

Abstract
The ubiquitous inositol 1,4,5-trisphosphate receptor (InsP3R) intracellular Ca2+ release channel is engaged by thousands of plasma membrane receptors to generate Ca2+ signals in all cells. Understanding how complex Ca2+ signals are generated has been hindered by a lack of information on the kinetic responses of the channel to its primary ligands, InsP3 and Ca2+, which activate and inhibit channel gating. Here, we describe the kinetic responses of single InsP3R channels in native endoplasmic reticulum membrane to rapid ligand concentration changes with millisecond resolution, using a new patch-clamp configuration. The kinetics of channel activation and deactivation showed novel Ca2+ regulation and unexpected ligand cooperativity. The kinetics of Ca2+-mediated channel inhibition showed the single-channel bases for fundamental Ca2+ release events and Ca2+ release refractory periods. These results provide new insights into the channel regulatory mechanisms that contribute to complex spatial and temporal features of intracellular Ca2+ signals.
Keywords: intracellular signalling, signal transduction, electrophysiology
Introduction
The ubiquitous endoplasmic reticulum (ER)-localized inositol 1,4,5-trisphosphate receptor (InsP3R) Ca2+ channel has a central role in regulating numerous cell physiological processes by modulating cytoplasmic free Ca2+ concentration ([Ca2+]i) through release of Ca2+ from the ER lumen in response to production of cytoplasmic InsP3 caused by extracellular stimuli (Taylor & Richardson, 1991; Berridge, 1993). InsP3R channels respond rapidly (on the order of milliseconds; Marchant & Taylor, 1998) to dynamic changes in the concentrations of their primary ligands—cytoplasmic Ca2+ and InsP3 (Foskett et al, 2007). However, kinetic information about single InsP3R channel responses to ligand concentration changes is lacking. Here, we record, for the first time to our knowledge and with millisecond resolution, the responses of single InsP3R channels in native ER membranes to controlled changes in [Ca2+]i and [InsP3] using rapid perfusion techniques (Hinkle et al, 2003) applied to cytoplasmic-side-out (cyto-out) nuclear membrane patches (Mak et al, 2005). The kinetics of activation and deactivation by [InsP3] and [Ca2+]i changes have shown novel high-affinity inhibitory Ca2+ binding and unexpected cooperativity between InsP3 and Ca2+. Such ligand regulation has not previously been accounted for in most quantitative models of InsP3-mediated Ca2+ signalling (Swillens et al, 1994, 1998; Tang et al, 1996; Falcke et al, 2000; Fraiman & Dawson, 2004; Sneyd & Falcke, 2005). The kinetics of Ca2+-mediated inhibition have now shown the single-channel bases for fundamental Ca2+ release events (Berridge, 1997) and Ca2+ release refractory periods (Fraiman et al, 2006). This study provides new insights into the channel regulatory mechanisms (Foskett et al, 2007) that contribute to complex spatial and temporal features of intracellular Ca2+ signals (Putney & Bird, 1993).
Results And Discussion
Channel activation and deactivation kinetics
Single-channel gating of homo-tetrameric InsP3R channels in native ER membranes was studied by applying patch-clamp techniques to outer membranes of nuclei isolated from Sf9 cells (Ionescu et al, 2006). In the nucleus-attached configuration, with the cytoplasmic aspect of the channel containing the binding sites for InsP3 and Ca2+ facing the pipette solution (Fig 1A), InsP3R channel activities were observed in the current traces (Fig 1D) when the pipette solution contained activating ligand concentrations—2 μM Ca2+ and 10 μM InsP3 (Ionescu et al, 2006). In excised nuclear membrane patches in the cyto-out configuration (Fig 1B), achieved here for the first time to our knowledge, InsP3R channels were not observed when the local perfusion solution lacked InsP3, but their presence was verified by observation of channel activities (Fig 1E) when the patches were perfused with a solution containing 10 μM InsP3 and 2 μM Ca2+ (Fig 1C).
Figure 1.

