

Abstract
The mitotic checkpoint is a mechanism that arrests the progression to anaphase until all chromosomes have achieved proper attachment to mitotic spindles. In cancer cells, satisfaction of this checkpoint is frequently delayed or prevented by various defects, some of which have been causally implicated in tumorigenesis. At the same time, deliberate induction of mitotic arrest has proved clinically useful, as antimitotic drugs that interfere with proper chromosome-spindle interactions are effective anticancer agents. However, how mitotic arrest contributes to tumorigenesis or antimitotic drug toxicity is not well defined. Here, we report that mitotic chromosomes can acquire DNA breaks during both pharmacologic and genetic induction of mitotic arrest in human cancer cells. These breaks activate a DNA damage response, occur independently of cell death, and subsequently manifest as karyotype alterations. Such breaks can also occur spontaneously, particularly in cancer cells containing mitotic spindle abnormalities. Moreover, we observed evidence of some breakage in primary human cells. Our findings thus describe a novel source of DNA damage in human cells. They also suggest that mitotic arrest may promote tumorigenesis and antimitotic toxicity by provoking DNA damage.
Introduction
Mitosis is often abnormal in cancer cells (1). Among the defects observed is frequent prolongation of prometaphase, which can occur when the spindle checkpoint arrests progression to anaphase (1). Several studies have now identified genetic abnormalities that provoke such arrest (2). Some of these, such as loss of hCDC4 or overexpression of MAD2, have been causally implicated in cancer (3, 4). At the same time, antimitotic chemotherapeutics that induce prolonged mitotic arrest and “mitotic slippage,” a process whereby prometaphase cells return to interphase without undergoing anaphase, can also elicit subsequent cell death or growth arrest (1). Therefore, evidence suggests that events occurring during mitotic arrest influence tumorigenesis and the toxicity of antimitotic chemotherapy. However, the nature of these events is not well defined (1). Interestingly, Wong and Stearns (5) have shown that human cells that had been presynchronized with double thymidine and the mictrotubule depolymerizer nocodazole later possessed foci of γ-H2AX, the phosphorylated form of histone H2AX that forms around sites of DNA breaks. We thus wondered whether DNA breaks might be generated during mitotic arrest in human cancer cells.
Materials and Methods
Cell lines and treatments
All cell lines were from the American Type Culture Collection, except BG-1 cells, which were from C. Moreno (Emory University, Atlanta, GA). Cancer cells were cultured in McCoy’s and IMR90 cells in DMEM. Cells were seeded at a density of 3 × 104/cm2 onto fibronectin-coated dishes or slides 24 h before the experiments. Nocodazole, paclitaxel, and monastrol were used at 200 nmol/L, 100 nmol/L, and 200 μmol/L, respectively, the minimum concentrations that inhibited cell division (data not shown). Q-VD-OPh was used at 50 μmol/L, the minimum concentration that inhibited apoptosis (data not shown). γ-Irradiation was done with a Cs-137 Gammacell. Stealth Select siRNAs targeted to CENP-E, control siRNA, and LipofectAMINE RNAiMax were obtained from, and used according to the instructions of, Invitrogen. All analyses were done 24 h after transfection.
Immunodetection
For immunocytochemistry, cells were fixed with 2% formaldehyde/PBS and permeabilized with −20°C methanol. Antibody incubations were 1 h at room temperature, and DNA was counterstained with Hoechst. Images were acquired with a Zeiss Axiovert 100M confocal microscope, except images of cells with spontaneous spindle defects, which were acquired with a Zeiss Axioskop 2 Plus microscope. For flow cytometry, cells were harvested by trypsinization and fixed overnight at −20°C in 70% ethanol. Antibody incubations were 1 h at room temperature, and DNA was counterstained with propidium iodide. Data were acquired using a FACSCalibur (Becton Dickinson) and analyzed with CellQuest. Immuno-blotting was done as previously described (6). Antibodies used and image quantification methods are detailed in Supplementary Methods.
Time-lapse imaging
Phase-contrast images of cells grown inside a 37°C, 5% CO2 chamber were automatically obtained at 6-min intervals in multiple locations using an Olympus IX81 microscope. All images were analyzed with Slidebook.
Cytogenetic analyses
Chromosome spreads were prepared using standard cytogenetic techniques; DNA was stained with 4′,6-diamidino-2-phenylindole; and images were obtained with a Zeiss Axioskop 2 Plus microscope. Scoring of chromosome aberrations was done according to the classification of Savage (7).
