gastrin family with the low renal uptake seen with CCK derivatives. Although 111In was selected as a suitable isotope for in vitro, biodistribution, and imaging studies, the ultimate goal was to explore the radiotherapeutic properties of this peptide and to develop an analog that could be labeled with a β-emitting radionuclide such as 90Y or 177Lu. A small library of peptide-DOTA and -DTPA conjugates based on the C-terminal structure of minigastrin was therefore prepared, and the biodistribution of these labeled peptides was compared with that of some of the radioligands described in nude mice bearing a variety of CCK-2–expressing tumors.
MATERIALS AND METHODS
Chemicals
Peptide conjugates were synthesized by either Cambridge Research Biochemicals or Pichem R&D. Identity and purity were confirmed by matrix-assisted laser desorption/ionization mass spectroscopy and reverse-phase high-performance liquid chromatography (HPLC). Indium (111In) chloride was purchased from Tyco Healthcare. All other reagents were purchased from Sigma-Aldrich.
Cell Lines and Animal Models
AGS human gastric tumor cells (European Collection of Cell Cultures [ECACC]) transfected with the CCK-2 receptor (AGS-CCK2R) were cultured at 37°C in RPMI 1640 media (Sigma-Aldrich) supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 0.4 mg/mL geneticin (Invitrogen) to maintain selection pressure. The cells were passaged 2 or 3 times weekly.
AR42J rat pancreatic tumor cells (ECACC), HT29 human colorectal tumor cells (ECACC), and CA20948 rat pancreatic tumor cells were maintained in serial passage in nude mice. For the biodistribution studies, the donor mice were sacrificed and tumors were excised. The tumor was finely minced, and 3-mm3 sections were implanted subcutaneously into the flank of male MF1 nude mice (Harlan) under anesthesia (Hypnorm/Hypnovel; Janssen/Roche). When measurable tumors were established, the mice were allocated to the biodistribution groups. Mean tumor size at this time was 300 mg, with a range of 100–500 mg.
Radiolabeling and Radiochemical Analysis
InCl3 (10 μL, 5–10 MBq in 0.05 M HCl), ammonium acetate (2 μL, 1 M, pH 5.5), monothioglycerol ([MTG] 5 μL, 0.5% in water) DOTA-peptide (10 μL, 1 mg/mL in 0.01 M phosphate-buffered saline [PBS], pH 7.2), and 28 μL water were mixed in a polypropylene vial. After heating (98°C, 15 min), ethylenediaminetetraacetic acid ([EDTA] 2.5 μL, 0.1 M) was added to quench the reaction, and the solution was diluted with PBS. Radiochemical purity was determined by reverse-phase HPLC (5-μm Jupiter 300 column, 250 × 4.6 mm inner diameter, Phenomenex; solvent A, 0.1% trifluoroacetic acid [TFA] in water; solvent B, 0.1% TFA in acetonitrile; flow rate, 1 mL/min; gradient, 0% solvent B for 5 min, then change to 60% solvent B over 20 min, then change to 100% solvent B over 3 min, and change back to 0% solvent B over 5 min) and instant thin-layer chromatography (2 mobile phases: 50 mM EDTA in 0.1 M sodium acetate and 3.5% [v/v] ammonia:methanol, 1:1).
Plasma Stability
Radiolabeled H2-Met (100 μL, 0.6 μg peptide, 4–5 MBq) was incubated (37°C, gently agitated) with either PBS or human blood from a healthy donor (500 μL). Aliquots were removed at 0, 1, 2, 4, and 24 h and centrifuged at 3,000g for 5 min. The plasma was removed, mixed with acetonitrile (plasma/acetonitrile, 2:3), and centrifuged (3,000g, 5 min). The supernatants and PBS samples (100 μL) were analyzed by reverse-phase HPLC as described.
Receptor Affinity Measurements
AGS-CCK2R tumor cells were harvested with 0.025% EDTA dissolved in PBS (pH 7.3). The cells were washed, counted, and resuspended into assay buffer (0.1% bovine serum albumin [Sigma-Aldrich] in PBS and 1 μM aprotinin (Trasylol; Bayer Plc.) at 5 × 106 cells per milliliter.
The test peptides were dissolved into assay buffer and serially diluted from 4 × 10−6 to 4 × 10−12 M. Triplicates (50 μL) of each peptide dilution were pipeted into 0.6-mL polypropylene Microfuge tubes along with 50 μL of 6 × 10−10 M 125I-G17 (PerkinElmer) and 100 μL of cell suspension.
The samples were incubated for 2.5 h at room temperature (RT) with end-to-end rotation before centrifugation at 13,000 rpm for 3 min. The supernatant was aspirated and the cell pellets were counted for 1 min on a γ-counter (Cobra II autogamma; Packard).
The data were transformed and analyzed using Prism software (GraphPad Software Inc.); the inhibition constant (Ki) and half of the maximum binding fraction (EC50) were determined by non-linear regression analysis.
Biodistribution Studies
Biodistribution studies were performed in a level 2 containment facility (Academic Unit of Cancer Studies, University of Nottingham, Nottingham, U.K.) according to guidelines of the Advisory Committee for Dangerous Pathogens, and in accordance with British Home Office regulations governing animal experimentation.
