

Summary
The zebrafish fat-free (ffr) mutation was identified in a physiological screen for genes that regulate lipid metabolism. ffr mutant larvae are morphologically indistinguishable from wild-type sibling larvae, but their absorption of fluorescent lipids is severely impaired. Through positional cloning, we have identified a causative mutation in a highly conserved and ubiquitously expressed gene within the ffr locus. The Ffr protein contains a Dor-1 like domain typical of oligomeric Golgi complex (COG) gene, cog8. Golgi complex ultrastructure is disrupted in the ffr digestive tract. Consistent with a possible role in COG-mediated Golgi function, wild-type Ffr-GFP and COG8-mRFP fusion proteins partially colocalize in zebrafish blastomeres. Enterocyte retention of an endosomal lipid marker in ffr larvae support the idea that altered vesicle trafficking contributes to the ffr mutant defect. These data indicate that ffr is required for both Golgi structure and vesicular trafficking, and ultimately lipid transport.
Introduction
Dietary fat consumption is known to be a risk factor in cardiovascular diseases, diabetes, and obesity (Hill et al., 2003; Joffe et al., 2001; McNeely et al., 2001). In recent years, significant effort has been applied to defining basic mechanisms whereby cholesterol and other lipids are absorbed and transported by small intestinal enterocytes. Despite recent advances, many features of this process remain undefined (Cao et al., 2003; Ehehalt et al., 2004a, 2004b; Field, 2001; Pohl et al., 2004; Tso and Fujimoto, 1991). Heritable diseases associated with alteration of lipid absorption can perturb either digestion and uptake of dietary lipids or their transport and subsequent export into the lymphatic system and bloodstream as lipoprotein particles or protein-bound lipids. Recently, causative mutations in genes that play a role in intracellular trafficking have been identified for several of these latter groups of disorders (Chang et al., 2005; Jones et al., 2003; Orso et al., 2000; Soutar et al., 2003).
In previous work, we showed that fluorescent reporters can serve as optical biosensors that can be used in high-throughput genetic screens to identify genes that regulate lipid metabolism (Farber et al., 2001). In a pilot screen using the quenched fluorescent phospholipid PED6, we identified a recessive lethal mutation in fat free (ffr) that alters processing of several classes of lipid compounds. ffr mutant larvae swallow normally but have limited absorption of phospholipids, cholesterol, and long-chain fatty acids compared with wild-type siblings. Initial biochemical analysis suggested that altered bile synthesis or secretion may account for this lipid absorption defect (Farber et al., 2001; Ho et al., 2003).
Here, we describe a more detailed phenotypic analysis of ffr mutant larvae and the positional cloning of the ffr gene. ffr encodes a conserved protein with no known function that is expressed in a wide range of eukaryotic organisms. The only recognizable feature of the protein is a small domain homologous to the Dor-1 region of a conserved gene that is a component of the COG complex (Ungar et al., 2002). This hetero-octomeric protein complex regulates Golgi structure and vesicle traffic between the Golgi and the endoplasmic reticulum (ER) in yeast and mammalian cells (Oka et al., 2004, 2005; Ram et al., 2002; Suvorova et al., 2002; Ungar et al., 2005; VanRheenen et al., 1998). Saccharomyces cerevisiae dor-1, a COG component, is essential for vesicle targeting to the Golgi (Whyte and Munro, 2001), whereas the mammalian Dor-1 ortholog Cog8 has been proposed to regulate intra-Golgi transport and secondary protein glycosylation (Ungar et al., 2002). Consistent with a role for Ffr in intracellular trafficking and Golgi function, we show that a Ffr-GFP fusion protein partially colocalizes with Cog8 and that an ApoA1 fusion protein is mislocalized in ffr-deficient zebrafish embryos. In addition, wild-type Ffr protein localizes in part to the TGN, while mutant Ffr protein, which has an intact Dor-1-like domain, is restricted to the TGN. Endosomal labeling experiments using the styryl dye AM1-43 also show that vesicular recycling is impaired in ffr mutants. These data, together with observations of altered intestinal Golgi ultrastructure in ffr mutants, support the idea that Ffr is an important mediator of intestinal lipid absorption through its effects on intracellular trafficking and protein secretion.
Results
Zebrafish ffr mutants exhibit digestive system defects
Homozygous ffr mutant larvae are morphologically indistinguishable from wild-type siblings at 5 days postfertilization (dpf), the onset of exogenous feeding. However, ffr mutants fail to process fluorescently labeled phosphatidylcholine (PC), long-chain fatty acid and cholesterol analogs ingested at this developmental stage (Farber et al., 2001). To better understand the lipid processing defect caused by the ffr mutation, we performed histological and ultrastructural analyses of the digestive organs of ffr mutant larvae.
Liver and biliary anatomies were characterized first, because of the important role that bile plays in dietary lipid absorption. Hepatocytes appear normal in histological sections and electron micrographs (data not shown). Immunostainings with an antibody directed against a cytokeratin biliary marker (Figure 1B) and with a mammalian canalicular marker, p-glycoprotein (Figure 1D), were markedly abnormal compared to wild-type (Figures 1A and 1C). Ultrastructural analysis showed paucity of intrahepatic bile ducts with degeneration of most remaining biliary epithelial cells (Figures 1E and 1F). These initial findings suggested that the ffr gene is essential for the maintenance or development of the intrahepatic biliary system.
Figure 1. Phenotypic analysis of ffr mutants.

A and B) Confocal projections through the liver of 5 dpf wild-type and ffr larvae processed for cytokeratin immunohistochemistry. These immunostainings reveal widespread biliary degeneration in ffr mutants (5 dpf).
C and D) Similar immunostainings of 5 dpf wild-type and ffr larvae processed for p-glycoprotein immunohistochemistry. These images of the wild-type and ffr liver show that canalicular architecture is also disrupted in the ffr mutants (5 dpf).
E and F) Transmission electron micrographs showed degenerating biliary epithelial cells (bc) (indicated by arrow) and canaliculi (indicated by arrowhead) within the liver of ffr mutants (F) compared with wild-type (E).
G and H) Electron micrographs of the pancreas reveal small zymogen granules in the ffr mutants (arrowhead; [H]) compared with wild-type (G).
I and J) Confocal image from the pancreas of a 5 dpf ffr mutant (J) processed for carboxypeptidase A immunohistochemistry (green) showed altered distribution of this digestive enzyme compared with wild-type (I). Nuclei stained with Dapi (blue).