Results and Discussion
To address whether human cancer cells acquire DNA damage during mitotic arrest, we first examined nocodazole-arrested HCT116 colon cancer cells for the presence of γ-H2AX foci. During 36 h of treatment, nocodazole produced a transient increase in mitotic index that peaked at 12 h and was followed by mitotic slippage (Supplementary Fig. S1). Whereas control prometaphase cells exhibited few γ-H2AX foci, nocodazole-arrested prometaphase cells showed a time-dependent increase in γ-H2AX foci per cell (Fig. 1A and B). A similar increase was observed after treatment with the microtubule stabilizer paclitaxel and the Eg5 mitotic kinesin inhibitor monastrol (Fig. 1A), indicating that agents that induce mitotic arrest through different mechanisms also induce γ-H2AX foci. Notably, cells exposed to drug treatment, but not yet in mitosis, displayed no increase in γ-H2AX foci (Supplementary Fig. S2), indicating that only cells that have entered mitosis exhibit the response.
Figure 1.
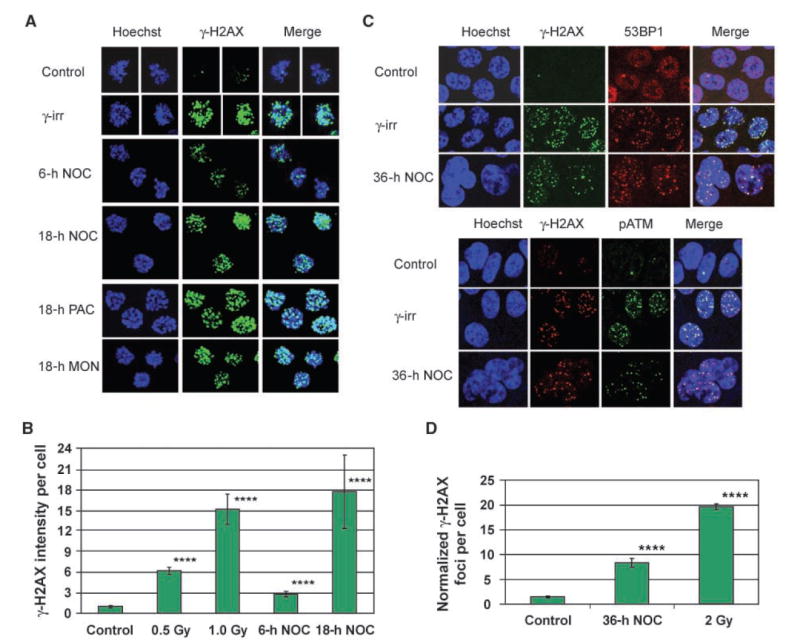
γ-H2AX foci accumulate during drug-induced mitotic arrest in HCT116 cells. A, maximum projection images of stacks of prometaphase cells stained for γ-H2AX. γ-irr, 30 min after 2-Gy γ-irradiation. NOC, nocodazole; PAC, paclitaxel; MON, monastrol. B, quantification of γ-H2AX pixel intensity per prometaphase cell. Columns, mean from at least 60 cells per sample taken from two independent experiments; bars, SE. ****, P < 0.0001, compared with control (t tests on log-transformed values, which were normally distributed). C, single focal planes of interphase cells costained for γ-H2AX and either 53BP1 or pATM. D, quantification of γ-H2AX foci per interphase cell. To adjust for cell cycle differences in DNA content, foci number was normalized to the DNA content of a 2N cell using the Hoechst signal. Columns, mean from at least 145 cells per sample taken from three independent experiments; bars, SE. ****, P < 0.0001, compared with control (Mann-Whitney tests). Specificity of the mouse anti–γ-H2AX antibody is shown in Supplementary Fig. S5.
We next determined whether these γ-H2AX foci recruit additional proteins involved in the DNA damage response. However, it has previously been shown in HeLa cells that whereas γ-H2AX foci form in irradiated mitotic cells, other DNA damage response proteins are only recruited once cells return to interphase (8). Because we also observed this lack of recruitment of DNA damage response proteins in mitotic HCT116 cells (data not shown), we examined the cells after 36 h of nocodazole treatment, at which point virtually all cells had undergone mitotic slippage (Supplementary Fig. S1). Indeed, these postmitotic cells had increased γ-H2AX foci (Fig. 1D), which recruited the DNA damage response protein 53BP1 and a phospho-activated form of ATM (pATM; Fig. 1C). Thus, γ-H2AX foci acquired during mitotic arrest, like those produced by γ-irradiation, recruit other DNA damage response proteins once cells have exited mitosis.