Two types of study were performed: for initial comparison of the peptide library, 100 μL (0.2 μg) of 111In-labeled peptide were injected intravenously into the tail vein of nude mice bearing AR42J, CA20948, or HT29-CCK2R tumors (n = 3 or 4). After 4 h, tumor, liver, kidney, pancreas, and a sample of blood were collected and processed as described below.
For the full biodistribution study on the final lead candidate, 100 μL of 111In-labeled peptide with or without 50 μg of blocking peptide were injected intravenously into the tail vein of the AR42J tumor-bearing mice (n = 3–6 per condition). After 1, 4, 24, 48, or 72 h, the mice were anesthetized and a 0.5-mL blood sample was obtained by cardiac puncture. Tumor, pancreas, kidneys, heart, stomach, spleen, small intestine, large intestine, liver, lung, and muscle were also sampled, each sample being weighed and counted on a γ-counter (Cobra II autogamma), together with a dilution of the injection sample. The percentage of injected dose per gram of tissue (%ID/g) was calculated for each tissue type.
Statistical Analysis
Statistical comparisons were performed on the mean values of replicate biodistributions performed at 4 h in AR42J-bearing mice. One-way ANOVA was first performed on the replicate mean and SD values obtained with each peptide to ensure that there was no statistically significant variation within the groups. A second ANOVA was then performed to compare the mean values between groups. The F statistic was calculated from the ratio of the within-group mean square values obtained for each peptide and converted to a probability value (FDIST) using Excel (Microsoft). P < 0.05 was considered to be statistically significant.
Statistical comparison of the blocked and unblocked biodistribution results was performed using an unpaired, 2-tailed t test (Excel; Microsoft). P < 0.05 was considered to be statistically significant.
Imaging
Nude mice bearing AR42J tumors were injected with 10 MBq (10 μg) of 111In-labeled H2-Met and imaged using a NanoSPECT/CT animal scanner (Bioscan) 4 h later under intramuscular anesthesia. SPECT images were obtained in 24 projections over 20 min using a 4-head scanner with 4 × 9 (1.4 mm) pinhole collimators in helical scanning mode and CT images with a 45-kVp x-ray source in 180 projections over 6 min. Images were reconstructed in a 256 × 256 matrix using proprietary Bioscan software and fused using PMOD (Mediso).
RESULTS
In designing our small peptide library, we postulated that the high renal uptake previously observed with the minigastrin analogs was largely due to the anionic pentaglutamate sequence in the structure. We therefore synthesized peptide conjugates in which this sequence was missing or replaced by a cationic sequence of several histidine residues. We explored the effect of substituting l- for d-residues in the N-terminal region of the peptides, and we also studied the effect of substituting the methionine residue near the C-terminus with either a norleucine residue or the oxidized sulfone analog of methionine. In all, we synthesized and studied 34 peptides with sequences related to that of minigastrin. After labeling these peptides with 111In, we compared their biodistribution with that of 3 comparators—described by Reubi et al. (10), Behe et al. (9), and Bernard et al. (15). We studied the biodistribution in 3 nude mouse models using tumors expressing differing levels of gastrin/CCK-2 receptors: AR42J (16), CA20948 (17), and HT29-CCK2R, a human colorectal cell line transfected with relatively low levels of gastrin receptors (Academic Unit of Cancer Studies). We chose to study the biodistribution of these radioligands 4 h after intravenous administration. We sampled a limited number of organs at this time point, but we were particularly interested in uptake in the tumor, uptake in the kidney, and that remaining in the blood.
The results obtained with a selected subset of the library are shown in Table 1. It can be seen that the tumor uptake of all peptides is highest in the model expressing the greatest number of receptors and lowest in that expressing the lowest number. In the AR42J model, the CCK-8 comparator showed lower tumor uptake than the minigastrin (MG0) comparator and the kidney uptake was considerably lower in concordance with published results. Removal of the pentagastrin sequence (MG11) resulted in a 20-fold reduction in kidney uptake from 60 %ID/g to around 3 %ID/g (P < 0.0004) and also reduced the tumor uptake by a factor of 3 (although this just failed to achieve statistical significance; P < 0.06). Replacement of the pentaglutamate sequence with a hexahistidine tag (and substitution of the DTPA chelator by DOTA) (H6-Met) resulted in a slight, but statistically not significant, further reduction in kidney uptake and a similar effect on tumor uptake. Interestingly, however, reducing the number of histidine residues from 6 to 2 (H2-Met) lowered the kidney uptake still further (P < 0.05), while actually increasing the tumor uptake (P < 0.05). In view of the importance in targeted radionuclide therapy of the relative uptake of the tumor with that of the critical organ, the kidney, we calculated the mean tumor-to-kidney ratio as a likely indicator of therapeutic potential. Even though MG0 showed the highest tumor uptake, it showed the lowest mean tumor-to-kidney ratio (0.04) because of its very high kidney uptake, CCK had a higher tumor-to-kidney ratio of 0.23, and MG11 was still higher at 0.31. However, the dihistidine analog showed the highest tumor-to-kidney ratio of 0.47.
TABLE 1.
