

Abstract
Angiotensin-converting enzyme (ACE) plays a central role in blood pressure regulation by producing the vasoconstrictor angiotensin II. When ACE knockout mice were studied, they presented with a complicated phenotype, including cardiovascular, reproductive, hematologic and developmental defects. The complexity of an ACE knockout mouse emphasizes the advantages and disadvantages of the classic knockout strategy. An animal lacking all ACE is very different from a wild type animal, and can be modeled as representing an extreme phenotype. To understand the role of ACE in a tissue and organ specific fashion, our group used targeted homologous recombination to create mouse models in which a promoter swapping strategy results in very restricted tissue patterns of ACE expression. Mice with ACE expression only in the heart, termed ACE 8/8 mice, present with atria enlargement and electrical conduction defects, but normal ventricular function. Mice with ACE expression only in monocytes and macrophages, termed ACE 10/10 mice, have a marked resistance to the growth of melanoma due to an enhanced immune response characterized by increased tumor specific CD8+ T cells and increased proinflammatory cytokines. These mice may define a new means of augmenting the immune response, potentially useful in human clinical situations. The promoter swapping strategy permits scientific investigation of questions unapproachable by other experimental approaches.
Introduction
It is now well accepted that the renin-angiotensin system plays an important role in blood pressure regulation and a variety of other physiologic systems. This is apparent from the characterization of knockout mice lacking angiotensin-converting enzyme (ACE), angiotensinogen or angiotensin II receptors.1 In particular, ACE knockout mice present with low blood pressure, reproductive defects, electrolyte abnormalities, anemia, and kidney defects.2,3
The complexity of an ACE knockout mouse emphasizes the advantages and disadvantages of the classic knockout strategy. An animal lacking all ACE is very different from a wild type animal, and in fact can be modeled as representing an extreme phenotype. While such a phenotype is interesting and illustrative of the many different roles of the renin-angiotensin system, it also has drawbacks. The total lack of a protein is not necessarily physiologic and the cascading complexity of secondary effects can have obfuscating results in terms of understanding the direct and primary role of a protein like ACE. For example, the lack of ACE leads to low blood pressure. This itself undoubtedly has secondary effects, which may lead to an altered physiology and can complicate studies on the role of ACE apart from blood pressure control. Others have realized the limitation of a total knockout and have approached genetic manipulation in mice through cre-lox systems in which there is tissue specific removal of the protein of interest. While this is a viable approach for many proteins, the wide physiologic regulation of renin expression suggested that a selective loss of ACE might have little physiologic consequence.
In order to understand the role of ACE in a tissue specific fashion, our group envisioned a mouse with ACE expression limited to selected tissue types. Thus, instead of creating an animal that only lacked ACE in one organ, our idea was to create a mouse model in which only the kidney or only the heart would express this protein. Our hope was that this approach would allow unique experimental insight into the tissue specific role of the renin-angiotensin system. In order to create such an animal, we adopted the approach of promoter manipulation. This concept is based on a view of a gene as being composed of two essential, but separate, components. The exons of a gene contain the coding information leading to the primary amino acid structure. In contrast, the promoter region contains the instructions as to when and where the protein will be made. Thus, our hypothesis was that by changing the promoter region, while leaving the coding regions intact, we would essentially reprogram the expression of ACE.
ACE 3/3 - Hepatic expression of ACE
In order to test this approach, our group first constructed a mouse (termed ACE 3/3) in which the albumin promoter was substituted for the endogenous somatic ACE promoter.4 More specifically, gene targeting was used to insert a specialized neomycin cassette downstream of the natural ACE promoter (Fig. 1). Both through physical distance and the presence of a strong transcriptional terminator in the neomycin cassette, this was designed to eliminate the influence of the intrinsic ACE promoter. Downstream of the neomycin cassette, we further positioned an albumin promoter which now controlled the expression of the ACE gene. In a sense, this approach models the translocations often seen in leukemias, in which gene rearrangement positions two different genes in juxtaposition. However in our case, the use of a well-defined promoter region allowed us to predict the expressional pattern of ACE in the resulting mouse.