Pharmacologic induction of mitotic arrest is accompanied by some degree of cell death in most cell lines, and γ-H2AX formation can participate in apoptosis (1, 9). We thus investigated whether γ-H2AX formed during mitotic arrest represents ongoing cell death. Live-cell videomicroscopy indicated that <1% of cells died in a 30-h period after release from a 6-h nocodazole arrest (Fig. 2B and Supplementary Movie 1), whereas 25% of cells died if continuously treated with the drug (Fig. 2B and Supplementary Movie 2). Given that the average γ-H2AX per cell is increased 2.5-fold after 6 h of nocodazole (Fig. 1B) but 99% of these cells survive following drug washout, acquisition of γ-H2AX foci must precede any eventual commitment to cell death. Consistent with this, only cells with apoptotic nuclear morphology, which comprised <10% of the total after 18 h nocodazole, exhibited caspase-3 cleavage, cytoplasmic localization of cytochrome c, or activation of BAX, whereas mitotic cells with normal nuclear morphology and abundant γ-H2AX foci displayed none of these events (Fig. 2A). Moreover, in contrast to the γ-H2AX foci seen in mitotic cells, apoptotic cells exhibited a distinct γ-H2AX staining pattern at the periphery of apoptotic chromatin (Fig. 2A). Finally, cotreatment of nocodazole-arrested cells with the caspase inhibitor Q-VD-OPh (10) produced a 98% inhibition of apoptotic cells but no inhibition of γ-H2AX foci (Fig. 2C). Taken together, these data indicate that whereas death indeed follows mitotic arrest in a fraction of HCT116 cells, such death is neither the cause of nor the obligatory conclusion to γ-H2AX foci acquired during mitotic arrest.
Figure 2.
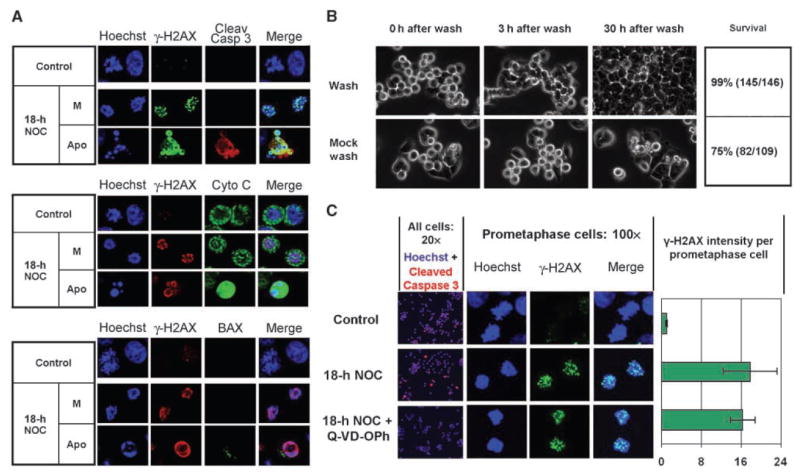
γ-H2AX foci acquired during mitotic arrest arise independently of cell death in HCT116 cells. A, single focal planes of cells stained for γ-H2AX and cleaved caspase-3 (top), cytochrome c (middle), or activated BAX (bottom). M, mitotic cells; Apo, apoptotic cells. Cleav Casp 3, cleaved caspase-3. Cyto C, cytochrome c. B, cells were treated with nocodazole for 6 h, washed with either drug-free or drug-containing medium, and returned to the chamber for an additional 30 h of filming. Shown are time-lapse images of cell fate at increasing times after either drug washout (Wash) or mock washout and continued culture in the drug (Mock wash). C, Q-VD-OPh inhibits cleaved caspase-3–positive apoptotic cells (20× column) but not γ-H2AX foci in prometaphase cells (100× column and chart). Columns, mean from at least 60 cells per sample taken from two independent experiments; bars, SE.
To explore the downstream consequences of this DNA damage, we examined cells for the presence of acquired chromosome aberrations. Whereas untreated cells harbored few spontaneous aberrations (Fig. 3A), cells that were grown in drug-free medium for 20 h following release from an 18-h nocodazole arrest exhibited a 6-fold increase in the frequency of chromosome breaks (Supplementary table). The majority were of the chromosome type, such as double minutes, acentric fragments, and dicentrics [Fig. 3, B (bottom inset) and C (all insets)], consistent with γ-H2AX data indicating that breaks arise during mitotic arrest, and thus before subsequent DNA replication. However, some chromatid-type aberrations were also observed (Fig. 3B, top inset), raising the possibility that additional DNA lesions, such as single-stranded breaks, base damages, or postreplication double-strand breaks, might be acquired by the cells (7, 11). Notably, aberrations were observed not only in tetraploid cells produced through mitotic slippage (Fig. 3C) but also in near-diploid cells generated from cell division following drug washout (Fig. 3B), indicating that breaks occur even in cells that recover from transient arrest. Thus, DNA breaks incurred during mitotic arrest manifest as structural karyotype changes in subsequent cell cycles.
