

Abstract
E153 is a respiratory deficient mutant of Saccharomyces cerevisiae with a mutation in the active site of the Sit4p protein phosphatase. Measurements of mitochondrial respiration and cytochromes indicate that the mutation suppresses glucose repression. The escape from catabolite repression is accompanied by a marked reduction of the transcriptional repressor Mig1p. The presence of normal levels of MIG1 mRNA in the mutant and its association with the polysome fraction suggests that depletion of Mig1p is the result of protein degradation. This study shows that in addition to phosphorylation by Snf1p, the transcriptional repressor activity of Mig1p is also regulated by a post-transcriptional Sit4p-dependent pathway. Our evidence suggests that this pathway involves turnover of Mig1p.
Keywords: Saccharomyces cerevisiae, SIT4, MIG1, catabolite repression, mitochondria, respiration
1. Introduction
Biogenesis of the mitochondrial respiratory chain is regulated by several distinct pathways in response to carbon source. The Hap complex is an important global transcriptional activator of a large number of nuclear genes including those coding for subunits of the respiratory complexes [1, 2] and TCA cycle enzymes [3]. Transcription of Hap-regulated genes fails to be activated in the presence of glucose leading to decreased respiration. Another catabolite-responsive pathway is controlled by the Mig1p/Ssn6p/Tup1p transcriptional repressor [4]. While a great deal is known about this pathway from studies of the SUC2 gene for sucrase [5–7], the genes involved in regulation of mitochondrial respiration are not well understood. For example, it is unclear how mutations in SSN6 and SNF1, the kinase responsible for inactivating Mig1p, produce a respiratory defect [8, 9].
Here we report on the properties of E153, a mutant previously assigned to complementation group G168 of a pet mutant collection [10]. The mutation in E153 has been localized to SIT4, encoding a PP2A type Ser/Thr protein phosphatase [11]. In agreement with phenotype of a sit4 reported by Jablonka et al [12], mutants with the E153 allele display a growth defect on non-fermentable carbon sources and fail to be repressed by glucose. We present evidence that the escape from catabolite repression is the result of increased turnover of Mig1p co-repressor, indicating that in addition to its phosphorylation by Snf1p, Mig1p is also regulated by turnover through a SIT4 dependent pathway.
2. Materials and Methods
2.1 Yeast strains and media
The yeast strains used in this study are listed in Table I. The compositions of the yeast growth media have been described elsewhere [14].
Table I.
Genotypes and sources of S. cerevisiae strains
| Strain | Genotype | Source |
|---|---|---|
| D273-10B/A1 | MATα met6 | [13] |
| W303-1A | MATa ade2-1 his3-11 leu2-3,112 trp1-1 ura3-1 | R.Rothsteina |
| W303-1B | MATα ade2-1 his3-11 leu2-3,112 trp1-1 ura3-1 | R.Rothsteina |
| a/aW303 | M7ATa/α ade2-1 his3-11 leu2-3,112 trp1-1 ura3-1 | R.Rothsteina |
| E153 | MATα met6 sit4H55Y | This study |
| aE153/U2 | MATa ura3-1 sit4H55Y | E153 × W303-1A |
| E153/A | MATα ade2-1 his3-11 leu2-3,112 trp1-1 ura3-1 sit4H55Y | E153 × W303-1A |
| a/aW303ΔSIT4 | MATa/α ade2-1 his3-11 leu2-3,112 trp1-1 ura3-1 SIT4/sit4::HIS3A | This study |
| WVZ | MATa ade2-1 his3-11 leu2-3,112 trp1-1 ura3::COX5a-lacZ(URA3) | [14] |
| E153VZ | MATa ade2-1 his3-11 leu2-3,112 trp1-1 ura3::COX5a-lacZ(URA3) sit4H55Y | This study |
Department of Genetics and Development, Columbia University, New York
2.2 Disruption of SIT4
The C-terminal 468 nucleotides of SIT4 (BglII to SphI site) were replaced with a 1 kb BamH1-SphI fragment containing the yeast HIS3 gene. This construct (pG168/ST8) was partially digested with XbaI, releasing a 2.2 kb fragment containing the sit4:: HIS3 allele. This DNA fragment was used to introduce a copy of the disrupted sit4 allele in the diploid strain W303 [15]. Dissections of tetrads from the heterozygous diploid transformants yielded only two viable His− spores from each tetrad, indicating SIT4 to be an essential gene in the W303 nuclear background.