Fig. 1.
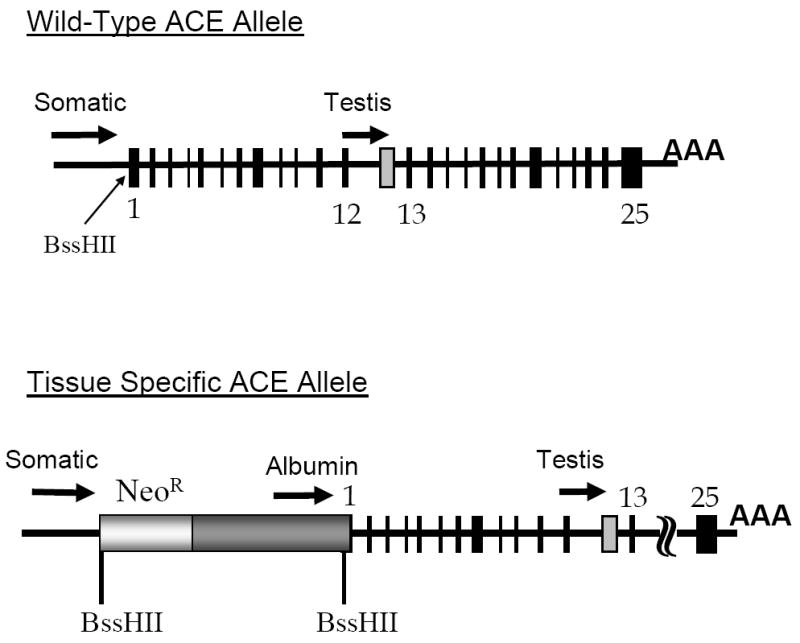
Tissue specific gene expression. The top portion of the figure shows the organization of the wild type ACE allele. The two promoter regions, indicated by arrows, give rise to the somatic and testis isozymes of ACE. Exons are indicated by black boxes. A testis specific exon is indicated in grey. The BssHII restriction site is positioned between the transcription and translation start sites. The bottom portion of the figure indicates our approach to targeted expression of ACE. A neomycin resistance cassette blocks the effects of the intrinsic ACE promoter. 3’ of this, a tissue specific promoter, such as the albumin promoter, is positioned to control ACE expression.
In the terminology adopted by our laboratory, the ACE 3 mouse has ACE expression driven by the albumin promoter. A mouse heterozygous for this mutation is termed ACE 3/wt, while a homozygous mouse is designated ACE 3/3. ACE expression was carefully evaluated in these mice by ACE activity assays, western blot analysis and immunohistochemistry. ACE 3/3 mice express abundant ACE on the surface of hepatocytes (a wild type mouse expresses virtually no ACE by such cells). In contrast, ACE 3/3 mice make no ACE in vascular tissues such as endothelium or the vascular adventitia. While a wild type mouse expresses abundant ACE in the lung due to the high content of endothelium, no lung ACE is made by ACE 3/3 mice. Thus, the reorganization of the ACE promoter markedly changed the tissue patterns of ACE expression.
What are the advantages and disadvantages of this approach? First, the ACE 3/3 mouse allowed us to ask important scientific questions that would not be approachable using classical knockout technology. In particular, the ACE 3/3 model addressed the question of whether vascular ACE was required for the normal control of blood pressure, or whether the presence of ACE at any location within the animal would suffice. Careful analysis showed that ACE 3/3 mice had a blood pressure indistinguishable from wild type or heterozygous animals. These studies are a dramatic demonstration that, given the normal ability to regulate renin expression, homeostasis can be maintained without the localized production of angiotensin II at the endothelial surface.