Figure 3.
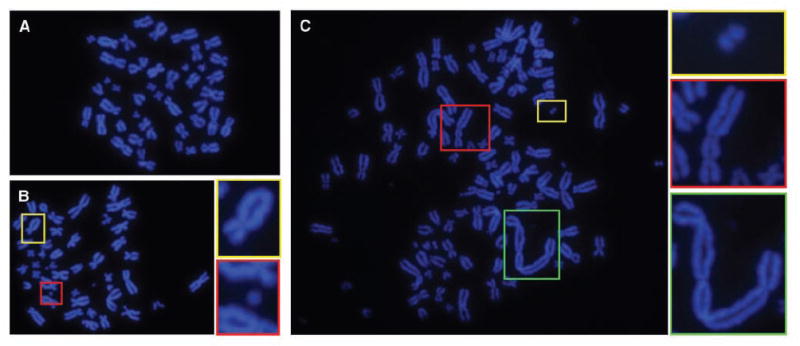
HCT116 cells released from mitotic arrest harbor chromosome aberrations. Treated cells were exposed to nocodazole for 18 h, washed, and released into drug-free medium for 19 h. Control cells were incubated in drug-free medium for 19 h. All cells were then exposed to nocodazole for 1 h to disassemble spindles for spreading, and chromosome spreads were prepared. A, a representative control HCT116 cell. B, a representative near-diploid cell produced through cell division following drug washout. Insets, a chromatid exchange (top) and a chromosome fragment (bottom). C, a representative tetraploid cell produced through mitotic slippage. Insets, a double-minute chromosome (top) and two dicentric chromosomes (middle and bottom).
We next asked whether a genetic induction of mitotic arrest might also provoke DNA damage. Because depletion of the kinetochore protein CENP-E disrupts chromosome-spindle interactions and induces transient mitotic arrest in human cancer cells (12-14), we examined HCT116 cells that had been transfected with siRNAs against this protein. Consistent with previous reports, CENP-E siRNAs produced an elevated mitotic index (Fig. 4B) and aberrant mitotic figures (Fig. 4C, left) as compared with control siRNA, indicating the induction of mitotic arrest. Moreover, CENP-E–depleted prometaphase cells exhibited an increase in γ-H2AX foci (Fig. 4C). Thus, genetic induction of mitotic arrest, like that elicited by antimitotic agents, provokes γ-H2AX foci in HCT116 cells.
Figure 4.
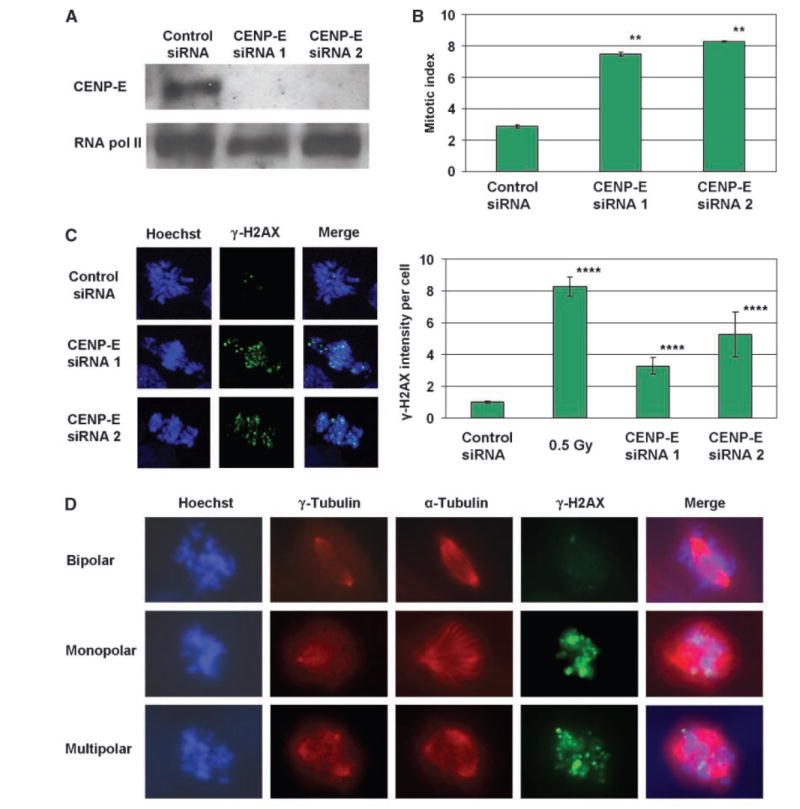
Prometaphase elevation of γ-H2AX in HCT116 cells occurs both on knockdown of CENP-E and in untreated cells with spontaneous spindle defects. A, CENP-E protein levels in cells transfected with two different CENP-E siRNAs or with control siRNA. RNA polymerase II was used as a loading control. Representative of two independent experiments. B, mitotic indices of siRNA-transfected cells. Columns, mean from two independent experiments; bars, SE. **, P < 0.01, compared with control (t tests). C, left, maximum projection images of stacks of siRNA-transfected prometaphase cells stained for γ-H2AX. Right, quantification of γ-H2AX pixel intensity per prometaphase cell after siRNA transfection. Columns, mean from at least 70 cells per sample taken from two independent experiments; bars, SE.****, P < 0.0001, compared with control (t tests on log-transformed values, which were normally distributed). D, single focal planes of untreated prometaphase cells stained for γ-H2AX, γ-tubulin, and α-tubulin.