2.3 Cloning of the sit4 allele from aE153/U2
Nuclear DNA from aE153/U2 was digested with SacII and NheI and fragments ranging from 1.5kb to 3 kb were ligated to YEp351 [16] digested with SacII and XbaI. The resultant library was amplified in E. coli and colonies containing the gene were identified by colony hybridization using a 32P labeled 469 bp probe from the 5′ coding region of SIT4.
2.4 Construction of a plasmid expressing HA-tagged Mig1p
A plasmid expressing HA-tagged Mig1p was constructed by replacing the NotI fragment containing GFP in pBM3315 [17] with a 111 bp 3-HA epitope tag from pBS-3HA [18]. The resultant plasmid pM9 was transformed into the wild type strain W303-1A and aE153/U2.
2.5 Miscellaneous
Standard procedures were used for plasmid construction, plasmid amplification in E. coli, and Southern and Northern analysis [19]. Yeast was transformed by the LiAc method of Schiestl and Gietz [20].
3. Results
3.1 Phenotype of mutants with the E153 allele
E153 is the only member of complementation group G168 of a pet1 mutant collection [10]. Like other respiratory deficient mutants, E153 has a severe growth defect on non-fermentable carbon sources.
Spectra of mitochondria from E153 or aE153/U2 (a meiotic segregant from a cross to W303-1A) grown to log phase in 10% YPD (repressed) were compared to those grown to stationary phase in 2% YPGal (derepressed). Mutant mitochondria from derepressed cells had partially reduced cytochromes b and a-a3, but not cytochrome c, which correlated with a two times lower NADH-oxidase activity (Fig. 1A). As expected, wild type cells grown to log phase in 10% glucose compared to cells grown in 2% galactose showed repressed levels of cytochromes (Fig. 1B). The concentrations of cytochromes in the mutant, however, were nearly the same under both growth conditions and were similar to the wild type grown on galactose (Fig 1B). The lack of repression of the mutant by glucose was also evident from the NADH- and succinate-oxidase activities of mitochondria from yeast grown in high glucose medium and harvested at log phase (repressed) and stationary phase when glucose was exhausted (derepressed). Both oxidase activities increased approximately 3-fold in wild type cells harvested in stationary phase (Table II). In contrast, independent of the growth phase, the oxidase activities of aE153/U2 were only slightly lower than those of derepressed wild type. The efficiency of ATP synthesis (P/O ratios) during succinate oxidation was also similar in the mutant and wild type (data not shown).
Fig. 1.

Effect of glucose on expression of mitochondrial respiratory chain components. (A) Spectra of mitochondrial cytochromes in the wild type D273-10B/A1 and the sit4 mutant aE153/U2. Mitochondria of cells grown in YPGal to early stationary phase were extracted with deoxycholate at a final protein concentration of 5 mg/ml and difference spectra of reduced vs oxidized extracts were recorded at room temperature as described previously [21]. The positions of the α absorption bands of cytochromes a,a3, cytochrome b, and cytochromes c and c1 are identified. The specific activities of NADH oxidase (NADH->O2) are expressed in μmoles O2/min/mg proteins (B) The wild type strain D273-10B/A1 and the sit4 mutant E153 were grown to log phase (A600 about 1) either on 10% glucose or 2% galactose. Spectra of mitochondrial cytochrome were obtained as above. (C) Expression of COX5a-lacZ fusions in wild type W303-1A and the sit4 mutant E153/A grown on different carbon sources. Yeast strains were inoculated from fresh YPGal plates into 10 ml of YPD, YPGal or YPEG and grown at 30°C for 24 hours. The YPEG medium required a larger inoculum of sit4 mutant because of its slow growth on glycerol. Cells were then transferred to 50 ml of the same medium so that after overnight growth the A600 were approximately 1. The cultures were harvested and β-galactosidase activities were assayed [23]. The specific activities (ΔA420/min/A600) reported are averages of three independent experiments.