Further analyses revealed additional ultrastructural defects throughout the ffr digestive system. Zymogen granules within acinar cells of the ffr exocrine pancreas were small (Figure 1H), and the digestive enzyme carboxypeptidase A was mislocalized (Figure 1J) in comparison to wild-type siblings (Figures 1G and 1I) indicating a defect in protein sorting and/or processing. The Golgi was also markedly dilated in ffr mutant intestinal enterocytes and surviving biliary epithelial cells at 5 dpf (Figures 2A–2D and 2G–2H). Less pronounced Golgi defects were present in intestinal enterocytes and biliary epithelial cells at earlier developmental stages (4 dpf; data not shown) and within the pancreatic exocrine cells (5 dpf; Figures 2E and 2F) as well as hepatocytes (5 dpf; data not shown). By contrast, Golgi morphology was normal in kidney epithelia, skeletal muscle and endocrine pancreas (data not shown). Together, these data suggested that the Ffr protein is essential for lipid processing and Golgi morphology within the digestive tract.
Figure 2. Ultrastructural analyses reveal abnormal Golgi apparatus in the ffr mutants.

A–D) Transmission electron micrographs show that intestinal Golgi architecture is disrupted in in 5 dpf ffr mutant larvae (B and D) compared to wild-type larvae (A and C). E–H) Golgi morphology is also disrupted in the 5 dpf ffr exocrine pancreas (F) and biliary cells (H), compared with wild-type (E and G) (5 dpf). n, nucleus; bd, bile duct lumen; arrowheads point to Golgi in all panels.
Dor-1 (COG8) homology in Ffr
To identify the ffr gene, a positional strategy was employed. Bulk segregant analysis using markers from the zebrafish genetic map placed the ffr locus on zebrafish chromosome 10. In subsequent steps, a critical region surrounding the ffr locus bounded by markers 1.1 cM (z54342) and 3.8 cM (z25936) from the ffr locus was defined. Additional studies identified a polymorphic marker within an EST (fc83b07) that was 0.06 cM (1 recombinant in 1732 meioses) from the ffr locus. Using this marker 5 BAC clones containing sequences adjacent to the ffr locus were identified. Based upon end sequences of these BAC clones, a fully sequenced BAC (dK56F11) predicted to contain eight genes was identified from the public zebrafish genome database (Figure 3A). Sequence analysis of the eight candidate cDNAs from ffr mutant and wild-type sibling larvae identified only a single point mutation within an open reading frame with high sequence homology to the human gene ANG2 (Lemmens et al., 1997) (Figure 3B). This C to T transition generated a premature stop codon in the zebrafish ang2 cDNA. Supporting the idea that zebrafish ang2 was in fact the ffr gene, 0 of 2000 ffr mutants were recombinant for a polymorphic marker identified within this predicted gene. Further supporting ffr as an ANG2 ortholog, the region adjacent to ffr is syntenic to human chromosome 11q13 (Figure 3D).
Figure 3. Positional cloning of the zebrafish ffr locus.
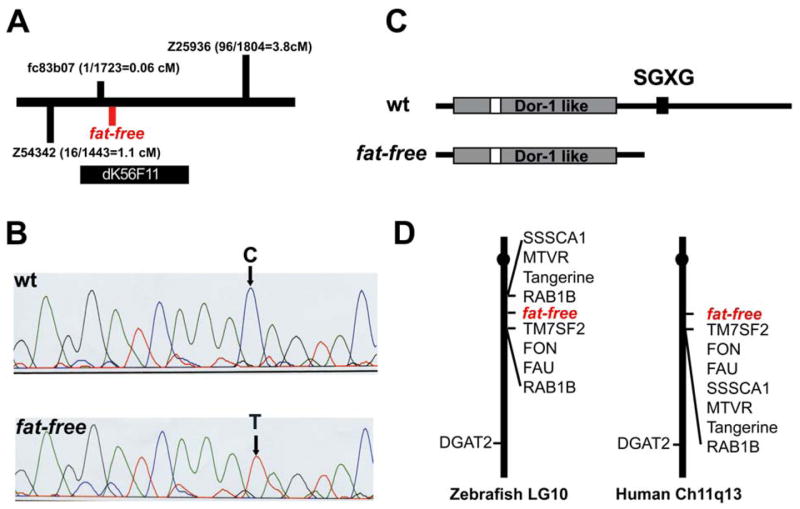
A) Schematic of the ffr locus. Marker fc83b07 is 0.06 cM from the ffr locus (1 recombinant out of 1732 meioses). A radiolabeled probe synthesized from fc83b07 identified five BAC clones. Mapping using the end sequences of these BAC clones indicated that clone dK56F11 spanned the entire ffr locus.
B) A single nucleotide mutation (C to T) that results in a premature stop codon was identified in ffr mutants (0 recombinants > 2000 meioses) within a predicted open reading of dK56F11.
C) Domain analysis indicates that Ffr has a Dor-1 like domain, a tyrosine phosphorylation site (Tyr-P), and a SGXG motif. The truncated mutant protein is predicted to be approximately half the length of wild-type Ffr.
D) ffr is located on zebrafish chromosome 10 in a region syntenic to a region of human chromosome 11q13.
A comparison of the cDNA and genomic sequences revealed that the ffr gene encodes a 2.5-kilobase transcript containing 11 exons. The predicted 842 amino acid zebrafish protein is well conserved compared with its invertebrate and mammalian orthologs and overall has 75% identity to human ANG2 (Figure 4). Analysis of the predicted amino acid sequence indicates that Ffr contains a Dor-1 like domain, a canonical tyrosine phosphorylation site, and a putative SGXG motif (a potential glycosaminoglycan attachment site) (Figure 3C).
Figure 4. Zebrafish Ffr protein is highly conserved from invertebrate to human.

A) The zebrafish Ffr protein sequence reveals 75% identity with human ortholog, and 46% identity with fly ortholog.
B) Dor-1 like domain (amino acids 41–384) was identified using a prosite motif scan (http://us.expasy.org/prosite/) and returned a hit to Dor-1 (amino acids 41–384, raw-score = −111.2, N-score = 11.150, E-value = 0.00015; pfam Dor1–Dor1-like family).
To confirm that the ffr gene is responsible for the mutant phenotype, “knockdown” experiments were performed using nonoverlapping antisense morpholino oligonucleotides (MOs) (Figure 5A). A morpholino designed to target the ffr translation initiation codon (MO-1) was lethal even when injected into 1- to 4-cell stage wild-type embryos at low concentrations (42–126 ng per embryo; data not shown). Several other MOs were designed to target either splice donor (MO-2; MO-5) sites or a splice acceptor (MO-4) site within newly transcribed zygotic ffr mRNA. These splice blocking MOs act to disrupt zygotic mRNA processing without interfering with maternal ffr mRNA translation. The effectiveness of splice junction targeting by MO4 was confirmed by RT-PCR (data not shown). Injection of MO2, MO4 and MO5 phenocopied the ffr lipid absorption defect in 80% of 5 dpf larvae (n = 20 from three experiments) (Figure 5E).
Figure 5. Phenocopy of the ffr mutation by targeted morpholino antisense oligonucleotides and rescue of the phenotype by ffr mRNA injection.