Using our approach, it is also easy to create compound heterozygous mice. One such animal is termed ACE 1/3 in which the “1” allele is an ACE null allele.5 Thus, the ACE 1/3 mouse has only one functional ACE allele, and this targets ACE expression to the liver. Similar to the ACE 3/3, the ACE 1/3 mouse has an absolutely normal blood pressure. The loss of a single ACE allele leads to compensatory up regulation of renin expression, an enhancement of circulating angiotensin II levels and a compensated physiologic state characterized by normal blood pressure.
There are other potential advantages of the promoter manipulation approach. For example, the hepatocytes of a wild type mouse produces virtually no ACE. Thus, it would be quite difficult to engineer an animal only producing ACE in the liver using the standard cre-lox approach. Since now, many promoter regions are well characterized, the promoter manipulation approach offers the possibility of designing mice, not only with specific expression patterns of a protein, but also with individual tissue expression levels that are higher or lower than that seen under natural physiologic conditions. In other words, swapping promoters provides a potential plasticity of expression that would be difficult to achieve with a standard cre-lox approach.
Our approach also has potential drawbacks. The mice we study were created using targeted homologous recombination in embryonic stem cells. This technology is intrinsically slower than that used in creating transgenic mice. However, as compared to a standard transgenic approach, we create mice in which ACE expression is limited to those tissues recognizing the promoter region driving ACE expression. This contrasts to standard transgenic methodology in which tissue over expression of a protein is found on top of the natural distribution of that protein. A final consideration is that the use of targeted homologous recombination creates a model with a known and easily verified genotype. In contrast, transgenic methodology often creates multimers of the transgene and can result in secondary genetic changes due to chromosomal breakage and rearrangement. Finally, our experience indicates that the major determinate of success or failure in using the promoter swapping approach is the strength and specificity of the new promoter chosen to control expression of the gene of interest. For example, our group used the γ-glutamyl transpeptidase promoter to create a mouse with ACE expression limited to the kidney.6 While some ACE was indeed made, the levels were so low (3% of normal or less) that the resulting mice essentially resembled knockout animals. The use of a different renal specific promoter, the KSP-cadherin promoter,7 gave a satisfactory result with strong renal specific expression of ACE (unpublished data). A we demonstrate below, the use of well-characterized and moderately strong or strong promoters results in experimental models that are both unique and startling in their phenotype.
ACE 8/8 - Cardiac ACE Expression
Our group used the cardiac specific α-myocin heavy chain (α-MHC) promoter to target ACE expression to the heart.8 The reason for creating this model was to isolate ACE and angiotensin II formation in the myocardium. This animal was designed to investigate the important question of whether increased levels of angiotensin II are deleterious to cardiac function. Homozygous mice with this mutation are called ACE 8/8.
The cardiac specific α-MHC promoter is often portrayed as only recognized by cardiomyocytes. In fact, the original paper characterizing the promoter also found some expression in lung tissue, specifically in the vascular smooth muscle cells of the pulmonary veins.9 This pattern was recapitulated in the ACE 8/8 mouse, in which ACE expression was markedly elevated in myocardium and there was some patchy expression in lung parenchyma. In contrast, these mice make no renal ACE and no ACE in vascular endothelium. Due to a low level of ACE expression in normal cardiac tissue, cardiac ACE expression in the ACE 8/8 mice was about 100-fold that of the wild type level. ACE 8/8 mice present with a cardiac phenotype characterized by atrial enlargement, marked reduction of connexin 43 expression and atrial-ventricular conduction defects (Fig. 2).10 While electrical burst pacing of ventricles induced no ventricular tachycardia in wild type mice, the same stimuli induced transient ventricular fibrillation in a majority of the ACE 8/8 mice. Despite a blood pressure only slightly less than normal, ACE 8/8 mice have enhanced mortality, presumably due to the conduction defects within the heart and ventricular arrhythmias. The localized production of ACE in the heart results in this tissue having an angiotensin II concentration of at least twice normal. Despite this, the ventricles of ACE 8/8 mice showed no hypertrophy and no real increase in fibrosis, and they were able to develop ventricular pressure profiles equivalent to wild type mice. One possible explanation for the different response of atrial and ventricular myocardium to ACE over-expression may lie in the intrinsic property of the α-MHC promoter, which is active in the atria starting from early embryonic development, but is expressed postnatally predominantly in the ventricles.11
Fig. 2.