Nuclear patch-clamp studies of single InsP3R channel responses to ligand concentration changes using rapid perfusion techniques applied to cyto-out membrane patches. (A,B) Schematic diagrams illustrating orientation of InsP3R channels in nucleus-attached and excised cyto-out configurations, respectively. In the nucleus-attached configuration, the cytoplasmic side of InsP3R channels (black) faces the pipette solution. When the cyto-out configuration is obtained, the cytoplasmic side of different InsP3R channels (purple) faces the bath solution. (C) Micrograph showing experimental set-up for rapid perfusion. B1 and B2 are square glass barrels used to deliver different bath solutions, to which the InsP3R channels in the cyto-out nuclear membrane patch at the tip of the micropipette P were alternately exposed. (D) Single-channel current trace showing InsP3R channel activity with 10 μM InsP3 and 2 μM [Ca2+]i in the pipette solution in the nuclear-attached configuration. Closed-channel baseline current levels are indicated by arrows in all current traces. (E–L) Single-channel current traces from cyto-out patches showing the responses of InsP3R channels to abrupt changes in ligand concentrations, indicated by coloured bars above the traces. Ligand concentrations were changed by switching the perfusing bath solutions, which also caused a change in the level of the closed-channel baseline current because of the different [KCl] in the perfusing solutions (supplementary information online). This allowed the time course of the perfusing solution switch to be monitored. Response latencies are denoted by grey bars below the traces. Cyto-out, cytoplasmic-side-out; InsP3R, inositol 1,4,5-trisphosphate receptor.
We first explored the kinetic responses of the channel to rapid changes in [InsP3]. The mean activation latency in response to a step change of [InsP3] from 0 to 10 μM in the constant presence of optimal (2 μM) [Ca2+]i was 500±60 ms (Fig 1E; number of solution switches (n)=458; number of cyto-out patches used (N)=14). Because this and all other activation and deactivation latencies showed non-gaussian distributions (Fig 2), non-parametric statistics were used in latency analyses (supplementary information online). A shorter (P=0.002) but still large latency (350±40 ms, n=168, N=4) was observed in response to a step change of [InsP3] from 0 to 100 μM, indicating that InsP3 binding and subsequent conformation changes required for channel opening both contribute significantly to the total activation latency. The mean deactivation latency in response to rapidly dropping [InsP3] from 10 to 0 μM in the constant presence of optimal [Ca2+]i (690±50 ms; Fig 1F, n=260, N=14) was similar (P=0.06) to that in response to [InsP3] dropping from 100 μM (700±70 ms, n=123, N=4), as expected. The latency of channel activation by InsP3 in the presence of 2 μM cytoplasmic Ca2+ (Ca2+i) is long compared with those observed for other ligand-activated channels in excitable cells (on the order of microseconds; Maconochie et al, 1994). It is also long compared with the latency of Ca2+ release resulting from the [InsP3] jump owing to photolysis of caged InsP3 in cells (Khodakhah & Ogden, 1995; Parker et al, 1996), or rapid perfusion of permeabilized cells or microsomes (Dufour et al, 1997). This is not surprising because in such experiments, the latency of Ca2+ release through a population of InsP3R channels was observed. In a system with a large number of InsP3R channels, the latency to first Ca2+ release will be significantly shorter than the activation latency for single InsP3R channels in response to the same ligand concentration change (supplementary information online). Furthermore, the non-gaussian distribution of single-channel InsP3 activation latency (Fig 2A) means that most of the InsP3 activation latencies observed were shorter than 60 ms even though the mean latency was 500 ms (discussed further in the supplementary information online).
Figure 2.

Latency histograms of InsP3R channels responding to changes in [InsP3] and [Ca2+]i. [InsP3] and [Ca2+]i are denoted by the coloured bars above each histogram. For each type of ligand concentration change investigated, both the linearly binned histogram (left) covering a limited latency range and the logarithmically binned histogram (right) showing the complete range of latencies over all times are shown. In each logarithmically binned histogram, each blue curve is the theoretical fit generated using one linear Markov chain scheme with the tabulated number of functionally indistinguishable conformations (supplementary information online). The black curve is the sum of the theoretical fits if several linear Markov chains were required. In each linearly binned histogram, the blue continuous curve is evaluated using the fastest linear Markov chain scheme and therefore is equivalent to the blue continuous curve in the corresponding logarithmically binned histogram. The range of the x-axis was selected so that the histogram includes at least 95% of the events described by the fastest linear Markov chain scheme. InsP3R, inositol 1,4,5-trisphosphate receptor.
We next explored the kinetics of Ca2+ regulation by applying rapid [Ca2+]i jumps in the continuous presence of a saturating concentration (10 μM) of InsP3. Channel activation by a jump from low (<10 nM) to optimal (2 μM) [Ca2+]i had a mean latency of 40±3 ms (Fig 1G, n=532, N=11), an order of magnitude faster than the response to InsP3 activation (P≪0.001). The latency for [Ca2+]i jumping to 300 μM was shorter (P≪0.001) but detectable (24±6 ms, n=94, N=3), indicating that Ca2+ binding and subsequent conformation changes contribute appreciably to the Ca2+ activation latency. Channel deactivation when Ca2+ was returned to <10 nM had a mean latency of 160±20 ms (Fig 1H, n=376, N=11), also significantly faster than channel deactivation owing to removal of InsP3 (P≪0.001). These results show that InsP3-bound channels respond rapidly to changes in [Ca2+]i, a property ideally suited for Ca2+-induced Ca2+ release (CICR) to couple InsP3R channels in Ca2+ signalling (Berridge, 1997).