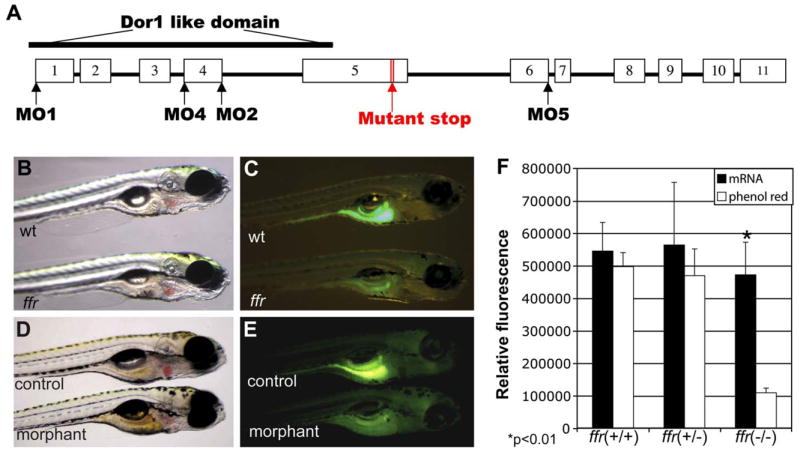
A) Genomic structure of the ffr gene. The white blocks represent the exons from 1–11. The morpholino antisense oligonucleotides were designed either targeting the AUG translation initiation codon (MO1) or at mRNA splice junctions (MO2, MO4, and MO5).
B and C). ffr mutants (6 dpf) exhibit altered lipid absorption. Lipid processing was visualized by labeling with fluorescent cholesterol (NBD-cholesterol, 2 hr, 3 μg/ml solubilized with fish bile). Gall bladder fluorescence is absent in the ffr larvae. The ffr larva in (B) was indistinguishable from wild-type.
D and E) Antisense targeting of ffr phenocopies the mutant phenotype. Embryos were injected with either water (control) or splice junction MOs (morphant) at the 1- to 4-cell stage. Larvae (5 dpf) were labeled with fluorescent cholesterol (NBD-cholesterol, 2 hr, 3 μg/ml solubilized with fish bile). Morphants exhibited decreased fluorescence in the digestive system.
F) ffr mRNA injections can restore fluorescent cholesterol labeling of mutant larvae (p < 0.01). The offspring of heterozygous ffr parents were injected with ffr mRNA and phenol red, or with water and phenol red (control) at the 1- to 4-cell stage. Injected larvae (5 dpf) were immersed in the embryo medium containing fluorescent cholesterol (2 hr, 3 mg/ml solubilized with fish bile). Each larva was then photographed prior to molecular genotyping. Fluorescence intensity was digitally quantified as described in the Experimental Procedures. Data represent mean 6 SEM.
Overexpression studies with in vitro transcribed, capped ffr mRNA, provided further evidence that mutation of the ffr gene disrupts lipid metabolism in zebrafish larvae. wild-type ffr mRNA was synthesized and injected into 1- to 4-cell stage embryos obtained from a cross of ffr heterozygotes. ffr mutant embryos (n = 5 of 20 injected larvae) were indistinguishable from their wild-type siblings following PED6 or fluorescent cholesterol labeling (Figure 5F). Given that exogenous mRNA is rapidly degraded following injection into zebrafish embryos, these data suggest that the Ffr protein has a long half-life in developing zebrafish.
Ffr localizes to the perinuclear region and TGN
In order to identify where the ffr gene is expressed, we first attempted to detect its expression by whole-mount RNA in situ hybridization. We also attempted to localize the protein using whole-mount immunostaining with the zebrafish Ffr antibody described above. However, the low signal/noise ratio make ffr RNA hybridization or Ffr immunostaining signal indistinguishable from the nonspecific control (data not shown). We therefore hypothesize that ffr is likely expressed at a low level in a widespread fashion. Indeed, our microarray data were consistent with the idea that ffr is expressed at low levels relative to β-actin in developing zebrafish embryos and larvae (Figure 6A). Further, we did detect ffr transcript ubiquitously expressed in adult zebrafish tissues by RT-PCR (Figure 6B).
Figure 6. ffr expression and protein localization.

A) Developmental profile of ffr expression. mRNA levels were determined by a zebrafish oligo microarray. Normalized data are expressed relative to β-actin and from 18 somite to 5 dpf stages. Data represent mean ± SEM, (n = 3–6 at each stage).
B) ffr is widely expressed in the adult zebrafish. Expression of ffr was determined by RT-PCR. Tissues: Pa, pancreas; Ht, heart; Ad, adipose tissue; Hd: head; Ms; muscle; In: intestine; Li: liver; Gb, gall bladder.
C) Human ffr and COG8 are widely expressed. Expression was assayed by RT-PCR. Tissues: Ht, heart; Br, brain; Pl, placenta; Lu, lung; Li, liver; SM, skeletal muscle; Ki, kidney; Pa, pancreas; Sp, spleen; In, small intestine; Co, colon; Le, leucocyte.
D–F) Ffr and COG8 partially colocalize in the Golgi of zebrafish blastomeres. Embryos were coinjected with ffr-gfp mRNA and cog8-mrfp mRNA at the 1- to 4-cell stage.
D) Expression of Ffr-GFP in the perinuclear region.
E) COG8-mRFP localizes primarily to the peripheral Golgi.
F) Merged image of (D) and (E). Arrowheads indicate colocalization of COG8 and Ffr.
G–I) Wild-type (in green) and mutant (in red) Ffr partially colocalize in the cells. (G) Expression of ffrWt-GFP. (H) Expression of ffr mut-mRFP. (I) Merged image of (G) and (H). Yellow indicates colocalization.
J–L) Mutant Ffr (in red) and clathrin light chain (in green) colocalization. (J) TGN marker (Clathrin, GFP; pClGFP) (Gaidarov et al., 1999) expression in a single enveloping layer cell. (K) ffr mut-mRFP expression in a single cell. (L) Merged image of (J) and (K). Yellow indicates truncated Ffr mutant localized in the TGN. The scale bar represents 5 μm.
Because Ffr has a Dor-1 like domain, we compared its expression and subcellular localization to COG8, another Dor-1 family protein that is reported to localize to the Golgi to regulate intra-Golgi transport (Ungar et al., 2002). We also examined the tissue distribution of human ffr. Human ffr and cog8 were expressed in all tissues tested (Figure 6C). Because both ffr and cog8 contain a Dor-1 domain, we hypothesized that they colocalize at the subcellular level. To test this hypothesis, mRNAs encoding a zebrafish Ffr-GFP fusion protein and a zebrafish COG8-mRFP fusion protein were coinjected into newly fertilized zebrafish embryos to overcome the low signal/noise ratio for ffr detection. Injected embryos were imaged so that the fusion protein could be localized within individual blastomeres using confocal microscopy. The Ffr-GFP fusion protein was present in the perinuclear region in a punctate pattern (Figure 6D) that partially colocalized with COG8-mRFP which likely localized within the Golgi (Ungar et al., 2002) (Figures 6E and 6F). We were able to clearly image the large enveloping layer cells (the outermost cell layer) that surround the embryo by confocal microscopy. Interestingly, mutant Ffr mRFP (Figures 6H and 6K) is restricted to the TGN as indicated by its partial colocalization with Clathrin-GFP (Figure 6J), an established Golgi marker (Gaidarov et al., 1999). Wild-type Ffr-GFP (Figures 6D and 6G) colocalizes with mutant Ffr -mRFP in the TGN. However, it is also is found in perinuclear regions (Figures 6D and 6G). Microinjection of the mRNA encoding the Ffr-GFP fusion protein did not rescue ffr mutant larvae (data not shown).