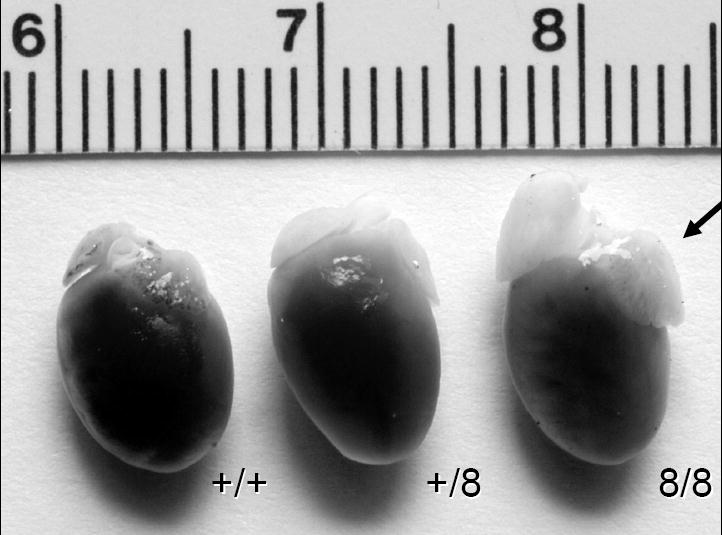
Heart of ACE 8/8. This figure shows the hearts from wild type (+/+), heterozygous (+/8) and homozygous ACE 8/8 mice (8/8). The arrow indicates the markedly enlarged atria present in the ACE 8/8 hearts. Ventricular size is not significantly different from wild type mice.
One of the questions addressed in this experimental model was the ability of the kidney to produce a concentrated urine in the absence of all renal ACE expression. An important caveat of this experiment is to examine a model in which blood pressure is not substantially reduced. Investigation of renal concentrating ability in ACE 8/8 mice showed that this was not markedly different from wild type mice.9 After 24-hour water restriction, ACE 8/8 mice produced urine with osmolalities substantially above 3,000 mOsm/L. Undoubtedly this reflects the fact that the kidney structure is normal in ACE 8/8 mice and that the presence of ACE in myocardium does allow for the generation of angiotensin II. This, plus the presence of many non-angiotensin II mediated systems contributing to a concentrated urine, produced a normal phenotype. Nonetheless, this experiment demonstrates that the local production of angiotensin II within the kidney is not necessary for achieving high urine osmolalities.
In terms of the cardiac phenotype of ACE 8/8 mice, we can provide a partial physiologic explanation. The reduction of cardiac connexin 43 levels is associated with conduction defects.10 As to how the over expression of cardiac ACE influences connexin expression, this is still under investigation. In particular, our group is creating ACE 1/8 mice, a compound heterozygous model in which one ACE allele is null while the second ACE allele targets ACE expression to myocardium. Such an animal should also present with elevated cardiac angiotensin II levels but will achieve this with lesser cardiac over expression of the ACE protein. Further, we are investigating ACE 8/8 mice that are also null for angiotensinogen protein expression. Such mice are genetically unable to produce angiotensin II, but can still produce other peptide substrates for ACE. These models are designed to investigate the specific role of angiotensin II in the ACE 8/8 model.