Biodistribution in Nude Mouse Xenografts of Selected Peptides, 4 Hours After Intravenous Administration
| %ID/g ± SD |
||||||
|---|---|---|---|---|---|---|
| Cell line | Peptide | Blood | Tumor | Kidney | n | Ratio |
| AR42J | H2-Met1 | 0.04 ± 0.01 | 0.87 ± 0.21 | 1.87 ± 0.60 | 15 | 0.47 |
| CCK-82 | 0.05 ± 0.00 | 0.49 ± 0.14* | 2.09 ± 0.85† | 4 | 0.23 | |
| H6-Met3 | 0.20 ± 0.03 | 0.59 ± 0.14* | 2.95 ± 0.60* | 8 | 0.20 | |
| MG04 | 0.06 ± 0.02 | 2.60 ± 0.65* | 60.45 ± 9.04* | 4 | 0.04 | |
| MG115 | 0.04 ± 0.01 | 0.92 ± 0.10† | 3.30 ± 0.35* | 5 | 0.28 | |
| H2-Nle6 | 0.03 ± 0.00 | 0.73 ± 0.14† | 2.40 ± 2.13† | 2 | 0.31 | |
| H2-Met(O)7 | 0.04 ± 0.00 | 0.12 ± 0.01 | 1.21 ± 0.03 | 1 | 0.10 | |
| CA20948 | H2-Met1 | 0.07 ± 0.02 | 0.59 ± 0.10 | 2.41 ± 0.91 | 7 | 0.24 |
| CCK-82 | 0.05 ± 0.01 | 0.46 ± 0.13 | 2.33 ± 0.68 | 4 | 0.20 | |
| H6-Met3 | 0.21 ± 0.01 | 0.55 ± 0.14 | 3.08 ± 0.77 | 9 | 0.18 | |
| MG115 | 0.03 ± 0.00 | 0.75 ± 0.05 | 4.11 ± 0.39 | 1 | 0.18 | |
| H6-Nle8 | 0.02 ± 0.00 | 0.15 ± 0.06 | 0.98 ± 0.18 | 1 | 0.16 | |
| H2-Nle6 | 0.03 ± 0.00 | 0.53 ± 0.08 | 1.73 ± 0.31 | 1 | 0.31 | |
| HT29-CCK2R | H2-Met1 | 0.08 ± 0.01 | 0.19 ± 0.05 | 2.44 ± 0.47 | 6 | 0.08 |
| CCK-82 | 0.05 ± 0.00 | 0.13 ± 0.03 | 1.95 ± 0.28 | 9 | 0.06 | |
| H6-Met3 | 0.26 ± 0.03 | 0.26 ± 0.04 | 3.47 ± 0.52 | 13 | 0.08 | |
| MG115 | 0.02 ± 0.00 | 0.07 ± 0.03 | 3.47 ± 0.34 | 1 | 0.02 | |
| H6-Nle8 | 0.02 ± 0.01 | 0.05 ± 0.00 | 1.01 ± 0.08 | 1 | 0.05 | |
Statistically significant (P < 0.05) with corresponding value for H2-Met.
Not statistically significant (P < 0.05) with corresponding value for H2-Met.
H2-Met = DOTA-HHEAYGWMDF-NH2.
CCK-8 = DOTA-dYMGWMDF-NH2.
H6-Met = DOTA-HHHHHH-EAYGWMDF-NH2.
MG0 = DTPA-eEEEEEAYGWMDF-NH2.
MG11 = DTPA-eAYGWMDF-NH2.
H2-Nle = DOTA-HHEAYGWMDF-NH2.
H2-Met(O) = DOTA-HHEAYGWM(sulfoxide)DF-NH2.
H6-Nle = DOTA-HHHHHHEAYGWMDF-NH2.
SD = mean SD achieved in all studies; ratio = tumor-to-kidney ratio (column 4 mean value/column 5 mean value); CCK-8 = cholecystokinin-8; CCK2R = CCK-2 receptor.
Results show mean values obtained from n replicate studies in groups of 3 or 4 animals.
In the CA20948 model, the tumor-to-kidney values were lower because the tumor uptake was reduced; however, the relative values were similar, with the tumor-to-kidney ratio of the H2-Met analog being the second highest. In the transfected HT-29 model, tumor-to-kidney ratios were further reduced but that of the H2-Met analog was still the greatest.
In view of the potential for oxidation of the methionine residue in the gastrin sequence, we studied the effect of substitution of Met by Nle in a manner similar to that used by Reubi et al. for CCK-8 (10). In both the hexahistidine and dihistidine analogs, this substitution resulted in a reduction in tumor uptake in all tumor models. We also studied the effect of oxidation of the methionine residue on tumor uptake. As can be seen from Table 1, the oxidized analog (H2-Met(O)) showed extremely low tumor uptake as well as a modest reduction in kidney retention.
Because the H2-Met analog showed the most promising pattern of biodistribution in this preliminary screen study, a full biodistribution analysis of this compound was undertaken in AR42J-bearing mice at time points from 1 to 72 h after injection. The results are shown in Figure 1. The blood clearance was extremely rapid, with close to background levels of activity in blood 4 h after administration. A gradual washout from all tissues can be seen, including tumor and kidney, although the washout rate from kidney is faster than that from tumor. Thus, although kidney uptake values exceed those in the tumor at 1 and 4 h, by 24 h, and thereafter, the tumor uptake is greater than that in the kidney. A blocking study was undertaken at 4 h to determine the extent to which tissue uptake is receptor mediated. A significant level of blockade (P < 0.0005) was observed in the blood, tumor, and the stomach but not in any other organ.