Ffr regulates vesicle recycling and protein sorting
Golgi defects within ffr mutant digestive organs resemble Golgi defects associated with mutations of COG complex genes (Ungar et al., 2002). Although protein secretion is reported to be normal in cells carrying COG gene mutations, protein glycosylation within the Golgi of these cells is disrupted (Kingsley et al., 1986). Lipid absorption defects in ffr mutants suggested that Golgi-associated vesicular trafficking might also be impaired in ffr mutant larvae. To test this hypothesis, larvae (5 dpf) were fed AM1-43, a fixable form of a styryl dye (FM1-43) commonly used to measure endocytosis (Brumback et al., 2004). Following AM1-43 labeling, embryos ingested a selective cyclodextran (ADVASEP-7) to remove noninternalized dye from the plasma membrane (Kay et al., 1999), thus allowing the fate of labeled enterocytes to be followed histologically. ffr enterocytes (Figures 7A, 7G, and 7I) had significantly more AM1-43-containing particles in their enterocytes than wild-type (Figures 7D, 7H, and 7J) larvae. In addition, AM1-43-derived fluorescence in ffr enterocytes was localized in enlarged perinuclear organelles suggestive of dilated Golgi apparati (Figure 7C). These results suggest that trafficking of endocytotic cargo is disrupted at the level of the Golgi in ffr mutant enterocytes.
Figure 7. ffr mutants exhibit altered vesicular trafficking and APOA1 targeting.

A–F) Histological cross-sections through the intestine of 5 dpf ffr (A–C) and wild-type (D–F) larvae. Red fluorescence shows AM1-43 accumulation in plasma membrane (arrows) and intracellular vesicles (arrow head) in ffr mutant and wild-type enterocytes. (B and E) Identical sections in (A) and (D) stained with 1% methylene blue in 1% sodium borate and 1% Azure B (1:1). (C and F) Composites of panels (A) and (B), and (D) and (E), respectively. Note accumulation of AM1-43 in the region of the Golgi of ffr enteroctyes (C). This is best appreciated in histological cross-sections through the intestine of 5 dpf ffr and wild-type larvae following AM1-43 ingestion and cyclodextran wash to remove noninternalized dye within the plasma membrane (G–J). (K–L) Following ffr “knockdown” by MO-1, the apoA1-mRFP fusion protein had a diffuse intracellular distribution (arrowhead in [L]), whereas in control embryos the apoA1-mRFP fusion protein was present within vesicles surrounding the nucleus (arrowhead). The scale bar represents 15 μm.
The Golgi complex is important not only for vesicular trafficking during endocytosis but also for protein sorting and secretion. To further assess Ffr function, we examined the processing of an apolipoprotein A1-mRFP fusion protein in ffr−/− zebrafish embryos. In vitro transcribed apoAI-mrfp mRNA was injected into 1- to 4-cell stage embryos alone and in combination with low doses of ffr MO-1. Following injections, embryos were fixed and processed for confocal microscopy. ApoA1-mRFP was diffusely distributed within the cytoplasm of ffr-deficient blastomeres (Figure 7L), whereas in wild-type embryos, ApoA1 was clustered intracellularly (Figure 7K). These data further suggest a role for Ffr in protein sorting.
Discussion
The absorption and transport of lipids by vertebrate cells is dependent upon genes that regulate vesicular trafficking. The Golgi complex receives and sorts vesicular cargo derived from endosomes and ER and thus plays a central role in these processes. In the intestine, the majority of dietary and biliary lipids traverse the Golgi within chylomicrons and very low-density lipoprotein particles that are formed in the ER or recycled from endosomes. From the Golgi, the lipoprotein particles are transported to the basolateral surface of the enterocyte and secreted into the lymphatic system (Albers and Cheung, 2004; Field, 2001).
Altered lipid metabolism is a central feature of heritable human diseases that disrupt intracellular vesicular trafficking. Tangier disease, an autosomal recessive disorder that arises from defective efflux of cellular cholesterol, is caused by mutations of the ABCA1 transporter, encoded by a gene that is essential for the cellular efflux of phospholipids and cholesterol via ApoAI (Soumian et al., 2005). Ultrastructural analyses suggest that the ABCA1 transporter also regulates Golgi morphology and function in ABCA1 knockout mice (Orso et al., 2000). Mutations in the Sar1-GTPase, a protein that facilitates ER to Golgi trafficking, have been reported in patients with heritable disorders that are associated with severe fat malabsorption (Jones et al., 2003). Here, we define a role in lipid metabolism for a previously described vertebrate gene of unknown function that is likely to be involved in intracellular vesicular trafficking and Golgi morphology in zebrafish.
Several lines of evidence support our identification of the zebrafish ang2 ortholog, which we have renamed fat-free, as the ffr gene. First, gene mapping experiments place the ffr mutant locus within close proximity to the ang2 gene. Second, predicted translation of the truncated protein encoded by the ffr gene coincided with the size of the Ffr protein in mutant embryos as determined by Western analyses. Third, knockdown of Ffr protein by antisense morpholinos generates a ffr phenocopy whereas injection of ffr mRNA rescued mutant larvae. Together, these data confirm that the zebrafish ang2 ortholog is in fact the ffr gene.
Although no human syndrome associated with altered lipid metabolism has been mapped within the vicinity of the human ffr ortholog, the established role of vesicle trafficking genes in human lipid metabolism and the prominent Golgi defects in ffr mutants argue that this protein is likely to play an important role in lipid transport in humans.
Ffr shares homology with vesicle trafficking proteins
Scanning the predicted ffr amino acid sequence for conserved sequence motifs (http://myhits.isb-sib.ch/cgi-bin/motif_scan), we found that the Ffr has a Dor-1 like domain (E = 0.00015) and a tyrosine phosphorylation site (amino acids 172–179), which are similar to yeast COG8. The human Ffr ortholog, ANG2, has previously been noted to have sequence similarities to yeast COG8 (Whyte and Munro, 2001). In yeast, the cog8 (dor-1) mutation is synthetically lethal when combined with mutations in genes that code for other proteins involved in the Golgi vesicular trafficking and secretion such as gyp1, hoc1, ypt6, and arl3 (Tong et al., 2004). Thus, it is conceivable that the Ffr protein regulates Golgi function through interaction with these or other genes, either on its own, or in conjunction with the COG complex. The partial colocalization of Ffr and Cog8 fusion proteins supports this idea, at least in part.