ACE 10/10 - ACE Over expression by Macrophages
Our group has recently reported a mouse model, called ACE 10/10, in which ACE is over expressed by monocytes and macrophages.12 The model is the result of positioning the c-fms promoter to control ACE expression. This promoter is recognized by monocytes and cells of a similar origin such as Kupffer cells and microglia.13 The ACE 10/10 model was originally created to study the origins of atherosclerosis. Over the last several years, the concept that atherosclerosis is the result of an aberrant inflammatory process within blood vessels has gained much credence. In fact, there is substantial evidence to suggest that inflammatory cell production of angiotensin II may play a role in this process.14,15 Using the c-fms promoter, we created the ACE 10/10 mice in which ACE expression is not only limited to monocytes and macrophages, but ACE levels are also markedly augmented in these cells types. Thus, ACE 10/10 mice are not particularly physiologic in that their monocytes and macrophages have a 15 to 80-fold increase of ACE expression, depending on which assay is used to measure ACE. Put another way, monocytes and macrophages from wild type mice produce very limited quantities of ACE; these tissues produce abundant ACE in the ACE 10/10 model. In contrast, ACE 10/10 mice express no ACE by endothelium or in renal tissues. Despite this, these mice have blood pressures and a renal function indistinguishable from wild type mice.
In order to investigate atherosclerosis in this model, it is necessary to breed the ACE 10 allele with animals that lack ApoE expression. The idea is to compare atherosclerotic development in ApoE knockout mice with ApoE knockout mice that are also ACE 10/10. These experiments are still in progress. However our group recently reported a very unexpected and exciting phenotype of the ACE 10/10 mice, a resistance to the growth of melanoma.
The B16 melanoma is a widely used model for tumor formation in mice. B16 cells were derived from a C57BL/6 background tumor.16 They are easily cultured in vitro, and when injected into the skin of a mouse, will produce a thumbnail sized tumor in approximately two weeks. Such a model is often used to investigate angiogenesis in mice. When ACE 10/10 mice were implanted with B16 melanoma, we noticed the unusual results shown in Figure 3. The growth of tumors in ACE 10/10 mice was substantially reduced as compared to littermate wild type mice. This phenotype was both pronounced and highly reproducible.
Fig. 3.
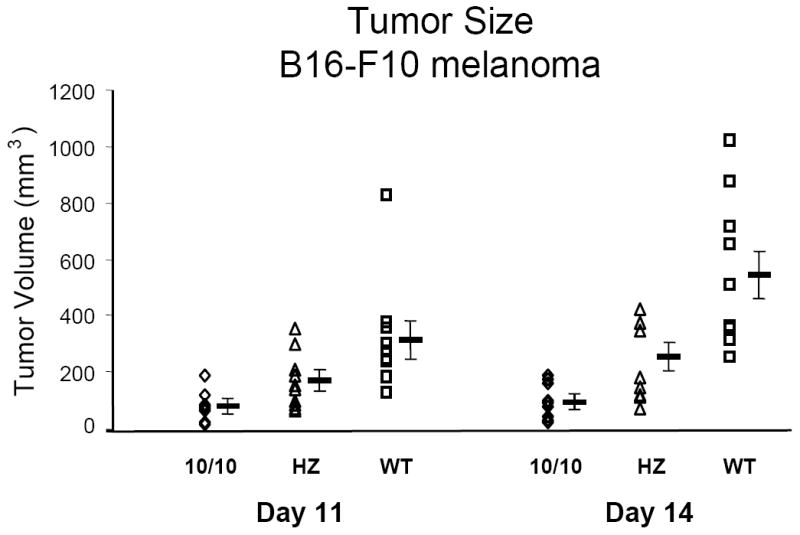
Melanoma growth in ACE 10/10 mice. 1 × 106 B16-F10 tumor cells were injected into the skin of ACE 10/10 homozygous (10/10), heterozygous (HZ) and wild type (WT) mice. At days 11 and 14 after injection, the size of the tumors was measured. Data points from individual mice, as well as the group means ± standard errors, are shown. At 14 days, ACE 10/10 mice (◊) have much smaller tumors than present in wild type mice (◻).