FIGURE 1.

Biodistribution of H2-Met in AR42J-bearing nude mice, between 1 and 72 h after intravenous administration. Greatest initial uptake is observed in tumor and kidney but washout from kidney is faster than that from tumor. Four-hour blocking data indicate biodistribution when 100 μg of unlabeled peptide were coadministered with radiolabeled peptide. Significant blocking effect (P < 0.0005) is seen in blood, tumor, and stomach, but no other organs. int = intestine.
A study to determine the effect of peptide dose on tissue uptake was also undertaken. Doses ranging from 0.02 to 50 μg were injected, and the uptake in receptor-positive and receptor-negative tissues was measured. The results are shown in Figure 2 and are expressed as the percentage change in uptake compared with the standard biodistribution dose of 0.2 μg (where the uptake for 0.2 μg is expressed as 100%). No effect on uptake can be seen in receptor-negative samples such as blood and kidney. In contrast, a dose–uptake relationship can be seen in receptor-positive tumor and stomach. Interestingly, however, little increase in tumor uptake is seen as the dose is reduced below the “standard” dose of 0.2 μg, whereas the uptake in the stomach continues to increase as the dose is further reduced, showing an increase in uptake of 50% above that obtained with the standard dose.
FIGURE 2.
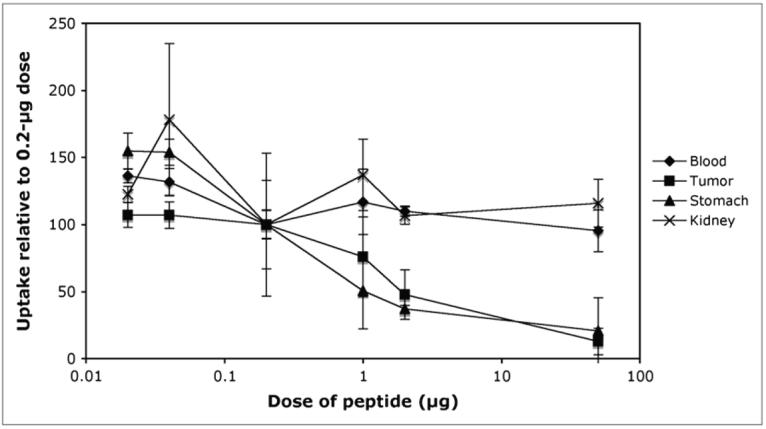
Effect of peptide dose on biodistribution. Results are expressed as percentage change in uptake compared with that obtained with “standard” dose of 0.2 μg. No significant dose effect was observed in any tissue other than tumor and stomach. Increasing the dose above 0.2 μg reduced uptake in both tissues. Decreasing the dose had no effect on tumor uptake but increased uptake in the stomach.
γ-Camera imaging of AR42J-bearing mice with this radioligand was also undertaken using a dedicated small-animal SPECT/CT scanner. The results obtained were entirely consistent with the biodistribution studies performed previously and described here and showed a modest level of tumor uptake, heterogeneously distributed within the tumor mass, and low levels of abdominal activity. A typical example of one of the images obtained is shown in Figure 3.
FIGURE 3.
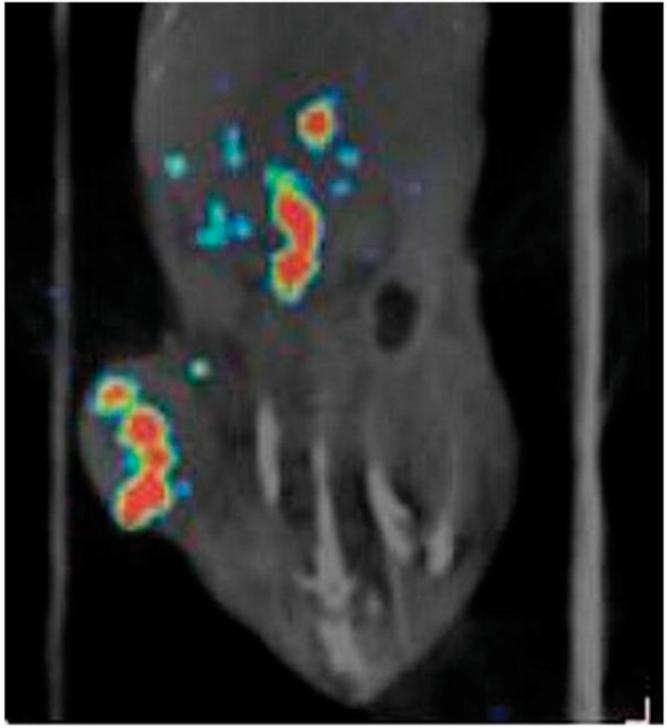
Fused SPECT/CT image shows biodistribution of 111In-H2-Met in AR42J-bearing nude mouse, 4 h after intravenous injection. Heterogeneous uptake can be observed in tumor with little uptake in central organs.