Table II.
Respiratory activities in wild type and mutant mitochondria
| Strain | Medium | Specific activity (μmoles O2/min/mg protein) | |||
|---|---|---|---|---|---|
| NADH oxidase | Succinate oxidase | ||||
| Log | Stationary | Log | Stationary | ||
| D273-10B/A1 | 10% glucose | 0.129 ± 0.003 | 0.375 ± 0.010 | 0.042 ± 0.005 | 0.159 ± 0.000 |
| aE153/U2 | 10% glucose | 0.311 ± 0.000 | 0.385 ± 0.011 | 0.113 ± 0.002 | 0.140 ± 0.000 |
Mitochondria were isolated from the parental wild type strain D273-10B/A1 and from the sit4 mutants aE153/U2 grown in 10% glucose (YPD). Part of the cultures grown in glucose were harvested in log phase and the remaineder in stationary phase. The NADH and succinate oxidase activities were measured at 24°C as described previously [22]. The values reported are averages of two assays with the ranges indicated.
The absence of glucose repression in the mutant was also evident from the β-galactosidase activities expressed from a fusion of the COX5a promoter (subunit 5a of cytochrome oxidase) to lacZ [14]. The wild type strain W303-1A with a chromosomal copy of the lacZ fusion expressed four times higher β-galactosidase activity in galactose and 8 times higher activity in glycerol than in glucose grown cells. Expression of COX5a-lacZ, however, was much less responsive to carbon sources in the mutant (Fig 1C). The β-galactosidase activity in the mutant grown in glucose was almost two times higher than wild type and was only marginally increased when the mutant was grown on non-repressive substrates such as galactose or glycerol. Similar results were obtained with CYC1-lacZ and KGD2-lacZ fusion constructs (data not shown). The increased β-galactosidase activity in the E153/A mutant grown on glucose and the decreased activity on galactose or glycerol, indicates that the mutation significantly reduced repression by glucose but also compromised derepression/activation by non-repressible substrates.
3.2 aE153/U2 is complemented by SIT4 and suppressed by ISF1
Respiratory competent clones were obtained by transformation of aE153/U2 with YEp24-based yeast genomic DNA library [24]. Plasmids isolated from the transformants indicated two distinct regions of DNA capable of restoring respiration in the mutant. Subcloning of the inserts of two representative plasmids (pG168/T1 and pSSG168/T1) identified SIT4 and ISF/MBR3 as the genes responsible for rescuing the mutant (Fig. 2). ISF1 conferred slow growth on glycerol consistent with it acting as a suppressor of the E153 allele. SIT4 fully rescued the growth defect of aE153/U2 suggesting that it was complementing the mutation of aE153/U2.
Fig. 2.