Remarkably, the mean activation latency in response to simultaneous jumps of [InsP3] (from 0 to 10 μM) and [Ca2+]i (from <10 nM to 2 μM) was 59±4 ms (Fig 1I, n=638, N=7)—much shorter than that for InsP3 activation in the continuous presence of optimal [Ca2+]i (P≪0.001) and comparable with the latency for Ca2+ activation in the continuous presence of saturating [InsP3]. If InsP3 binding was required before Ca2+ can bind, then the activation latency for a simultaneous jump of the two ligands should be comparable with that for InsP3 in the presence of optimal [Ca2+]i. These results indicate that there is no requirement for strict sequential InsP3 and Ca2+ binding to activate the channel, invalidating the assumption used in many models that Ca2+ can only bind to InsP3-bound channels (Tang et al, 1996; Marchant & Taylor, 1997; Sneyd & Falcke, 2005). The activation latencies observed here are also incompatible with the independent binding of InsP3 and Ca2+ to activate the channel that is assumed in many other models (Swillens et al, 1994, 1998; Tang et al, 1996; Hirose et al, 1998; Fraiman & Dawson, 2004; Sneyd & Falcke, 2005), because independence predicts that the activation latency for simultaneous [InsP3] and [Ca2+]i jumps should be longer than the activation latencies for individual [InsP3] or [Ca2+]i jumps. Instead, the kinetic data are consistent with an unexpected negative cooperative interference, in which high-affinity Ca2+ binding to a channel not bound to InsP3 retards subsequent activation of the channel by InsP3.
The mean deactivation latency of 69±5 ms for simultaneous removal of InsP3 (from 10 μM to 0) and Ca2+ (from 2 μM to <10 nM) (Fig 1J, n=458, N=7) was significantly shorter than the deactivation latencies for removal of either InsP3 (P≪0.001) or Ca2+ (P=0.002) alone. This result implies that unbinding of InsP3 and Ca2+ from the InsP3R is neither sequential nor independent.
Channel activation and deactivation latency distributions
The distributions of InsP3R channel activation and deactivation latencies showed distinct deficits of short latencies (linearly binned histograms in Fig 2A–F) when compared with a single exponential distribution, similar to those observed during activation of voltage-gated channels (Patlak & Horn, 1982; Hoshi et al, 1994). Such activation latency deficits can be well accounted for by a linear Markov chain scheme connecting C1—the closed inactive conformation adopted by the channel before the ligand concentration change—to O—the first open conformation adopted by the channel after the jump in ligand concentration—through one or more intervening closed conformations: C1↔C2↔…↔Cm↔O, where m (⩾2) is the total number of functionally indistinguishable closed conformations in the chain. The deficits of short deactivation latencies can be similarly accounted for by linear Markov chain schemes connecting the initial active open channel conformation to the first inactive closed conformation through several intervening, functionally indistinguishable active conformations (Fig 2; supplementary information online).
The logarithmically binned histogram (Sigworth & Sine, 1987) showing the latency distribution over all timescales of a channel activation or deactivation process described by a linear Markov chain scheme has a peak shape with a single maximum at the dominant time constant. This peak becomes narrower as the number of functionally indistinguishable conformations (m) present in the linear chain increases. However, logarithmically binned histograms of most InsP3R channel activation and deactivation latencies show multiple maxima or broad shapes that cannot be described with a single linear Markov chain (Fig 2A,B,D–F). These can be satisfactorily described by branched Markov chain schemes such as the one shown below: 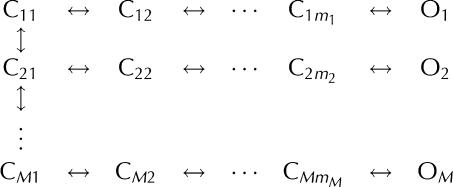 in which the channel responding to the ligand concentration change passes through M conformation transition pathways, each corresponding to a linear Markov chain that generates a single peak in the logarithmically binned latency histogram (Fig 2; supplementary information online).
in which the channel responding to the ligand concentration change passes through M conformation transition pathways, each corresponding to a linear Markov chain that generates a single peak in the logarithmically binned latency histogram (Fig 2; supplementary information online).