Golgi defects are well described in Tangier disease enterocytes and mononuclear phagocytes despite the fact that ABCA1 protein, which is targeted in this disorder, has been reported to principally localize to plasma membrane and the endosomal system (Neufeld et al., 2001). Thus, it is conceivable that the Ffr protein functions in some other non-Golgi capacity. Extra-Golgi localization of the Ffr-GFP fusion protein within perinuclear vesicles and the nucleus of zebrafish embryonic cells is consistent with such a role for Ffr.
Ffr is required for Golgi structure, protein sorting, and vesicle trafficking
Several lines of evidence show that the Ffr protein is required for normal Golgi structure and function. First, ultrastructural analyses identified dilated juxtanuclear cisternae in ffr mutant enterocytes. Their location strongly supports the identification of the cisternae as Golgi, as opposed to a multivesicular body or other endosomal organelle that are typically located in the apical regions of enterocytes and have a distinctive ultrastructural appearance (Fransen et al., 1985; Klumperman et al., 1991). Second, mutant Ffr fusion protein colocalized with clathrin-GFP (Gaidarov et al., 1999), an established Golgi marker, while the mutant and wild-type Ffr fusion proteins colocalized with each other. Third, lipids such as cholesterol and phospholipids, that are transported as part of lipoprotein particles are metabolized far less efficiently by ffr mutant larvae than short chain fatty acids whose absorption is not dependent upon proper lipoprotein particle assembly (Sheridan, 1988). This is important because lipoprotein assembly in enterocytes is dependent upon normal Golgi function.
The mechanism of Golgi biogenesis and maintenance is complex. A leading model, the cisternal maturation model, posits that secretory cargo transits the stacks of Golgi cisternae that mature in situ. As cisternae mature, resident Golgi proteins are recycled to developing cisternae via retrograde transport within coat protein complex I (COPI) vesicles, or via intercisternal tubules or simple diffusion (Glick and Malhotra, 1998). A central question to arise from this study is whether Golgi abnormalities and subsequent alterations of lipid metabolism in ffr mutants arise from a defect of anterograde or retrograde Golgi transport. Such defects could derive from the disruption of the Ffr protein function within the Golgi itself or from an effect on the biogenesis of vesicles that traverse the Golgi complex. The partial colocalization of Ffr-GFP and Cog8-mRFP fusion proteins in zebrafish blastomeres is consistent with the hypothesis that Ffr functions in concert with the COG complex to regulate retrograde Golgi transport (Ram et al., 2002). Colocalization of the mutant Ffr mRFP fusion protein with wild-type Ffr-GFP (Figure 6I) and Cla-thrin-GFP, a well-established TGN marker (Figure 6L), supports the hypothesis that wild-type Ffr localizes to the TGN. In preliminary experiments, the wild-type Ffr-GFP fusion protein did not rescue ffr mutants. However, we have yet to determine whether the Ffr-GFP mRNA encodes a functional protein.
The observation that ApoA1-mRFP was improperly sorted by ffr morpholino injected blastomeres (Figure 7L) indicates that Ffr is required for correct protein sorting and anterograde vesicular trafficking. This observation is consistent with mislocalization of carboxypeptidase A in the pancreatic exocrine cells of ffr mutants (Figure 1J). At this time we can only speculate about the cause of protein missorting in ffr deficient cells. However, the finding that AM1-43 was readily endocytosed by ffr enterocytes but accumulated in the Golgi, suggests that ApoA1-mRFP mis-localization in ffr deficient embryos arises from defective trafficking of the ApoA1-mRFP fusion protein through the Golgi rather than its internalization from the plasma membrane (Banerjee et al., 1997).
Ffr is required for normal intestinal lipid metabolism
Widespread ffr expression in developing and adult zebrafish suggests that Ffr plays a role in most tissues. Thus, it is surprising that ffr mutants have a pronounced defect of digestive organ function, whereas the development and presumed function of other tissues is normal. Several explanations may account for these findings. First, Ffr may be essential for cellular trafficking in other organs. Histological and ultrastructural assays may not be sensitive enough to document these defects. Second, the ffr mutation may not completely abolish the function of the truncated Ffr protein. This possibility coupled with the late decline in ffr expression and the large amount of maternal ffr transcripts could explain organ-specific defects in ffr mutants. Third, if Ffr functions in sorting specific classes of proteins, such as apolipoproteins and peptidases (which are both mislocalized in ffr-deficient embryos), it is conceivable that the digestive organ Golgi defects are more pronounced due to the higher demand for sorting these proteins (classes) in these organs. Fourth, the Drosophila COG5 mutation only exhibits Golgi defects in spermatocytes (Farkas et al., 2003), providing an example of a mutation in an ubiquitously expressed COG gene that results in a tissue specific defect.
Altered lipid metabolism in ffr mutant larvae is likely to arise from defects in multiple organs. Biliary degeneration undoubtedly impairs bile secretion and secondarily the emulsification of dietary lipids and their absorption by enterocytes. Golgi defects are likely to interfere with the transport of lipids in lipoprotein particles by enterocytes into the lymphatics. Consistent with these proposed defects, ffr mutants processed a fluorescent short chain fatty acid analog normally (Farber et al., 2001). This latter finding is noteworthy because short chain fatty acids absorbed by enterocytes are bound to albumin and secreted directly into the portal bloodstream rather than being incorporated into lipoprotein particles and secreted into the lymphatic system (after traversing the Golgi). Normal processing of short chain fatty acids may imply that Golgi-mediated lipid transport, rather than luminal absorption, is the primary defect within the ffr mutant intestine. However, it is conceivable that altered Golgi function in ffr mutants affects localization of transporters specifically required for cholesterol and phospholipids absorption (Albers and Cheung, 2004; Field, 2001).
In conclusion, we have identified a mutation in zebrafish, ffr, which causes impaired lipid processing in the digestive system. The positional cloning and in vivo analyses showed Ffr is a new member of the Dor-1 like family. Altered Golgi morphology in ffr mutants indicates this protein plays an important role in Golgi structure, Golgi-associated protein secretion or vesicular recycling. The expanded subcellular distribution of Ffr fusion protein compared with another Dor-1 family member, COG8, suggest additional cellular roles for Ffr protein.
Experimental procedures
Histology and immunohistochemistry
Wild-type and ffr mutant larvae were fixed in 4% paraformaldehyde at 4°C overnight or in MeOH/DMSO (4/1) for 2 hr at room temperature. In some cases, specimens were washed and embedded in glycol methacrylate (JB-4 Plus, Polysciences). Sections and immunohistochemistry were performed as previously described (Lorent et al., 2004; Pack et al., 1996).