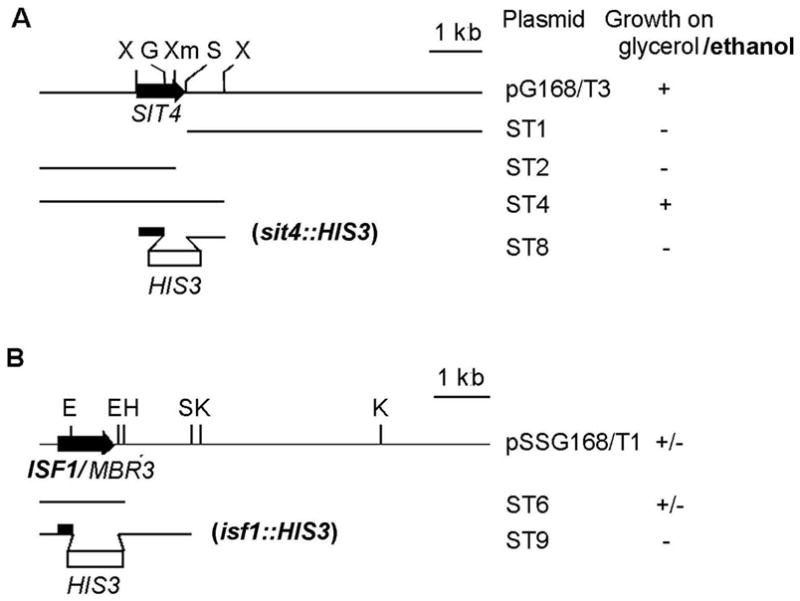
Restriction maps and subclones of plasmids with (A) SIT4 and (B) ISF1. All the inserts were cloned in multicopy plasmids. The locations of restriction sites EcoRI, (E); HindIII, (H); BglII, (G); KpnI, (K); SphI, (S); XbaI, (X); XmaI, (Xm) are marked. The genes are depicted by the solid arrows and the disruptors by the open bars. Growth of the transformants on rich glycerol/ethanol medium is indicate by the + (wild type growth), +/− (two times slower than wild type), − (mutant growth).
The phenotype of sit4 null mutants differs depending on the genetic background [25]. Null mutations in SIT4 are lethal in W303. In the D273-10B background sit4 mutants are viable but respiratory incompetent. The failure of the E153 mutant allele to rescue the lethality of a sit4 null mutation in W303 (data not shown) suggests that the H55Y mutation abolished phosphatase activity.
Sit4p was previously localized in the nucleocytoplasm [26]. This was confirmed in the present study as the protein was not detected with a polyclonal antibody against a GST-Sit4p fusion protein in mitochondria. The abundance of Sit4p was the same in wild type or the mutant and was not altered when grown under repressed or derepressed conditions in rich glucose or in galactose (not shown).
3.3 Mig1p depletion in sit4 mutants
The loss of glucose repression of mitochondrial respiration in sit4 mutants (Fig. 1, Table II) suggested that Sit4p might be involved in the catabolite repression pathway that uses the Ssn6p/Tup1p/Mig1p repressor complex. Transcriptional repression by this complex is determined by the phosphorylation status of Mig1p, which in the phosphorylated state, dissociates from the complex and relocates from the nucleus to the cytoplasm, thereby releasing the transcriptional block. Dephosphorylation of Mig1p could potentially affect its interaction with the Ssn6p/Tup1p, its binding to DNA, and the ability of the complex to repress transcription [17, 27].
Mig1p levels and phosphorylation status were examined in vivo in yeast expressing a C-terminally HA-tagged protein (Mig1p-HA). The cellular abundance of Mig1p-HA was reduced at least 50 fold in the sit4 mutant, although its phosphorylation was not affected (Fig. 3A). Most of Mig1p was dephosphorylated in either wild type or mutants cells grown in high glucose media but was phosphorylated in cells grown in galactose (Fig. 3A). The reduced concentration of Mig1p in the sit4 mutant was confirmed by fluorescence microscopy of cells expressing a GFP-Mig1p fusion protein. The fluorescence in wild type cells grown in glucose was localized in the nucleus. The nuclear accumulation of GFP-Mig1p was not evident in wild type cells grown in galactose, presumably because of its relocation to the cytoplasm. Consistent with the Westerns, no fluorescence was detected in the sit4 mutant grown either in glucose or galactose (data not shown).
Fig. 3.