Receptor Affinity Measurements
Receptor binding assays using transfected AGS-CCK2R were undertaken on the peptide analogs to compare the results of the biodistribution with an in vitro measurement of receptor affinity. The results are shown in Table 2. Similar measures of affinity of approximately 3 nM were found for CCK-8, MG0, and MG11, and insertion of histidine residues into the sequences had little effect on binding. Substitution of Nle for Met in the sequence appeared to result in a modest reduction in binding affinity, whereas oxidation of the methionine residue effectively abolished any specific receptor interaction.
TABLE 2.
Receptor Binding Affinity Measurements on Selected Peptides Using 125I-G17 as Radioligand
| Peptide | EC50/Ki (nM) |
|---|---|
| DOTA-CCK8 | 3.1/2.1 |
| DOTA-H6-Met | 3.1/2.1 |
| DOTA-H2-Met | 5.7/3.9 |
| DOTA-H2-Met(O) | 1,300.0/900.0 |
| DOTA-H6-Nle | 5.7/3.8 |
| DOTA-H2-Nle | 16.7/11.3 |
| MG0 | 2.5/1.7 |
| MG11 | 3.4/2.4 |
Radiolabeling
The labeling of DOTA-peptides with trivalent metals such as 111In is now well established in the radiopharmaceutical literature. The conjugate is normally heated with the radionuclide at a slightly acidic pH in the presence of a weak chelator such as acetate (18). It is usual to perform the reaction at a temperature around 100°C to maximize the rate of complexation but it is important to consider the effects of this heating process on the peptide itself. While labeling some of the peptides in our library, we discovered that an extended period of heating (100°C for 30 min) resulted in degradation of the peptide, seen by the appearance of additional peaks on the HPLC chromatogram. In the case of peptides containing one or more methionine residues, some of these peaks were caused by oxidation of the thio-ether side chain. For most of these peptides, we discovered that we could reduce the temperature of the reaction to 70°C and still achieve labeling yields of >90%—the specification we set ourselves during this phase of the study. However, the introduction of oligohistidine sequences into the peptide significantly affected the outcome of the labeling procedure, reducing the labeling yield to ≤60%. To identify the cause of this effect we explored the effect of adding a simple hexahistidine peptide to DOTA-peptides that lacked any histidine in their sequence. This had no effect—thus, simply mixing an oligohistidine peptide with the DOTA-minigastrin conjugate still resulted in a very high labeling efficiency, but inserting the same oligohistidine into the linear peptide sequence lowered the labeling yield very significantly. To overcome this phenomenon, we found that we had to heat the His-containing peptides to 100°C to routinely achieve high labeling efficiencies. This, however, reintroduced the problem of oxidation of methionine residues, and it was clear, from the biodistribution results and the receptor affinity measurements that oxidation of the peptide effectively abrogates its binding to the gastrin receptor. To overcome this problem, we explored the use of several potential antioxidants during the labeling procedure. We found that the most effective, clinically acceptable antioxidant was MTG. The effect of adding MTG to the reaction mixture is shown in Figure 4, which shows reverse-phase HPLC chromatograms of our lead candidate peptide labeled in the absence and presence of 0.04 M MTG. The reduction in the level of the oxidized peptide peak at retention time (Rt) = 19.5 min can clearly be seen.
FIGURE 4.
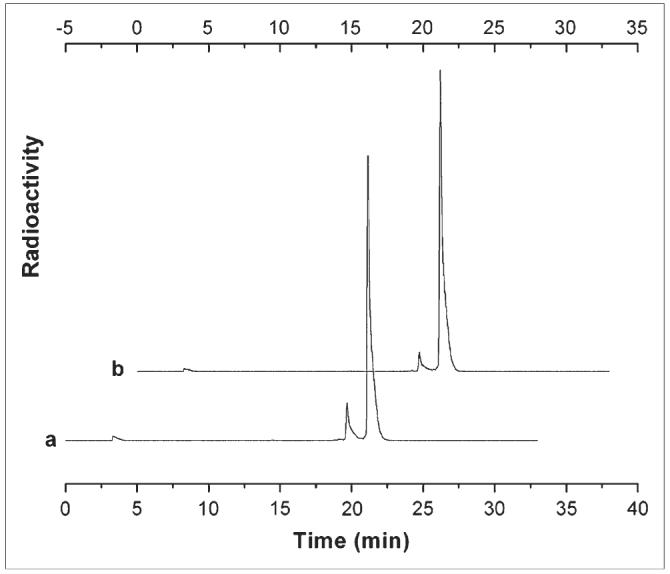
HPLC chromatograms of 111In-H2-Met prepared with and without addition of antioxidant MTG, where trace a contained no MTG (resulting in 12.3% oxidized peptide, Rt = 19.6 min) and trace b contained 0.04 mg MTG (resulting in 5.9% oxidized peptide, Rt = 19.2 min).
Plasma Stability
To ensure that the radiolabeled peptide remains intact in vivo long enough to target the gastrin receptors, we studied the effect of incubation in human blood in vitro. 111In-H2-Met was incubated with fresh human blood at 37°C for up to 24 h and then analyzed by reverse-phase HPLC. More than 95% of intact peptide remained after a 4-h incubation in human blood at 37°C. After 24 h in human blood, 62% of intact peptide remained (compared with 98% in PBS).