Kinetic schemes accounting for channel latencies
The observed response latencies for single InsP3R channels point to complex and seemingly paradoxical interactions between InsP3 and Ca2+ activation of the channel. The long activation latencies in response to jumps in [InsP3] in the constant presence of optimal [Ca2+]i compared with the activation latencies in response to simultaneous jumps in [InsP3] and [Ca2+]i indicate that Ca2+ can bind with high affinity to the channel in the absence of InsP3. However, unlike the high-affinity Ca2+ binding that activates the channel along with InsP3 binding that has been established from steady-state studies (Foskett et al, 2007), this Ca2+ binding interferes negatively with the activation of the channel by InsP3. It does this by predisposing the channel to activate through slower conformation transition pathways or by reducing the rate of channel activation through available conformation transition pathways in the branched Markov chain scheme (cf. Fig 2A,E).
Conversely, the significantly more rapid deactivation of the channel in response to simultaneous drops in [InsP3] and [Ca2+]i compared with channel deactivation caused by drops in either [InsP3] or [Ca2+]i alone indicates that the InsP3 and the high-affinity Ca2+ sites have positive cooperative coupling in an activated channel. Occupation of the InsP3 or Ca2+ sites causes the channel to deactivate through slower conformation transition pathways in the branched Markov chain (cf. Fig 2B,D,F), thereby stabilizing the channel in active conformations.
Fig 3A depicts a kinetic scheme that can account for these observations qualitatively, assuming that the high-affinity inhibitory Ca2+ sites are distinct from the high-affinity activating Ca2+ sites. Ca2+ binding to these inhibitory Ca2+ sites precludes InsP3 binding to the channel, and vice versa. Pre-exposure to optimal [Ca2+]i in the absence of InsP3 retards InsP3 activation of the channel because InsP3 cannot bind to the channel until Ca2+ dissociates from the inhibitory sites, even after the jump in [InsP3]. In this scheme, the activating Ca2+ sites and the InsP3 sites show a positive allosteric interaction in an active channel. This is a result of the different channel deactivation rates by various pathways in the branched Markov chain generated by multiple InsP3 and Ca2+ binding to the tetrameric channels, shown partly in Fig 3A.
Figure 3.

Kinetic schemes accounting for the observed InsP3R channel response latencies. (A) A qualitative model stipulating distinct high-affinity inhibitory and activating Ca2+ sites in each InsP3R channel. C and O represent closed inactive and active channel conformations. Channel conformations with different ligand binding states are explicitly shown in the scheme by superscript and subscript numbers in different colours: number of occupied high-affinity Ca2+ inhibitory sites in red, occupied high-affinity activating Ca2+ sites in black and occupied InsP3 sites in green (as indicated by the key at the top right). Ligand binding and dissociation are indicated by arrows: InsP3 binding in green, Ca2+ binding to inhibitory sites in red and Ca2+ binding to activating sites in black. Yellow arrows represent conformation changes. Because the channel is a tetrameric unit, more than one Ca2+ can bind to either the activating or inhibitory sites. Those ligand-binding states are not shown in the scheme for clarity. Multiple InsP3 binding to the channel (dashed lines) is shown to demonstrate that the scheme can accommodate branched Markov chains. (B) A qualitative allosteric model stipulating only one kind of high-affinity Ca2+ site in an InsP3R channel. Occupation of the InsP3 binding sites causes the channel to adopt the C and D conformations instead of the A and B conformations, so that Ca2+ binding to the high-affinity Ca2+ site becomes activating instead of inhibitory. Unlike (A), only conformation changes are shown in this kinetic scheme. InsP3 and Ca2+ binding to the channel, and the ligand-binding state of the channel conformations are omitted for clarity.