Effect of the sit4 mutation on expression and phosphorylation of Mig1p. (A) The wild type strain W303-1A and the sit4 mutant aE153/U2 were each transformed with pM9 containing HA-tagged MIG1. The two transformants were grown in minimal selective media in the presence of glucose (Glu) or galactose (Gal) to log phase (A600 = 0.5 – 1). Cells were harvested, cooled with ice and mixed with an equal volume of ice cold 2M NaOH containing 1% β-mercaptoethanol (β-ME). After 10 min lysis on ice, the suspension was neutralized with 6M HCl and centrifuged at 20,000 × g for 15 min at 4°C. The denatured protein pellets were suspended in 1% SDS, 25 mM Tris-Cl, pH 7.5, 1% β-ME, heated at 95 °C for 5 min and clarified by centrifugation at 20,000 × g for 15 min. Supernatants (30 μg protein) were separated on an 8% SDS-PAGE gel, transferred to nitrocellulose, and processed with rabbit anti HA antibody (Covance Inc.) as in Fig. 3A. The migration of the unphosphorylated (HA-Mig1p), phosphorylated (HA-Mig1p-P) protein, and of molecular weight standards are indicated in the margins. (B) W303-1A and aE153/U2 were grown to log phase in 10% glucose and 2% galactose media. PolyA- enriched RNA (3 μg) was separated on a 1% agarose gel, blotted to DBM paper [28], and probed with a 32P-labeled DNA fragment containing the entire MIG1 reading frame. The blot was stripped of the probe and rehybridized to a 32P-labeled actin probe. (C) Overnight cultures of D273-10B/A1 (WT) and aE153/U2 were diluted to an A600 0.1 in 100 ml liquid YPD medium and grown at 30°C with vigorous shaking to a density of A600 =1.0. Cycloheximide was added to a final concentration of 50 μg/ml, and the suspension was poured into a centrifuge bottle containing an equivalent volume of ice. Cell were harvested, washed twice with lysis buffer (10 mM Tris-Cl, pH7.4, 100 mM NaCl, 30 mM MgCl2, 200 μg/ml heparin, 50 mg/ml cycloheximide and 2 mM PMSF) and suspended in the same buffer. The washed cells were mixed with 0.75 volumes of glass beads (0.45 mm) and the mixture was vortexed 6 times for 20 seconds, with 1 min ice cooling intervals. The homogenates were cleared by centrifugation at 3,000 × g for 5 min followed by 10,000 × g for 10 min. Cell extract equivalent to 25 A254 units were loaded on 5 ml of a linear 7–47 % sucrose gradient prepared in the presence of 50 mM Tris-acetate pH 7.0, 50 mM NH4Cl, 12 mM MgCl2, 1 mM DTT, 50 mg/ml cycloheximide, and 200 μg/ml heparin. The gradients were centrifuged at 39,000 rpm for 90 min in a Beckman SW65 rotor at 4°C. Fourteen equal fractions were collected with a Brandel Model 184 Fractionator connected to Pharmacia UV MII monitor equipped with a A254 filter. Samples (135 μl) of each fraction were diluted with 346 μl H2O and 60 μl 10% SDS, extracted with 520 μl water-saturated phenol and the aqueous phase (500 μl) was precipitated in the presence of 30 μl 5M NaCl and 1.5 ml ethanol. The RNA pellets were dried, suspended in loading buffer and one half used for Northern analysis as in part (B). The 80S monosome and polysome peaks are marked. The distribution of the MIG1 mRNA is shown under the A254 tracing of each gradient. Fractions 4 and 5 have a high background but no specific hybridization signals.
The near complete absence of Mig1p in the sit4 mutant indicated that this phosphatase functions in a pathway that regulates either transcription of MIG1 or translation/stability of the protein. MIG1 mRNA levels of the wild type and the sit4 mutant were not significantly different nor were they affected by carbon sources used to grow cells (Fig. 3B). The Northern results suggested that depletion of Mig1p in the mutant occurs post-transcriptionally either as a result of lower translation or increased turnover of the protein.
Translation of Mig1p was examined by comparing the mRNA in fractionated polysomes from wild type and the sit4 mutant. The similarity in the distribution of MIG1 mRNA in fractionated polysomes of the two strains (Fig. 3C) favors the notion that the Mig1p deficit in the sit4 mutant is not due to reduced translation. Rather it suggests increased turnover as the mechanism by which Mig1p levels are regulated by Sit4p. However, because turnover of Mig1p was not measured directly an alternative mechanism cannot be entirely rule out.