Our group spent substantial effort characterizing the enhanced tumor resistance of ACE 10/10 mice. For example, different B16 tumor cell substrains were tested. Also, the ACE 10/10 phenotype was placed into partially outbred, as well as genetically inbred mouse genetic backgrounds. In all instances, mice over expressing monocyte and macrophage ACE had enhanced resistance to melanoma. One of the most important questions was whether the enhanced tumor resistance was due to the presence of ACE in monocytes/macrophages or the absence of ACE by endothelium. We investigated this using two approaches. First, ACE 3/3 mice were tested for tumor growth. As discussed above, these mice also lack endothelial ACE. In this model, tumor growth was indistinguishable from wild type mice. Second, ACE 10/10 mice were treated with ACE inhibitors. If the resistance of this strain to tumor growth was due to the lack of ACE in endothelium, then further inhibition of ACE should have no effect. What we found was that ACE 10/10 mice treated with an ACE inhibitor showed substantially more pronounced tumor growth than present in non-treated ACE 10/10 animals. In fact, ACE 10/10 mice treated with an ACE inhibitor showed no statistical difference in tumor growth from wild type mice treated with an ACE inhibitor. Taken together, these data strongly suggest that it is the presence of ACE in the ACE 10/10 model that is responsible for the enhanced tumor resistance.
One of the most important experiments we performed was bone marrow transplantation into wild type mice. In this experiment, wild type C57BL/6 mice were lethally irradiated and then salvaged by bone marrow transplantation with bone marrow from ACE 10/10 or litter mate wild type mice. This experiment was performed after substantial back breeding of ACE 10/10 mice to the C57BL/6 strain. After allowing two months for the bone marrow to repopulate, these mice were challenged with B16 melanoma. Thus, this experiment examined a population of animals with normal endothelial expression of ACE, where the only difference was bone marrow transplantation from the ACE 10/10 or wild type mice. Again, the result was very clear-cut, with those animals receiving the ACE 10/10 bone marrow showing substantially smaller tumors than animals receiving the wild type bone marrow.12
Our group investigated the role of angiotensin II in this phenomenon using two approaches. In a genetic approach, angiotensinogen knockout mice were bred so that they were either wild type or ACE 10/10 in terms of ACE expression. Despite the inability of all these mice to produce angiotensinogen (or angiotensin II), those animals with the ACE 10/10 phenotype, showed enhanced resistance to melanoma growth. In a second approach, AT1 receptor antagonists were used. Here again, ACE 10/10 mice receiving AT1 receptor antagonists showed no difference in the growth of tumor, as compared to untreated ACE 10/10 mice. Thus, both a genetic and a pharmacological approach suggest no major role of angiotensin II in the ACE 10/10 phenotype.
Our group has now collected substantial evidence that the tumor resistance of ACE 10/10 mice is associated with a difference in the immune response of these mice, as compared to wild type animals. These data include the finding that ACE 10/10 mice develop greater numbers of CD8+ cytotoxic T lymphocytes directed at tumor antigens. Further, there is substantial evidence that the cytokine levels in ACE 10/10 mice are different from those observed in wild type animals. Specifically, ACE 10/10 cells demonstrate enhanced expression of the pro-inflammatory cytokine IL-12 and decreased expression of the anti-inflammatory cytokine IL-10. This pattern is observed in response to tumor cells, as well as after stimulation with agents such as lipopolysacharide (LPS).
In retrospect, perhaps it is not surprising that mice expressing the peptidase ACE in macrophages and other antigen presenting cells have a different immune response. These cell populations are responsible for stimulating the immune response by presenting small peptides, bound to MHC class I proteins, on their cell surface. This leads to the hypothesis that the over expression of ACE modifies the ability of these cells to process and present such peptides, perhaps resulting in an enhanced immune stimulation. In fact, there is now little doubt that cells from ACE 10/10 mice show an enhanced ability to present peptides (unpublished data).