Map cross and mutant collections
The male *AB founder fish were mutagenized with ethylnitrosourea (ENU) and screened for lipid processing as previously described (Driever et al., 1996; Farber et al., 2001). The F2 heterozygous ffr were out crossed to TLF strain to create a map cross. The offspring of heterozygous ffr parents were labeled with PED6 and screened at 5 dpf to identify mutant larvae. Genomic DNA was isolated from individual mutant larvae and utilized for positional cloning efforts.
Bulk segregant and linkage analysis
The ffr locus was initially mapped to linkage group 10 (marker z6183) using bulk segregant analysis (Rawls et al., 2003) performed using two samples of pooled genomic DNA obtained from 20 wild-type and 20 ffr mutant larvae. More than 2000 mutants were collected to perform the subsequent linkage and recombinant analyses. Potential markers were tested using the pooled DNA to determine if they were polymorphic using high resolution 2% metaphor agarose gels. Specific polymorphic markers were then amplified with PCR using genomic DNA of individual mutant larvae. The total number of recombinants (typically revealed as a doublet following PCR) were used to calculate the meiotic distance of specific markers from the ffr locus (Dicarprio and Hastings, 1976). In some cases, the PCR products generated by fluorescent primers for a particular marker were run on acrylamide gels and scanned (GeneScan, ABI 377) to identify polymorphisms that cannot be resolved using agarose (<10 bp). All informative markers were mapped on radiation hybrid panels (LN54 and T51) even if their location was previously determined by other laboratories to more precisely order markers in reference to the ffr critical region.
Cloning of zebrafish ffr, COG8, and ApoAI
The zebrafish ffr was cloned using primers 5′-ACA TCT GTC ATT CAT TCA TTT AAT AAT TC-3′ and 5′-CTA TCC CCT CTC ACA GAT GAC TTC-3′. The zebrafish COG8 was cloned using primers 5′-ATG GCT GCC GTA GAC GTG GAA GA-3′ and 5′-ATA ATA ATC TTC ACA AAA CAA AGT TGT TG-3′. The ApoAI was cloned using primers 5′-A TGA AAT TCG TGG CTC TTG CA-3′ and 5′-TGC CTG GAT GGC CTT GGC-3′. All three PCR products were cloned using PCRIITOPO (Invitrogen).
Morpholino injection and phenotypic rescuing
Embryos at 1- to 4-cell stage were injected (1–2 nl) with mRNA or MOs using glass microelectrodes fitted to a gas pressure injector (PL1-100, Harvard Apparatus). Electrodes were pulled (P-97, Flaming/Brown) and filled with a stock solution of MOs targeted to the ffr gene (1–2 mM, Gene Tools) or mRNA of ffr (1 mg/ml). Phenol red solution (0.2% final concentration) was added to all injection solutions to visualize injected embryos. MO4 injected morphants were collected at 4 dpf to isolate total RNA (TriZol). After reverse transcription, PCR was performed with primers 5′-AGA AGA TGG AGG ATG AAA TGG A-3′ and 5′-AAC AGG AAC TGC AGT TTT CGG AGA A-3′. For rescue experiments, embryos from heterozygous ffr parents were injected with either water or ffr mRNA at 1–4 cell stages and then labeled either with fluorescent cholesterol or PED6 at 5 dpf. Individual zebrafish larvae were then photographed to capture total gut fluorescence, which was digitally quantified (Image Quant TL, Amersham Biosciences Corp) and then genotyped using PCR with a closely linked marker (Z54342).
Domain analysis and transcriptional profiling
Domain analysis was performed using the prosite database of protein families and domains (http://us.expasy.org/prosite/). Transcriptional profiling was performed by the Thomas Jefferson University Microarray Facility at the Kimmel Cancer Center. The spotted array contains 16,399 oligos (Compugen Inc.,). More than 100 β-actin oligos that serve as positive controls were on each chip. Total RNA of 4 dpf and 5 dpf wild-type and ffr larvae were extracted by TriZol (Invitrogen). Gene expression was determined using biotin-labeled and in vitro-transcribed antisense RNA generated from the total RNA template. Each chip was scanned and quantified using a ScanArray Express laser scanner (Perkin Elmer). The signals on the oligo microarray were normalized by the median and regularized t test was performed to determine significant differences between the wild-type and ffr mutants.
RT-PCR for zebrafish and human ffr and human COG8
The human cDNA from a variety of tissues was commercially obtained (BD Biosciences) and used to examine ffr and COG8 expression by PCR. ffr primers were 5′-CAC CAT CCG GAA GAT GAA GAA CGA-3′ and 5′-AAC GAG GGC AGG TGT TGG TAC TG-3′, and COG8 primers were 5′-TCA TGG ACA CCT GTG TCC GGA AC-3′ and 5′-TGG CAC GGT ACT GGG TGA TGA TAT-3′. Adult zebrafish were dissected to collect different tissues and total RNA was prepared (TriZol). The first strand cDNA was prepared by reverse transcription. PCR was performed using primers 5′-ATT ACT CCC GGG GTC GGC AGT G-3′ and 5′-TGT GAG CGT GAG GAT GTA GGA GAT-3′.
Plasmid constructions and mRNA preparation
The pT3Ts plasmid was obtained from Dr. S. Ekker (University of Minnesota). Plasmid containing monomeric red fluorescent protein (mRFP) was obtained from Dr. Tsien (Campbell et al., 2002) and was subcloned into the pT3Ts plasmid to make pT3mRFP. Zebrafish ffr transcript was subcloned between the SpeI and BstEII sites of pT3GFP. Zebrafish COG8 and ApoAI were subcloned into pT3mRFP to make COG8-mRFP and APOAI -mRFPconstructs. mRNA of zffr-GFP, zCOG8-mRFP and zAPOAI-mRFP was prepared by mMES-SAGE mMACHINE (Ambion Inc).
Subcellular localization of ffr, COG8 and ApoAI
1- to 4-cell stage embryos were coinjected with mRNA of GFP or mRFP fusion constructs of ffr, COG8, and ApoAI. PBD Living Colors TM fluorescent proteins plasmid vectors that mark nucleus, Golgi, ER, and plasma membrane (BD Biosciences) were also injected into embryos at 1- to 4-cell stage. TGN marker pCleGFP was provided by Dr. J. Keen (Thomas Jefferson University). Embryos (24 hpf) were then fixed in 4% paraformaldehyde overnight at 4°C and then transferred to PBS. Confocal images were taken using a Zeiss LSM 510 META.
AM1-43 feeding experiments
5 dpf wild-type and ffr mutants were immersed into embryo medium containing 20 mM AM1-43 (Biotium, Inc.) for 2 hr at 28°C. Then the larvae were transferred into embryo medium containing ADVASEP-7 (a sulfonated β-cyclo-dextrin, Biotium, Inc.) (100 μM for 1 hr at 28°C). The larvae were fixed in 4% paraformaldehyde overnight at 4°C, embedded in plastic, and sectioned (1 μM). Some sections were labeled with 1% methylene blue in 1% sodium borate and 1% Azure B (1:1). Confocal images were taken using a Zeiss LSM510 META.