Alternatively, a model stipulating that only one high-affinity Ca2+ site is present in each InsP3R monomer is also qualitatively consistent with all the kinetic responses observed here. The model, a simplified version of a quantitative model proposed to account for steady-state InsP3 and Ca2+i regulation of channel activity (Mak et al, 2003), assumes only basic allosteric interactions between the two ligands that arise solely from preferential binding of InsP3 and Ca2+ to various channel conformations. In its simplest form (Fig 3B), the model hypothesizes that the channel can adopt four different conformations. In the absence of InsP3, it is energetically favourable for the channel to adopt two conformations, one active (A) and one inactive (B), with Ca2+ binding to the high-affinity sites stabilizing the inactive B conformation. Thus, in the absence of InsP3, the high-affinity Ca2+ sites are actually inhibitory. InsP3 binding to the channel energetically stabilizes the other two conformations, also one active (C) and one inactive (D). Ca2+ binding to the high-affinity sites stabilizes the active C conformation relative to the inactive D conformation. Consequently, the high-affinity Ca2+ sites are activating when the channel is InsP3-bound. Thus, because of the different relative affinities of the high-affinity Ca2+ sites in the four channel conformations, InsP3 binding to the channel changes the nature of the high-affinity Ca2+ sites from inhibitory to activating. This transformative effect of InsP3 on the nature of the high-affinity Ca2+ binding sites can account for the seemingly paradoxical channel response latencies measured here. Because Ca2+ binding to the high-affinity sites in the absence of InsP3 stabilizes the inactive B conformation, which has low affinity for InsP3, pre-exposure of the channel to 2 μM Ca2+i slows down binding of InsP3 to the channel and consequent activation kinetics compared with activation by simultaneous [Ca2+]i and [InsP3] jumps. Conversely, occupation of the high-affinity Ca2+ and InsP3 sites stabilizes an active channel in the C conformation with high affinity for InsP3 and Ca2+. This accounts for the shorter mean latency for deactivation by simultaneous removal of InsP3 and Ca2+i compared with that owing to removal of either Ca2+i or InsP3 alone. The model hypothesizes that the channel can adopt the different conformations in any ligand-binding state, and InsP3 binding and Ca2+ binding regulate channel activity by affecting the probability of the channel being in various conformations. Thus, active and inactive conformations are connected by many branched pathways through various ligand binding and conformation changes.
Channel activity level after activation
It has been speculated that spontaneous changes in channel activity after the initial activation might account for biphasic Ca2+ release kinetics and graded release of Ca2+ from intracellular stores in response to incremental levels of stimulation (Muallem et al, 1989; Meyer & Stryer, 1990; Marchant & Taylor, 1998). However, no change in single-channel Po was observed after the InsP3R was activated by jumps in [InsP3], [Ca2+]i or both simultaneously (Fig 4). Thus, behaviours akin to ‘adaptation' observed in the ryanodine receptor (Fill et al, 2000) are unlikely to account for InsP3R-mediated graded release of Ca2+.
Figure 4.

Steady level of InsP3R channel activity observed after channel activation by abrupt jumps in ligand concentration. (A) Current traces of InsP3R channels after activation by step change in [InsP3] (top), [Ca2+]i (middle) and both simultaneously (bottom). (B) Averaged time course of Po (in 0.1 s bins) of one InsP3R channel after step change of [Ca2+]i at t=0 in one membrane patch. A total of 72 [Ca2+]i jumps were averaged. InsP3R, inositol 1,4,5-trisphosphate receptor.
Channel inhibition and recovery kinetics
The steady-state gating activity of the InsP3R is biphasically regulated by [Ca2+]i, indicating that the channel also has low-affinity inhibitory Ca2+ binding sites (Foskett et al, 2007). Ca2+ inhibition can provide a negative feedback mechanism to either terminate or prevent Ca2+ release as the local [Ca2+]i is raised by InsP3R-mediated Ca2+ release. This can have significant roles in generating Ca2+ spikes and oscillations, and in creating highly localized Ca2+ signals in subcellular compartments by preventing runaway Ca2+ release due to CICR. Switching [Ca2+]i from 2 μM (optimal) to 300 μM (inhibitory; Ionescu et al, 2006) in the presence of 10 μM InsP3 inhibited channel gating (Fig 1K). This shows that InsP3 binding does not render the low-affinity inhibitory Ca2+ sites inaccessible for Ca2+ binding, disproving previous claims (Adkins & Taylor, 1999). The mean latency for Ca2+ inhibition (290±40 ms, n=186, N=7) was substantially longer than that for Ca2+ activation (P≪0.001).
The mean latency for channel recovery from Ca2+ inhibition was 2.4±0.2 s (Fig 1L, n=197, N=7). This remarkably long latency defines a refractory period between successive Ca2+ release events mediated by the same InsP3R channel or channel cluster. A refractory period has been invoked to account for mutual annihilation of colliding intracellular Ca2+ waves (Lechleiter et al, 1991) and the periodicity of Ca2+ oscillations (Parker & Ivorra, 1990), but its molecular basis has remained unknown. The observed latency is in quantitative agreement with the mean refractory periods (∼2.5 s) between two successive spontaneous InsP3R-mediated elementary Ca2+ release events (puffs) observed in the presence of saturating [InsP3] (Fraiman et al, 2006). This result indicates that the latency for channel recovery from Ca2+ inhibition, rather than other mechanisms such as lingering high [Ca2+]i in the vicinity of the Ca2+ release sites after a Ca2+ release event, is responsible for the refractory behaviour of Ca2+ release sites observed in vivo (Ilyin & Parker, 1994).