4. Discussion
The altered catabolite repression response of the sit4 mutant described in this study is evidenced by the derepressed cytochrome spectra and respiratory activities of mitochondria from cells grown in high glucose. The escape from glucose repression was confirmed by microarray analysis of total cellular mRNAs in cells grown under repressed and derepressed conditions (www.columbia.edu/cu/biology/faculty/tzagoloff/sit4data). Average values of mRNAs functioning in pathways previously reported to be subject to glucose repression through the Ssn6p/Tup1p/Mig1p co-repressor complex (sugar transport, carbohydrate metabolism), were higher in the mutant than in wild type grown on glucose (Fig. 4), indicating that the effect of the sit4 mutation is general. This finding points to a novel Sit4p-dependent transduction pathway involved in catabolite regulation.
Fig. 4.

The Ssn6p/Tup1p complex has been shown to recruit Mig1p to the promoter regions of glucose repressed genes by interacting with a GC-rich consensus site in their promoter regions [29]. The almost complete absence of Mig1p in the sit4 mutant helps to explain the lack of repression of the mitochondrial respiratory chain constituents as well as other gene products regulated by this pathway. The association of MIG1 mRNA with the polysomes fraction of the mutant favors turnover of Mig1p as the chief mechanism for its depletion. This mechanism appears to operate independent of the regulatory pathway involving Snf1p kinase-dependent phosphorylation of Mig1p (Fig. 5) [9, 27]. Mig1p could be degraded by protease regulated by Sit4p or Mig1p itself may be more susceptible to proteolysis in the sit4 mutant [30]. Dephosphorylation of cytoplasmic Mig1p has been proposed to be catalyzed by the Glc7p/Reg1p phosphatase [27] making it unlikely that Sit4p is the catalytic subunit of the phosphatase responsible for the activation of Mig1p. Despite the low amounts of Mig1p in the mutant, the ratio of phosphorylated and unphosphorylated Mig1p was about the same as in wild type. Although this suggests that both forms of Mig1p are degraded, it is not excluded that only one form is susceptible and the loss of the other occurs as a result of its phosphorylation or dephosphorylation.
Fig. 5.

While the depletion of Mig1p accounts for the lack of catabolite repression, it does not explain why sit4 mutants are unable to grow on non-fermentable carbon sources. The latter property is not related to Mig1p depletion as mig1null mutants are respiratory competent and are able to grow on rich ethanol-glycerol media. The respiratory deficient phenotype of the sit4 mutant suggests that Sit4p also affects some other pathway that targets mitochondrial respiration. Jablonka et al [12] found the sit4 mutant to accumulate glycogen and have suggested that they may have futile glycogen cycle that could lead to a depeletion of metabolites needed for ethanol utilization. Sit4p may also function in a pathway that affects transcription of gene regulated by the Hap complex. This might explain the partial rescue of the sit4 mutant by over-expression of ISF/MBR, which has been reported to suppress the respiratory defect of hap2, 3, and 4 mutants [32].
Acknowledgments
This research was supported by NIH Research Grants HL2274 (to A. T.), NIH CA77811 and The Robert A. Welch Foundation, I-0642 (to R. A. B.). We thank Andrey Shtanko for technical assistance, Dr. Mark Johnston for providing the plasmid pBM3315 and Dr. Charles Di Como for plasmid pBS-3HA.