Acknowledgments
We thank Dr. Charles Mansback for his helpful discussion. We thank M. Pearson, M. Molloy, R. DeRose, Y. Deng, K. Luesse, and A. Lee for their technical support. We thank Dr. Changgong Liu (Kimmel Cancer Center Microarray Facility) for assistance with microarrays. We also want to thank Michael Supanski for assistance with the preparation of histological sections and Neelima Shah for expert technical assistance with the electron microscopy. S.Y.H. was supported by American Heart Association (0325731U) and National Research Service Award individual postdoctoral fellowship (F32 DK066726-01). This work was supported by US National Institutes of Health Grants (DK060369 to S.A.F. and M.P. and DK54942 to M.P.) and by a Pew’s Scholar’s Award to S.A.F.
References
- Albers JJ, Cheung MC. Emerging roles for phospholipid transfer protein in lipid and lipoprotein metabolism. Curr Opin Lipidol. 2004;15:255–260. doi: 10.1097/00041433-200406000-00004. [DOI] [PubMed] [Google Scholar]
- Banerjee D, Rodriguez M, Rajasekaran AK. Rapid movement of newly synthesized chicken apolipoprotein AI to trans-Golgi network and its secretion in Madin-Darby canine kidney cells. Exp Cell Res. 1997;235:334–344. doi: 10.1006/excr.1997.3687. [DOI] [PubMed] [Google Scholar]
- Brumback AC, Lieber JL, Angleson JK, Betz WJ. Using FM1–43 to study neuropeptide granule dynamics and exocytosis. Methods. 2004;33:287–294. doi: 10.1016/j.ymeth.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci USA. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Burn P, Shi Y. Properties of the mouse intestinal acyl-CoA:monoacylglycerol acyltransferase, MGAT2. J Biol Chem. 2003;278:25657–25663. doi: 10.1074/jbc.M302835200. [DOI] [PubMed] [Google Scholar]
- Chang TY, Reid PC, Sugii S, Ohgami N, Cruz JC, Chang CC. Niemann-Pick type C disease and intracellular cholesterol trafficking. J Biol Chem. 2005;280:20917–20920. doi: 10.1074/jbc.R400040200. [DOI] [PubMed] [Google Scholar]
- Dicarprio L, Hastings PJ. Gene conversion and intragenic recombination at the SUP6 locus and the surrounding region in Saccharomyces cerevisiae. Genetics. 1976;84:697–721. doi: 10.1093/genetics/84.4.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driever W, Solnica-Krezel L, Schier AF, Neuhauss SC, Malicki J, Stemple DL, Stainier DY, Zwartkruis F, Abdelilah S, Rangini Z, et al. A genetic screen for mutations affecting embryogenesis in zebrafish. Development. 1996;123:37–46. doi: 10.1242/dev.123.1.37. [DOI] [PubMed] [Google Scholar]
- Ehehalt R, Jochims C, Lehmann WD, Erben G, Staffer S, Reininger C, Stremmel W. Evidence of luminal phosphatidylcholine secretion in rat ileum. Biochim Biophys Acta. 2004a;1682:63–71. doi: 10.1016/j.bbalip.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Ehehalt R, Wagenblast J, Erben G, Lehmann WD, Hinz U, Merle U, Stremmel W. Phosphatidylcholine and lysophosphatidylcho-line in intestinal mucus of ulcerative colitis patients. A quantitative approach by nanoElectrospray-tandem mass spectrometry. Scand J Gastroenterol. 2004b;39:737–742. doi: 10.1080/00365520410006233. [DOI] [PubMed] [Google Scholar]
- Farber SA, Pack M, Ho SY, Johnson ID, Wagner DS, Dosch R, Mullins MC, Hendrickson HS, Hendrickson EK, Halpern ME. Genetic analysis of digestive physiology using fluorescent phospholipid reporters. Science. 2001;292:1385–1388. doi: 10.1126/science.1060418. [DOI] [PubMed] [Google Scholar]
- Farkas RM, Giansanti MG, Gatti M, Fuller MT. The Drosophila Cog5 homologue is required for cytokinesis, cell elongation, and assembly of specialized Golgi architecture during spermatogenesis. Mol Biol Cell. 2003;14:190–200. doi: 10.1091/mbc.E02-06-0343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field FJ. Regulation of intestinal cholesterol metabolism. In: Mansbach CM, Tso P, Kuksis A, editors. Intestinal Lipid Metabolism. New York: Kluwer Academic; 2001. pp. 235–255. [Google Scholar]
- Fransen JA, Ginsel LA, Hauri HP, Sterchi E, Blok J. Immuno-electronmicroscopical localization of a microvillus membrane disaccharidase in the human small-intestinal epithelium with monoclonal antibodies. Eur J Cell Biol. 1985;38:6–15. [PubMed] [Google Scholar]
- Gaidarov I, Santini F, Warren RA, Keen JH. Spatial control of coated-pit dynamics in living cells. Nat Cell Biol. 1999;1:1–7. doi: 10.1038/8971. [DOI] [PubMed] [Google Scholar]
- Glick BS, Malhotra V. The curious status of the Golgi apparatus. Cell. 1998;95:883–889. doi: 10.1016/s0092-8674(00)81713-4. [DOI] [PubMed] [Google Scholar]
- Hill JO, Wyatt HR, Reed GW, Peters JC. Obesity and the environment: where do we go from here? Science. 2003;299:853–855. doi: 10.1126/science.1079857. [DOI] [PubMed] [Google Scholar]
- Ho SY, Pack M, Farber SA. Analysis of small molecule metabolism in zebrafish. Methods Enzymol. 2003;364:408–426. doi: 10.1016/s0076-6879(03)64023-1. [DOI] [PubMed] [Google Scholar]
- Joffe BI, Panz VR, Raal FJ. From lipodystrophy syndromes to diabetes mellitus. Lancet. 2001;357:1379–1381. doi: 10.1016/S0140-6736(00)04616-X. [DOI] [PubMed] [Google Scholar]
- Jones B, Jones EL, Bonney SA, Patel HN, Mensenkamp AR, Eichenbaum-Voline S, Rudling M, Myrdal U, Annesi G, Naik S, et al. Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nat Genet. 2003;34:29–31. doi: 10.1038/ng1145. [DOI] [PubMed] [Google Scholar]
- Kay AR, Alfonso A, Alford S, Cline HT, Holgado AM, Sakmann B, Snitsarev VA, Stricker TP, Takahashi M, Wu LG. Imaging synaptic activity in intact brain and slices with FM1–43 in C. elegans, lamprey, and rat. Neuron. 1999;24:809–817. doi: 10.1016/s0896-6273(00)81029-6. [DOI] [PubMed] [Google Scholar]