In contrast to the lack of short latencies in channel activation and deactivation, latency distributions of channel inhibition by Ca2+ and recovery (Fig 2G,H) have no appreciable deficits of short events. This indicates that the active conformation adopted by the channel in optimal (2 μM) [Ca2+]i is in effect connected directly to an inactive conformation through the binding of a single Ca2+ to one inhibitory low-affinity site. More remarkably, even in the presence of 300 μM Ca2+i, the channel is in an inactive conformation that is connected directly to an active conformation through the dissociation of a single Ca2+, despite the tetrameric channel structure with possibly four equivalent inhibitory Ca2+ sites. Thus, Ca2+ inhibition of the channel can be described by a simple two-state kinetic scheme: O↔I. This is consistent with the observation that Ca2+ inhibition of steady-state gating of Sf9 InsP3R in the presence of 10 μM InsP3 showed a Hill coefficient of approximately 1 (Ionescu et al, 2006). It is possible that the multiple Ca2+ inhibitory sites have strong negative cooperativity so that binding of Ca2+ to one of them prevents Ca2+ from binding to the remaining vacant sites.
Kinetics of single-channel Ca2+ release burst
In cells, Ca2+ released by an individual channel raises [Ca2+]i to inhibitory (∼100 μM) levels in the microdomain surrounding it. Our kinetic results predict that this Ca2+ will feed back to inhibit it and terminate release after a burst of activity. [Ca2+]i jumps from low sub-activating (<10 nM) levels directly to inhibitory (300 μM) levels in the presence of 10 μM InsP3 produced just such a burst of channel activity. This is due to Ca2+ first binding to the high-affinity sites to activate the channel before the low-affinity inhibitory sites become occupied (Fig 5A). Mean activation and inhibition latencies of channel activity bursts were 24±6 ms (Fig 5B, n=94, N=3) and 470±90 ms (Fig 5C, n=70, N=3), respectively. The mean single-channel burst duration was 470±100 ms (Fig 5D, n=50, N=3). The time course of these channel activity bursts represents the kinetics of the fundamental Ca2+ release event—the basic building block from which local concerted Ca2+ release by clusters of InsP3R channels (puffs) and global propagating InsP3R-mediated Ca2+ signals (waves) are generated through CICR (Berridge, 1997).
Figure 5.

Kinetics of InsP3R channel activity bursts in response to [Ca2+]i changes between sub-activating (<10 nM) and inhibitory (300 μM) levels in the presence of 10 μM InsP3. (A) A typical current trace of InsP3R channel activity burst in response to [Ca2+]i increasing from <10 nM to 300 μM. The burst activation and termination latencies are denoted by the dark and light grey bars, respectively. (B–D) Logarithmically binned histograms of burst activation and termination latencies, and burst durations, respectively. The latency distributions were fitted (black curves) using more than one linear Markov chain schemes (blue curves). The number of functionally indistinguishable conformations in each scheme is tabulated next to the corresponding blue curve. Short latencies of burst termination were not observed in (C) because when Ca2+ binds rapidly to the inhibitory sites before it binds to the activating sites, no channel activity follows. (E,F) Typical current traces showing different InsP3R channel responses to rapid decrease in [Ca2+]i from 300 μM to <10 nM.
A considerable fraction (9 out of 103) of [Ca2+]i jumps from <10 nM to 300 μM failed to generate a burst (although [Ca2+]i jumps before and after those jumps did generate bursts), substantially greater than that (7 out of 607) for [Ca2+]i jumps from <10 nM to 2 μM. Thus, Ca2+ binding to the high-affinity activating sites and low-affinity inhibitory sites is not sequential, and the failure of [Ca2+]i jumps (<10 nM to 300 μM) to generate an activity burst reflects the non-zero probability of Ca2+ binding to an inhibitory site before the activating sites becomes occupied. The mean latency for recovery from Ca2+ inhibition is over an order of magnitude longer than the mean latency of deactivation by Ca2+i removal (P≪0.001). Therefore during most [Ca2+]i drops (from 300 μM to <10 nM), no channel activity was observed as the channels were deactivated by Ca2+ dissociation from the high-affinity activating sites before they recovered from Ca2+ inhibition (Fig 5E). However, in 6 out of 94 [Ca2+]i drops, the channel recovered from Ca2+ inhibition first, generating a brief burst of activity before it was deactivated by Ca2+i unbinding (Fig 5F). This indicates that Ca2+ dissociation from the inhibitory and activating sites is also not sequential.