DISCUSSION
One of the most important requirements of a radiopharmaceutical for peptide receptor radionuclide therapy (PRRT) is a high therapeutic index that translates into the relative radiation doses received by target and critical nontarget tissues, respectively. In practice, this limits the amount of radioactivity administered to that which will deliver acceptable radiation doses to the normal life-sustaining organs and tissues of the patient. In the case of neuropeptides labeled with metallic radionuclides, the dose-limiting organs are the kidneys. Most, if not all, of such tracers show significant levels of renal retention and their therapeutic application has resulted in significant renal toxicity (19). In the case of 90Y-labeled octreotide analogs, the consequence of the kidney uptake is that the administered activity of 90Y must be limited to that which would deliver a dose of around 25 Gy to the kidneys (20). Because tumors that receive a higher radiation dose are more likely to show a response to PRRT (21), this clearly represents the most critical limitation to the application of radionuclide therapy with radiolabeled octreotide derivatives.
Thus, if radiolabeled gastrin-derived peptides are to be successfully used for PRRT, it is essential that the level of renal uptake and retention be kept as low as possible. The purpose of the study described in this article was to explore the extent to which changes in the structure of gastrin and CCK-related peptides affected their uptake in target and nontarget tissues. We synthesized a library of 34 different peptide sequences that shared either the gastrin or CCK-8 C-terminal sequences or analogs in which the methionine residue was substituted by the nonoxidizable norleucine residue. Toward the N-terminus, the peptides were extended with a variety of amino acids, including l- and d-isomers of basic and acidic residues, and finally capped with either a DTPA or DOTA chelating group. We included in this library several previously published compounds that had shown promising results in either animal models or clinical studies. These peptides were radiolabeled with 111In and their biodistribution was studied in tumor-bearing nude mouse models 4 h after administration as described. Plasma stability studies and receptor binding assays were performed on the peptides with the most promising biodistribution.
Few differences were observed in peptide uptake in most tissues. All of the peptides were cleared very rapidly from the blood and were excreted almost solely through the renal system. In view of the above discussion, we were mainly interested in the relative uptake of the receptor-positive tumors and the kidney. These results are summarized in Table 1. None of the peptides studied had a higher tumor uptake than that shown by the labeled minigastrin analog (MG0) described by Behr et al. Peptides with the CCK C-terminal sequence showed a lower tumor uptake than those with the gastrin C-terminus. Peptides with the gastrin C-terminus but lacking the pentaglutamate motif showed an intermediate tumor uptake. Labeled minigastrin (MG0) showed by far the highest kidney uptake, and this was profoundly reduced when the pentaglutamate sequence was removed. Insertion of histidine residues adjacent to the N-terminus reduced the renal uptake further without any significant effect on tumor uptake. The reason for this reduced kidney retention is not entirely clear. The pKa of the cyclic amine in the histidine side chain is pH 6.0. Therefore, at the physiologic pH of 7.4, the great majority of the residues will be uncharged and should not significantly affect the overall charge of the peptide in the blood. However, the pH of the urine in the proximal tubules may be significantly more acid, and this may reduce the net negative charge of the peptide at the point at which tubular reabsorption occurs. On the other hand, it may be that the histidine residues induce some physical interference of a recognition system responsible for the peptide reabsorption.
For each peptide the mean tumor-to-kidney ratio was calculated. The peptide that showed the highest tumor-to-kidney ratio had the sequence DOTA-HHEAYGWMDF-amide (H2-Met). The same trend of results was observed in all 3 tumor models explored. An extended biodistribution study was therefore performed on this peptide. The results are shown in Figure 1. It can be seen that receptor targeting is very rapid, with maximum tumor uptake apparent at the first sampling time of 1 h after injection. Thereafter, a washout of activity is seen in all tissues. Blood activity declines significantly by 4 h so that tumor-to-blood levels at this time are 50:1. A small degree of gastrointestinal excretion is seen but the great majority is cleared by renal excretion. A significant degree of renal retention is observed but the rate of washout from the kidneys is somewhat faster than that from the tumor so that tumor-to-kidney ratios gradually rise from a minimum of 0.6 at 4 h to 1.4 at 72 h. Coinjection of unlabeled peptide to block receptor sites resulted in a significant reduction of uptake by tumor and stomach, but not by any other samples, indicating that receptor-mediated uptake occurred only in these 2 tissues. A dose-ranging study was performed to determine the effect of peptide dose on biodistribution. Doses of 5-, 10-, and 100-fold greater and 5- and 10-fold less than the 1-μg dose used for the rest of the biodistribution studies were explored. No significant effect was observed in any tissue other than the tumor and stomach. Increasing the dose above 1 μg reduced the uptake in both tissues. Decreasing the dose had no effect on tumor uptake but increased the uptake in the stomach. Such an effect might result from either a different binding affinity to the mouse (stomach) or rat (tumor) strains of receptor or, perhaps, a lower concentration of receptors in the stomach, resulting in partial saturation at lower peptide concentrations. If this finding were reproduced in clinical studies, it may result in a therapeutic advantage from the use of intermediate rather than minimal doses of peptide.