Footnotes
Abbreviations: pet mutant, respiratory deficient mutant of yeast with a mutation in a nuclear gene; PCR, polymerase chain reaction, SDS; sodium dodecyl sulfate; PAGE, polyacrylamide gel electrophoresis; PMSF, phenylmethylsulfonyl fluoride.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Olesen J, Hahn S, Guarente L. Yeast HAP2 and HAP3 activators both bind to the CYC1 upstream activation site, UAS2, in an interdependent manner. Cell. 1987;51:953–961. doi: 10.1016/0092-8674(87)90582-4. [DOI] [PubMed] [Google Scholar]
- 2.Schneider JC, Guarente L. Regulation of the yeast CYT1 gene encoding cytochrome c1 by HAP1 and HAP2/3/4. Mol Cell Biol. 1991;10:4934–4942. doi: 10.1128/mcb.11.10.4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Repetto B, Tzagoloff A. Structure and regulation of KGD2, the structural gene for yeast dihydrolipoyl transsuccinylase. Mol Cell Biol. 1990;10:4221–4232. doi: 10.1128/mcb.10.8.4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trumbly RJ. Glucose repression in the yeast Saccharomyces cerevisiae. Mol Microbiol. 1992;6:15–21. doi: 10.1111/j.1365-2958.1992.tb00832.x. [DOI] [PubMed] [Google Scholar]
- 5.Neigeborn L, Carlson M. Genes affecting the regulation of SUC2 gene expression by glucose repression in Saccharomyces cerevisiae. Genetics. 1984;108:845–858. doi: 10.1093/genetics/108.4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams FE, Varanasi U, Trumbly RJ. The CYC8 and TUP1 proteins involved in glucose repression in Saccharomyces cerevisiae are associated in a protein complex. Mol Cell Biol. 1991;11:3307–3316. doi: 10.1128/mcb.11.6.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vallier LG, Carlson M. Synergistic release from glucose repression by mig1 and ssn mutations in Saccharomyces cerevisiae. Genetics. 1994;137:49–54. doi: 10.1093/genetics/137.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Celenza JL, Carlson M. Structure and expression of the SNF1 gene of Saccharomyces cerevisiae. Mol Cell Biol. 1984;4:54–60. doi: 10.1128/mcb.4.1.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Treitel MA, Kuchin S, Carlson M. Snf1 protein kinase regulates phosphorylation of the Mig1 repressor in Saccharomyces cerevisiae. Mol Cell Biol. 1998;18:6273–6280. doi: 10.1128/mcb.18.11.6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tzagoloff A, Dieckmann CL. PET genes of Saccharomyces cerevisiae. Microbiol Rev. 1990;54:211–225. doi: 10.1128/mr.54.3.211-225.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arndt KT, Styles CA, Fink GR. A suppressor of a HIS4 transcriptional defect encodes a protein with homology to the catalytic subunit of protein phosphatases. Cell. 1989;56:527–537. doi: 10.1016/0092-8674(89)90576-x. [DOI] [PubMed] [Google Scholar]
- 12.Jablonka W, Guzman S, Ramirez J, Montero-Lomeli M. Deviation of carbohydrate metabolism by the SIT4 phosphatase in Saccharomyces cerevisiae. Biochim Biophys Acta. 2006;1760:1281–1291. doi: 10.1016/j.bbagen.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 13.Tzagoloff A, Akai A, Foury F. Assembly of the mitochondrial membrane system XVI. Modified form of the ATPase proteolipid in oligomycin-resistant mutants of Saccharomyces cerevisiae. FEBS Lett. 1976;65:391–395. doi: 10.1016/0014-5793(76)80154-8. [DOI] [PubMed] [Google Scholar]
- 14.Myers AM, Crivellone MD, Koerner TJ, Tzagoloff A. Characterization of the yeast HEM2 gene and transcriptional regulation of COX5 and COR1 by heme. J Biol Chem. 1987;262:16822–166829. [PubMed] [Google Scholar]
- 15.Rothstein RJ. One-step gene disruption in yeast. Methods Enzymol. 1983;101:202–211. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- 16.Hill JE, Myers AM, Koerner TJ, Tzagoloff A. Yeast. 1986;2:163–167. doi: 10.1002/yea.320020304. [DOI] [PubMed] [Google Scholar]