Conclusions
The rapid kinetic responses of single InsP3R channels in native ER membranes to [InsP3] and [Ca2+]i changes observed here have provided new insights into the molecular mechanisms of channel activation and inhibition by these ligands. These measurements have shown that there is no requirement for sequential InsP3 and Ca2+ binding during channel activation or deactivation, but binding of the two ligands is nevertheless not independent. A complex interaction involving both positive and negative cooperativity exists between InsP3 and high-affinity Ca2+ binding sites, which was not anticipated in most theoretical models of the InsP3R. The kinetics of high [Ca2+]i inhibition of channel activity have provided insights into the molecular basis of fundamental Ca2+-release events and the refractory period that accounts for [Ca2+]i oscillation frequency and wave annihilation. These single-channel kinetic data thus provide novel quantitative details for future construction of realistic models to account for the role of InsP3R channel kinetics in generating cytoplasmic Ca2+ signals (Shuai et al, 2007). The approaches described here can also be used in future studies to define the kinetics of other types of InsP3R channel regulation, and facilitate better understanding of [Ca2+]i signalling by the ubiquitous InsP3 pathway.
Methods
Single-channel patch-clamp studies of InsP3R channels in a native membrane environment using isolated nuclei from cultured insect Ispodoptera frugiperda (Sf9) cells were performed in cyto-out excised patch configuration. The cytoplasmic side of the nuclear membrane patch and of any InsP3R channels therein was exposed to different perfusing solutions containing various ligand concentrations during a continuous patch-clamp recording using a rapid perfusion set-up. Activation latency is the duration between the solution switch time and the first observed channel opening event in response to the solution switch. Similarly, deactivation latency is the duration between solution switch time and the last observed channel closing. The analysis allows latencies >5 ms to be clearly and consistently resolved. Current traces from cyto-out membrane patches containing one to several (⩽10) active InsP3R channels were selected for latency analysis (supplementary information online).
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
supplementary Information
Acknowledgments
We thank P. De Weer, C. Deutsch, F.T. Horrigan, T. Hoshi and E.M. Ostap for comments on the manuscript and W.J. Bruno for help with statistical analysis of the data. This work was supported by National Institutes of Health grants GM074999 to D.-O.D.M., MH059937 to J.K.F. and GM65830 to J.E.P.
References
- Adkins CE, Taylor CW (1999) Lateral inhibition of inositol 1,4,5-trisphosphate receptors by cytosolic Ca2+. Curr Biol 9: 1115–1118 [DOI] [PubMed] [Google Scholar]
- Berridge MJ (1993) Inositol trisphosphate and calcium signalling. Nature 361: 315–325 [DOI] [PubMed] [Google Scholar]
- Berridge MJ (1997) Elementary and global aspects of calcium signalling. J Physiol 499: 291–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour JF, Arias IM, Turner TJ (1997) Inositol 1,4,5-trisphosphate and calcium regulate the calcium channel function of the hepatic inositol 1,4,5-trisphosphate receptor. J Biol Chem 272: 2675–2681 [DOI] [PubMed] [Google Scholar]
- Falcke M, Tsimring L, Levine H (2000) Stochastic spreading of intracellular Ca2+ release. Phys Rev E 62: 2636–2643 [DOI] [PubMed] [Google Scholar]
- Fill M, Zahradnikova A, Villalba-Galea CA, Zahradnik I, Escobar AL, Gyorke S (2000) Ryanodine receptor adaptation. J Gen Physiol 116: 873–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foskett JK, White C, Cheung KH, Mak D-OD (2007) Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87: 593–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraiman D, Dawson SP (2004) A model of IP3 receptor with a luminal calcium binding site: stochastic simulations and analysis. Cell Calcium 35: 403–413 [DOI] [PubMed] [Google Scholar]
- Fraiman D, Pando B, Dargan S, Parker I, Dawson SP (2006) Analysis of puff dynamics in oocytes: interdependence of puff amplitude and interpuff interval. Biophys J 90: 3897–3907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkle DJ, Bianchi MT, Macdonald RL (2003) Modifications of a commercial perfusion system for use in ultrafast solution exchange during patch clamp recording. Biotechniques 35: 472–476 [PubMed] [Google Scholar]