During radiolabeling development of the peptide library, we discovered that peptides that contained oligohistidine sequences adjacent to the DOTA chelating group were more difficult to label than those lacking this sequence. As the result of this phenomenon, it was found necessary to increase the incubation temperature from 70°C to 100°C. Unfortunately, this initially resulted in an increase in the level of oxidation of the methionine residue in the peptide; however, this could be overcome by the addition of the antioxidant MTG during the labeling procedure. The presence of this oxidizable residue will present a formulation challenge in the preparation of labeled peptides for clinical trials of this compound, as oxidation effectively abrogates receptor binding and, unfortunately, substitution of the methionine residue with the nonoxidizable norleucine lowers both in vitro receptor binding affinity and tumor uptake in vivo.
Radiolabeled gastrin analogs have previously been shown to be relatively stable in mouse and human serum but are rapidly degraded in the kidney and appear as small metabolites in the urine (12,14) (22). A recent report by Fröberg et al. identified differences in the rate of breakdown of different CCK-2 receptor imaging agents in vivo in patients (23), indicating the potential importance of radioligand stability. We studied the rate of degradation of H2-Met in fresh human whole blood and found no breakdown of the peptide after 4 h of incubation in vitro, although this does not preclude the possibility of breakdown occurring in vivo.
Although the tumor-to-kidney ratios achieved with H2-Met were the greatest among this group of peptides studied, they are much lower than those generally achieved by octreotide analogs in similar models. For example, in parallel biodistribution studies performed with 111In-DOTA-Tyr-3-octreotate (DOTA-TATE), we found tumor-to-kidney ratios of 1 and 1.8 in the AR42J and CA20948 models compared with 0.47 and 0.24 for H2-Met. This finding is entirely the result of higher tumor uptake rather than lower kidney retention, as the renal uptake of 111In-DOTA-TATE at 4 h was 9 %ID/g compared with around 2 %ID/g for H2-Met. The lower tumor uptake in these mouse models of the gastrin analogs may be a result of lower receptor expression or other, as yet unidentified, causes; however, their behavior in clinical practice is of more significant importance. A study comparing octreotide and minigastrin scintigraphy showed that in >20% of the cases, gastrin uptake exceeded that of octreotide (24). Moreover, preliminary PRRT studies with 90Y-minigastrin did show some evidence of efficacy, albeit with renal toxicity (7).
ACKNOWLEDGMENTS
We gratefully acknowledge financial support from Cancer Research U.K. and Aphton Inc. Expert statistical advice was provided by Dr. Joan Morris (Reader in Medical Statistics, St. Bartholomew's and the Royal London School of Medicine and Dentistry) and expert assistance in animal imaging was provided by Ciara Finucane.
REFERENCES
- 1.Reubi JC, Waser B. Unexpected high incidence of cholecystokinin-B/gastrin receptors in human medullary thyroid carcinomas. Int J Cancer. 1996;67:644–647. doi: 10.1002/(SICI)1097-0215(19960904)67:5<644::AID-IJC9>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 2.Reubi JC, Waser B. Concomitant expression of several peptide receptors in neuroendocrine tumours: molecular basis for in vivo multireceptor tumour targeting. Eur J Nucl Med Mol Imaging. 2003;30:781–793. doi: 10.1007/s00259-003-1184-3. [DOI] [PubMed] [Google Scholar]
- 3.Reubi JC, Schaer JC, Waser B. Cholecystokinin (CCK)-A and CCK-B/gastrin receptors in human tumors. Cancer Res. 1997;57:1377–1386. [PubMed] [Google Scholar]
- 4.Behr TM, Jenner N, Radetzky S, et al. Targeting of cholecystokinin-B/gastrin receptors in vivo: preclinical and initial clinical evaluation of the diagnostic and therapeutic potential of radiolabelled gastrin. Eur J Nucl Med. 1998;25:424–430. doi: 10.1007/s002590050241. [DOI] [PubMed] [Google Scholar]
- 5.Behr TM, Behe M, Angerstein C, et al. Cholecystokinin-B/gastrin receptor binding peptides: preclinical development and evaluation of their diagnostic and therapeutic potential. Clin Cancer Res. 1999;5(10 suppl):3124s–3138s. [PubMed] [Google Scholar]
- 6.Behe M, Kluge G, Becker W, Gotthardt M, Behr TM. Use of polyglutamic acids to reduce uptake of radiometal-labeled minigastrin in the kidneys. J Nucl Med. 2005;46:1012–1015. [PubMed] [Google Scholar]