Previous work by others has developed the concept that macrophages can be activated in a way that either promotes the immune response (characterized by enhanced killing of tumors) or, following a different developmental pathway, reduces the immune response (a state promoting tumor growth).17,18 The proinflammatory macrophages are often termed ‘classically activated macrophages’ or ‘M1’ macrophages, while macrophages thought to suppress the immune response are often termed ‘alternatively activated’ or ‘M2’ macrophages. The phenotype of cells seen in ACE 10/10 mice (increased numbers of cytotoxic lymphocytes, increased IL-12 and decreased IL-10) is precisely the phenotype observed in the classically activated cells.
Future Perspectives
In this article, we laid the scientific basis for why our group created mice with restricted tissue expression of ACE. The major motivation was to investigate the tissue specific role of the reninangiotensin system and to ask scientific questions that could not be approached using another methodology. One of the major dividends of this approach has been the unexpected observation that the over expression of ACE in monocytes and macrophages influences the immune response to implanted melanoma. We wish to emphasize that ACE 10/10 mice markedly over express ACE in immune cells; conclusions from these mice cannot be retrofitted to human patients taking ACE inhibitors. Nonetheless, our findings in ACE 10/10 mice open the possibility of a new means of addressing tumor therapy. At the very least, they represent an understudied area of immunology, namely, how the manipulation of antigen presenting cell peptidase expression can influence the immune response. It is our hope that, as we understand this phenomena further, we will develop the ability to further modify the immune response against cancer, and also perhaps against chronic viral infections. If this dream becomes a reality, it would be another example of a novel experimental approach leading to unexpected findings with important clinical ramifications. At the very least, our studies emphasize the physiologic importance of the renin-angiotensin system and the utility of this system in the study of a variety of biological phenomena. The finding that the manipulation of ACE expression can influence the immune response is only the latest interesting finding in which the renin-angiotensin system has served to elucidate diverse biological questions. During the 109 years that this system has been studied, it has provided insight into questions from the regulation of gene expression to the clinically important areas of heart failure and blood pressure control.
Acknowledgments
The work was supported by NIH grants R37 DK039777, and R01 DK051445. HDX is supported by a Scientist Development grant from the American Heart Association. PL is supported by a Postdoctoral Fellowship from the American Heart Association. SB is supported by grants from Fondation pour la Recherche Medicale (FRM) and Fondation Bettencourt. SF is supported by a Beginning Grant-In-Aid from the American Heart Association and by F99 HL88000 from the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bernstein KE, Xiao HD, Frenzel K, Li P, Shen XZ, Adams JW, Fuchs S. Six Truisms Concerning ACE and the Renin-Angiotensin System. Circ Res. 2005;96:1135–1144. doi: 10.1161/01.RES.0000169536.73576.66. [DOI] [PubMed] [Google Scholar]
- 2.Krege JH, John SWM, Langenbach LL, Hodgin JB, Hagaman JR, Bachman ES, Jennette JC, O’Brien DA, Smithies O. Male-female differences in fertility and blood pressure in ACE-deficient mice. Nature. 1995;375:146–148. doi: 10.1038/375146a0. [DOI] [PubMed] [Google Scholar]
- 3.Esther CR, Howard TE, Marino EM, Goddard JM, Capecchi MR, Bernstein KE. Mice lacking angiotensin-converting enzyme have low blood pressure, renal pathology, and reduced male fertility. Lab Invest. 1996;74:953–966. [PubMed] [Google Scholar]