- Hirose K, Kadowaki S, Iino M (1998) Allosteric regulation by cytoplasmic Ca2+ and IP3 of the gating of IP3 receptors in permeabilized guinea-pig vascular smooth muscle cells. J Physiol 506: 407–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW (1994) Shaker potassium channel gating. I: transitions near the open state. J Gen Physiol 103: 249–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilyin V, Parker I (1994) Role of cytosolic Ca2+ in inhibition of InsP3-evoked Ca2+ release in Xenopus oocytes. J Physiol 477: 503–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionescu L, Cheung KH, Vais H, Mak D-OD, White C, Foskett JK (2006) Graded recruitment and inactivation of single InsP3 receptor Ca2+-release channels: implications for quantal Ca2+ release. J Physiol 573: 645–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodakhah K, Ogden D (1995) Fast activation and inactivation of inositol trisphosphate-evoked Ca2+ release in rat cerebellar Purkinje neurones. J Physiol 487: 343–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechleiter J, Girard S, Peralta E, Clapham D (1991) Spiral calcium wave propagation and annihilation in Xenopus laevis oocytes. Science 252: 123–126 [DOI] [PubMed] [Google Scholar]
- Maconochie DJ, Zempel JM, Steinbach JH (1994) How quickly can GABAA receptors open? Neuron 12: 61–71 [DOI] [PubMed] [Google Scholar]
- Mak D-OD, McBride SM, Foskett JK (2003) Spontaneous channel activity of the inositol 1,4,5-trisphosphate (InsP3) receptor (InsP3R). Application of allosteric modeling to calcium and InsP3 regulation of InsP3R single-channel gating. J Gen Physiol 122: 583–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak D-OD, White C, Ionescu L, Foskett JK (2005) Nuclear patch clamp electrophysiology of inositol trisphosphate receptor Ca2+ release channels. In Methods in Calcium Signaling Research, Putney JW Jr (ed) pp 203–229. Boca Raton, FL, USA: CRC Press [Google Scholar]
- Marchant JS, Taylor CW (1997) Cooperative activation of IP3 receptors by sequential binding of IP3 and Ca2+ safeguards against spontaneous activity. Curr Biol 7: 510–518 [DOI] [PubMed] [Google Scholar]
- Marchant JS, Taylor CW (1998) Rapid activation and partial inactivation of inositol trisphosphate receptors by inositol trisphosphate. Biochemistry 37: 11524–11533 [DOI] [PubMed] [Google Scholar]
- Meyer T, Stryer L (1990) Transient calcium release induced by successive increments of inositol 1,4,5-trisphosphate. Proc Natl Acad Sci USA 87: 3841–3845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muallem S, Pandol SJ, Beeker TG (1989) Hormone-evoked calcium release from intracellular stores is a quantal process. J Biol Chem 264: 205–212 [PubMed] [Google Scholar]
- Parker I, Ivorra I (1990) Inhibition by Ca2+ of inositol trisphosphate-mediated Ca2+ liberation: a possible mechanism for oscillatory release of Ca2+. Proc Natl Acad Sci USA 87: 260–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker I, Yao Y, Ilyin V (1996) Fast kinetics of calcium liberation induced in Xenopus oocytes by photoreleased inositol trisphosphate. Biophys J 70: 222–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patlak J, Horn R (1982) Effect of N-bromoacetamide on single sodium-channel currents in excised membrane patches. J Gen Physiol 79: 333–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney JW Jr, Bird GS (1993) The inositol phosphate–calcium signaling system in nonexcitable cells. Endocr Rev 14: 610–631 [DOI] [PubMed] [Google Scholar]
- Shuai J, Pearson JE, Foskett JK, Mak D-OD, Parker I (2007) A kinetic model of single and clustered IP3 receptors in the absence of Ca2+ feedback. Biophys J 93: 1151–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth FJ, Sine SM (1987) Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys J 52: 1047–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneyd J, Falcke M (2005) Models of the inositol trisphosphate receptor. Prog Biophys Mol Biol 89: 207–245 [DOI] [PubMed] [Google Scholar]
- Swillens S, Combettes L, Champeil P (1994) Transient inositol 1,4,5-trisphosphate-induced Ca2+ release: a model based on regulatory Ca2+-binding sites along the permeation pathway. Proc Natl Acad Sci USA 91: 10074–10078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swillens S, Champeil P, Combettes L, Dupont G (1998) Stochastic simulation of a single inositol 1,4,5-trisphosphate-sensitive Ca2+ channel reveals repetitive openings during ‘blip-like' Ca2+ transients. Cell Calcium 23: 291–302 [DOI] [PubMed] [Google Scholar]
- Tang Y, Stephenson JL, Othmer HG (1996) Simplification and analysis of models of calcium dynamics based on IP3-sensitive calcium channel kinetics. Biophys J 70: 246–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CW, Richardson A (1991) Structure and function of inositol trisphosphate receptors. Pharmacol Ther 51: 97–137 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary Information