- 7.Behe M, Behr TM. Cholecystokinin-B (CCK-B)/gastrin receptor targeting peptides for staging and therapy of medullary thyroid cancer and other CCK-B receptor expressing malignancies. Biopolymers. 2002;66:399–418. doi: 10.1002/bip.10356. [DOI] [PubMed] [Google Scholar]
- 8.Behr TM, Behe MP. Cholecystokinin-B/gastrin receptor-targeting peptides for staging and therapy of medullary thyroid cancer and other cholecystokinin-B receptor-expressing malignancies. Semin Nucl Med. 2002;32:97–109. doi: 10.1053/snuc.2002.31028. [DOI] [PubMed] [Google Scholar]
- 9.Behe M, Becker W, Gotthardt M, Angerstein C, Behr TM. Improved kinetic stability of DTPA-dGlu as compared with conventional monofunctional DTPA in chelating indium and yttrium: preclinical and initial clinical evaluation of radiometal labelled minigastrin derivatives. Eur J Nucl Med Mol Imaging. 2003;30:1140–1146. doi: 10.1007/s00259-003-1178-1. [DOI] [PubMed] [Google Scholar]
- 10.Reubi JC, Waser B, Schaer JC, et al. Unsulfated DTPA- and DOTA-CCK analogs as specific high-affinity ligands for CCK-B receptor-expressing human and rat tissues in vitro and in vivo. Eur J Nucl Med. 1998;25:481–490. doi: 10.1007/s002590050247. [DOI] [PubMed] [Google Scholar]
- 11.de Jong M, Bakker WH, Bernard BF, et al. Preclinical and initial clinical evaluation of 111In-labeled nonsulfated CCK8 analog: a peptide for CCK-B receptor-targeted scintigraphy and radionuclide therapy. J Nucl Med. 1999;40:2081–2087. [PubMed] [Google Scholar]
- 12.Aloj L, Caraco C, Panico M, et al. In vitro and in vivo evaluation of 111In-DTPAGlu-G-CCK8 for cholecystokinin-B receptor imaging. J Nucl Med. 2004;45:485–494. [PubMed] [Google Scholar]
- 13.von Guggenberg E, Behe M, Behr TM, Saurer M, Seppi T, Decristoforo C. 99mTc-Labeling and in vitro and in vivo evaluation of HYNIC- and (Nalpha-His) acetic acid-modified [D-Glu1]-minigastrin. Bioconjug Chem. 2004;15:864–871. doi: 10.1021/bc0300807. [DOI] [PubMed] [Google Scholar]
- 14.Nock BA, Maina T, Behe M, et al. CCK-2/gastrin receptor-targeted tumor imaging with 99mTc-labeled minigastrin analogs. J Nucl Med. 2005;46:1727–1736. [PubMed] [Google Scholar]
- 15.Bernard BF, Béhé M, Breeman WAP. Preclinical evaluation of minigastrin analogs for CCK-B receptor targeting [abstract] Cancer Biother Radiopharm. 2003;18:281. [Google Scholar]
- 16.Svoboda M, Dupuche MH, Lambert M, Bui D, Christophe J. Internalization-sequestration and degradation of cholecystokinin (CCK) in tumoral rat pancreatic AR 4-2 J cells. Biochim Biophys Acta. 1990;1055:207–216. doi: 10.1016/0167-4889(90)90034-b. [DOI] [PubMed] [Google Scholar]
- 17.Bernard BF, Krenning E, Breeman WA, et al. Use of the rat pancreatic CA20948 cell line for the comparison of radiolabelled peptides for receptor-targeted scintigraphy and radionuclide therapy. Nucl Med Commun. 2000;21:1079–1085. doi: 10.1097/00006231-200011000-00015. [DOI] [PubMed] [Google Scholar]
- 18.Breeman WA, De Jong M, Visser TJ, Erion JL, Krenning EP. Optimising conditions for radiolabelling of DOTA-peptides with 90Y, 111In and 177Lu at high specific activities. Eur J Nucl Med Mol Imaging. 2003;30:917–920. doi: 10.1007/s00259-003-1142-0. [DOI] [PubMed] [Google Scholar]
- 19.Cybulla M, Weiner SM, Otte A. End-stage renal disease after treatment with 90Y-DOTATOC. Eur J Nucl Med. 2001;28:1552–1554. doi: 10.1007/s002590100599. [DOI] [PubMed] [Google Scholar]
- 20.Teunissen JJ, Kwekkeboom DJ, de Jong M, Esser JP, Valkema R, Krenning EP. Endocrine tumours of the gastrointestinal tract: peptide receptor radionuclide therapy. Best Pract Res Clin Gastroenterol. 2005;19:595–616. doi: 10.1016/j.bpg.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 21.Kwekkeboom DJ, Teunissen JJ, Bakker WH, et al. Radiolabeled somatostatin analog [177Lu-DOTA0,Tyr3]octreotate in patients with endocrine gastroenteropancreatic tumors. J Clin Oncol. 2005;23:2754–2762. doi: 10.1200/JCO.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 22.Behr TM, Jenner N, Behe M, et al. Radiolabeled peptides for targeting cholecystokinin-B/gastrin receptor-expressing tumors. J Nucl Med. 1999;40:1029–1044. [PubMed] [Google Scholar]
- 23.Fröberg A, Nock B, Maina T, et al. Comparison of three radiolabelled peptides for CCK 2-receptor scintigraphy in medullary thyroid carcinoma. Eur J Nucl Med Mol Imaging. 2006;33(supp 2):S109. doi: 10.1007/s00259-009-1098-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gotthardt M, Boermann OC, Behr TM, Behe MP, Oyen WJ. Development and clinical application of peptide-based radiopharmaceuticals. Curr Pharm Des. 2004;10:2951–2963. doi: 10.2174/1381612043383502. [DOI] [PubMed] [Google Scholar]