- 4.Cole J, Quach DL, Sundaram K, Corvol P, Capecchi MR, Bernstein KE. Mice lacking endothelial angiotensin-converting enzyme have a normal blood pressure. Circ Res. 2002;90:87–92. doi: 10.1161/hh0102.102360. [DOI] [PubMed] [Google Scholar]
- 5.Cole J, Khokhlova N, Sutliff RL, Adams JW, Disher KM, Zhao H, Capecchi MR, Corvol P, Bernstein KE. Mice lacking Endothelial Angiotensin-Converting Enzyme (ACE): Normal Blood Pressure with Elevated Angiotensin II. Hypertension. 2003;41:313–321. doi: 10.1161/01.hyp.0000050650.52007.83. [DOI] [PubMed] [Google Scholar]
- 6.Sepulveda AR, Huang SL, Lebovitz RM, Lieberman MW. A 346-base pair region of the mouse gamma-glutamyl transpeptidase type II promoter contains sufficient cis-acting elements for kidney-restricted expression in transgenic mice. J Biol Chem. 1997;2272:11959–67. doi: 10.1074/jbc.272.18.11959. [DOI] [PubMed] [Google Scholar]
- 7.Igarashi P, Shashikant CS, Thomson RB, Whyte DA, Liu-Chen S, Ruddle FH, Aronson PS. Ksp-cadherin gene promoter. II. Kidney-specific activity in transgenic mice. Am J Physiol. 1999;277:F599–610. doi: 10.1152/ajprenal.1999.277.4.F599. [DOI] [PubMed] [Google Scholar]
- 8.Xiao HD, Fuchs S, Campbell DJ, Lewis W, Dudley SC, Jr, Kasi VS, Hoit BD, Keshelava GT, Zhao H, Capecchi MR, Bernstein KE. Mice with cardiac restricted angiotensin converting enzyme (ACE) have atrial enlargement and sudden death. Am J Path. 2004;165:1019–1032. doi: 10.1016/S0002-9440(10)63363-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Subramaniam A, Jones WK, Gulick J, Wert S, Neumann J, Robbins J. Tissue-specific regulation of the alpha-myosin heavy chain gene promoter in transgenic mice. J Biol Chem. 1991;266:24613–24620. [PubMed] [Google Scholar]
- 10.Kasi VS, Xiao HD, Shang LL, Langberg J, Gallego CJ, Bernstein KE, Dudley SC., Jr Cardiac restricted angiotensin converting enzyme overexpression causes conduction defects, inducible arrhythmias, and ion channel dysregulation. Am J Physiol. 2007 doi: 10.1152/ajpheart.00684.2006. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lyons GE, Schiaffino S, Sassoon D, Barton P, Buckingham M. Developmental regulation of myosin gene expression in mouse cardiac muscle. J Cell Biol. 1990;111:2427–2436. doi: 10.1083/jcb.111.6.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen XZ, Li P, Weiss D, Fuchs S, Xiao HD, Adams JW, Williams IR, Capecchi MR, Taylor WR, Bernstein KE. Mice with enhanced macrophage angiotensin converting enzyme are resistant to melanoma. Am J Path. 2007;170:2122–2134. doi: 10.2353/ajpath.2007.061205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sasmono RT, Oceandy D, Pollard JW, Tong W, Pavli P, Wainwright BJ, Ostrowski MC, Himes SR, Hume DA. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood. 2003;101:1155–6. doi: 10.1182/blood-2002-02-0569. [DOI] [PubMed] [Google Scholar]
- 14.da Cunha V, Tham DM, Martin-McNulty B, Deng G, Ho JJ, Wilson DW, Rutledge JC, Vergona R, Sullivan ME, Wang YX. Enalapril attenuates angiotensin II-induced atherosclerosis and vascular inflammation. Atherosclerosis. 2005 Jan;178(1):9–17. doi: 10.1016/j.atherosclerosis.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 15.Egido J, Ruiz-Ortega M. Anti-inflammatory Actions of Quinapril. Cardiovasc Drugs Ther. 2007;21:211–20. doi: 10.1007/s10557-007-6019-1. [DOI] [PubMed] [Google Scholar]
- 16.Stephenson EM, Stephenson NG. Karyotype analysis of the B16 mouse melanoma with reassessment of the normal mouse idiogram. J Natl Cancer Inst. 1970;45:789–800. [PubMed] [Google Scholar]
- 17.Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23:344–6. doi: 10.1016/j.immuni.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 18.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–64. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]