- 17.De Vit MJ, Waddle JA, Johnston M. Regulated nuclear translocation of the Mig1 glucose repressor. Mol Biol Cell. 1997;8:1603–1618. doi: 10.1091/mbc.8.8.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tyers MG, Tokiwa R, Nash, Futche B. The Cln3-Cdc28 kinase complex of S. cerevisiae is regulated by proteolysis and phosphorylation. EMBO J. 1992;11:1773–1784. doi: 10.1002/j.1460-2075.1992.tb05229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maniatis T, Fritsch EF, Sambrook J. Molecular cloning: a laboratory manual. Cold spring Harbor Laboratory; Cold Spring Harbor, N. Y.: 1982. [Google Scholar]
- 20.Schiestl RH, Gietz RD. High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr Genet. 1989;16:339–346. doi: 10.1007/BF00340712. [DOI] [PubMed] [Google Scholar]
- 21.Tzagoloff A, Akai A, Needleman RB. Assembly of the mitochondrial membrane system. Characterization of nuclear mutants of S cerevisiae with defects in respiratory enzymes and ATPase. J Biol Chem. 1975;250:8228–8235. [PubMed] [Google Scholar]
- 22.Barrientos A, Korr D, Tzagoloff A. Assembly of cytochrome oxidase: Suppression of shy1 mutants by MSS51, a yeast model for Leigh’s syndrome. EMBO J. 2002;2:43–52. doi: 10.1093/emboj/21.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guarente L. Yeast promoters and lacZ fusions designed to study expression of cloned genes in yeast. Methods Enzymol. 1983;101:181–191. doi: 10.1016/0076-6879(83)01013-7. [DOI] [PubMed] [Google Scholar]
- 24.Botstein D, Davis RW. Principles and practice of recombinant DNA research with yeast. In: Strathern JN, Jones EW, Broach JR, editors. The molecular biology of the yeast Sacchromyces cerevisiae: metabolism and gene expression. Cold spring Harbor Laboratory Press; Cold Spring Harbor, N.Y.: 1982. pp. 607–636. [Google Scholar]
- 25.Sutton A, Freiman R. The Cak1p protein kinase is required at G1/S and G2/M in the budding yeast cell cycle. Genetics. 1997;147:57–71. doi: 10.1093/genetics/147.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jablonowski D, Fichtner L, Stark MJ, Schaffrath R. The yeast elongator histone acetylase requires Sit4-dependent dephosphorylation for toxin-target capacity. Mol Biol Cell. 2004;15:1459–1469. doi: 10.1091/mbc.E03-10-0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Treitel MA, Carlson M. Repression by SSN6-TUP1 is directed by MIG1, a repressor/activator protein. Proc Natl Acad Sci USA. 1995;92:3132–3136. doi: 10.1073/pnas.92.8.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alwine JC, Kemp DJ, Stark GR. Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes. Proc Natl Acad Sci USA. 1977;74:5350–5354. doi: 10.1073/pnas.74.12.5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nehlin JO, Ronne H. Yeast MIG1 repressor is related to the mammalian early growth response and Wilms’ tumour finger proteins. EMBO J. 1990;9:2891–2898. doi: 10.1002/j.1460-2075.1990.tb07479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singer T, Haefner S, Hoffmann M, Fischer M, Ilyina J, Hilt W. Sit4 phosphatase is functionally linked to the ubiquitin-proteasome system. Genetics. 1998;164:1305–1321. doi: 10.1093/genetics/164.4.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gancedo JM. Yeast carbon catabolite repression. Microbiol Mol Biol Rev. 1998;62:334–361. doi: 10.1128/mmbr.62.2.334-361.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daignan-Fornier B, Nguyen CC, Reisdorf P, Lemeignan B, Bolotin-Fukuhara M. MBR1 and MBR3, two related yeast genes that can suppress the growth defect of hap2, hap3 and hap4 mutants. Mol Gen Genet. 1994;243:575–583. doi: 10.1007/BF00284206. [DOI] [PubMed] [Google Scholar]