- Kingsley DM, Kozarsky KF, Segal M, Krieger M. Three types of low density lipoprotein receptor-deficient mutant have pleiotropic defects in the synthesis of N-linked, O-linked, and lipid-linked carbohydrate chains. J Cell Biol. 1986;102:1576–1585. doi: 10.1083/jcb.102.5.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumperman J, Boekestijn JC, Mulder AM, Fransen JA, Ginsel LA. Intracellular localization and endocytosis of brush border enzymes in the enterocyte-like cell line Caco-2. Eur J Cell Biol. 1991;54:76–84. [PubMed] [Google Scholar]
- Lemmens I, Merregaert J, Van de Ven WJ, Kas K, Zhang CX, Giraud S, Wautot V, Buisson N, De Witte K, Salandre J, et al. Construction of a 1.2-Mb sequence-ready contig of chromosome 11q13 encompassing the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1 Genomics. 1997;44:94–100. doi: 10.1006/geno.1997.4872. [DOI] [PubMed] [Google Scholar]
- Lorent K, Yeo SY, Oda T, Chandrasekharappa S, Chitnis A, Matthews RP, Pack M. Inhibition of Jagged-mediated Notch signaling disrupts zebrafish biliary development and generates multi-organ defects compatible with an Alagille syndrome phenocopy. Development. 2004;131:5753–5766. doi: 10.1242/dev.01411. [DOI] [PubMed] [Google Scholar]
- McNeely MJ, Edwards KL, Marcovina SM, Brunzell JD, Motulsky AG, Austin MA. Lipoprotein and apolipoprotein abnormalities in familial combined hyperlipidemia: a 20-year prospective study. Atherosclerosis. 2001;159:471–481. doi: 10.1016/s0021-9150(01)00528-7. [DOI] [PubMed] [Google Scholar]
- Neufeld EB, Remaley AT, Demosky SJ, Stonik JA, Cooney AM, Comly M, Dwyer NK, Zhang M, Blanchette-Mackie J, Santamarina-Fojo S, Brewer HB., Jr Cellular localization and trafficking of the human ABCA1 transporter. J Biol Chem. 2001;276:27584–27590. doi: 10.1074/jbc.M103264200. [DOI] [PubMed] [Google Scholar]
- Oka T, Ungar D, Hughson FM, Krieger M. The COG and COPI complexes interact to control the abundance of GEARs, a subset of Golgi integral membrane proteins. Mol Biol Cell. 2004;15:2423–2435. doi: 10.1091/mbc.E03-09-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Vasile E, Penman M, Novina CD, Dykxhoorn DM, Ungar D, Hughson FM, Krieger M. Genetic analysis of the subunit organization and function of the conserved oligomeric golgi (COG) complex: studies of COG5- and COG7-deficient mammalian cells. J Biol Chem. 2005;280:32736–32745. doi: 10.1074/jbc.M505558200. [DOI] [PubMed] [Google Scholar]
- Orso E, Broccardo C, Kaminski WE, Bottcher A, Liebisch G, Drobnik W, Gotz A, Chambenoit O, Diederich W, Langmann T, et al. Transport of lipids from golgi to plasma membrane is defective in tangier disease patients and Abc1-deficient mice. Nat Genet. 2000;24:192–196. doi: 10.1038/72869. [DOI] [PubMed] [Google Scholar]
- Pack M, Solnica-Krezel L, Malicki J, Neuhauss SC, Schier AF, Stemple DL, Driever W, Fishman MC. Mutations affecting development of zebrafish digestive organs. Development. 1996;123:321–328. doi: 10.1242/dev.123.1.321. [DOI] [PubMed] [Google Scholar]
- Pohl J, Ring A, Ehehalt R, Herrmann T, Stremmel W. New concepts of cellular fatty acid uptake: role of fatty acid transport proteins and of caveolae. Proc Nutr Soc. 2004;63:259–262. doi: 10.1079/PNS2004341. [DOI] [PubMed] [Google Scholar]
- Ram RJ, Li B, Kaiser CA. Identification of Sec36p, Sec37p, and Sec38p: components of yeast complex that contains Sec34p and Sec35p. Mol Biol Cell. 2002;13:1484–1500. doi: 10.1091/mbc.01-10-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawls JF, Frieda MR, McAdow AR, Gross JP, Clayton CM, Heyen CK, Johnson SL. Coupled mutagenesis screens and genetic mapping in zebrafish. Genetics. 2003;163:997–1009. doi: 10.1093/genetics/163.3.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan MA. Lipid dynamics in fish: aspects of absorption, transportation, deposition and mobilization. Comp Biochem Physiol B. 1988;90:679–690. doi: 10.1016/0305-0491(88)90322-7. [DOI] [PubMed] [Google Scholar]
- Soumian S, Albrecht C, Davies AH, Gibbs RG. ABCA1 and atherosclerosis. Vasc Med. 2005;10:109–119. doi: 10.1191/1358863x05vm593ra. [DOI] [PubMed] [Google Scholar]
- Soutar AK, Naoumova RP, Traub LM. Genetics, clinical phenotype, and molecular cell biology of autosomal recessive hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2003;23:1963–1970. doi: 10.1161/01.ATV.0000094410.66558.9A. [DOI] [PubMed] [Google Scholar]
- Suvorova ES, Duden R, Lupashin VV. The Sec34/Sec35p complex, a Ypt1p effector required for retrograde intra-Golgi trafficking, interacts with Golgi SNAREs and COPI vesicle coat proteins. J Cell Biol. 2002;157:631–643. doi: 10.1083/jcb.200111081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, et al. Global mapping of the yeast genetic interaction network. Science. 2004;303:808–813. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- Tso P, Fujimoto K. The absorption and transport of lipids by the small intestine. Brain Res Bull. 1991;27:477–482. doi: 10.1016/0361-9230(91)90145-a. [DOI] [PubMed] [Google Scholar]
- Ungar D, Oka T, Brittle EE, Vasile E, Lupashin VV, Chatterton JE, Heuser JE, Krieger M, Waters MG. Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J Cell Biol. 2002;157:405–415. doi: 10.1083/jcb.200202016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungar D, Oka T, Vasile E, Krieger M, Hughson FM. Subunit architecture of the conserved oligomeric Golgi complex. J Biol Chem. 2005;280:32729–32735. doi: 10.1074/jbc.M504590200. [DOI] [PubMed] [Google Scholar]
- VanRheenen SM, Cao X, Lupashin VV, Barlowe C, Waters MG. Sec35p, a novel peripheral membrane protein, is required for ER to Golgi vesicle docking. J Cell Biol. 1998;141:1107–1119. doi: 10.1083/jcb.141.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte JR, Munro S. The Sec34/35 Golgi transport complex is related to the exocyst, defining a family of complexes involved in multiple steps of membrane traffic. Dev Cell. 2001;1:527–537. doi: 10.1016/s1534-5807(01)00063-6. [DOI] [PubMed] [Google Scholar]