Interestingly, while carrying out γ-H2AX immunocytochemistry, we noticed that occasional untreated mitotic HCT116 cells bore characteristics of prolonged mitotic arrest: a prometaphase arrangement of chromosomes and abundant γ-H2AX foci. We thus wondered whether these cells might contain spontaneous spindle defects, which can disrupt mitotic progression (15, 16). Of 4,000 mitotic cells examined, prometaphase cells with marked elevation of γ-H2AX foci comprised 2.2% of the total, and 69% of these contained either monopolar or multipolar spindles (Fig. 4D). In contrast, only 5.3% of prometaphase cells with minimal γ-H2AX foci contained mitotic spindle abnormalities. Thus, the frequency of prometaphase γ-H2AX elevation was increased 13-fold in cells with spontaneous spindle defects (χ2 P < 0.0001). Of note, prophase cells with nascent monopolar or multipolar spindles did not possess elevated γ-H2AX foci (Supplementary Fig. S3), suggesting that γ-H2AX–intense mitotic cells acquire their foci during, and not before, prometaphase. These data indicate that prometaphase acquisition of γ-H2AX foci can occur spontaneously in HCT116 cells and may arise during mitotic arrest provoked by spindle abnormalities.
Finally, we explored the extent to which this phenomenon occurs in other human cells. Of eight colorectal cancer lines (including HCT116) exposed to nocodazole, six exhibited a γ-H2AX increase in some or most mitotic cells (Supplementary Fig. S4). Increased γ-H2AX was also observed in HeLa cervical cancer cells and BG-1 ovarian cancer cells (Supplementary Fig. S4). Thus, DNA damage during mitotic arrest is a common occurrence in human cancer cells. Additionally, we observed a modest γ-H2AX increase in IMR90 primary human fibroblasts (Supplementary Fig. S4), suggesting that even nontransformed human cells can acquire some DNA damage during mitotic arrest. However, the degree of γ-H2AX induction in primary cells was lower than in the majority of cancer lines, suggesting that malignant cells may be more prone to this damage.
Our study shows that many human cells acquire DNA damage during mitotic arrest. While the mechanism responsible for this damage remains an area for future work, our results have several important implications. First, although unscheduled DNA breaks are known to derive from sources such as replication, oxidative stress, and exogenous mutagens (17), it was hitherto unknown that such breaks could accumulate during mitotic arrest. As such, our findings describe a novel source of DNA damage. Second, because cancer cells frequently contain mitotic defects that induce arrest (1, 18), and because DNA breaks can promote tumorigenesis (17), our findings suggest that one way mitotic arrest may promote tumorigenesis is through DNA damage. Along these lines, it is interesting to note that inactivation of the tumor suppressor hCDC4 and oncogenic overexpression of MAD2 are accompanied not only by prolongation of mitosis and aneuploidy but also by evidence of DNA damage (3, 4). Our results suggest that breaks acquired during mitotic arrest could contribute to the damage observed in these systems. In this way, mitotic arrest may be a source of structural, as well as numerical, chromosomal instability in human cells.
At the same time, this and other studies indicate that cells that endure prolonged or complete inhibition of mitosis exhibit reduced reproductive capacity (1). Indeed, although cell death was not the compulsory fate of HCT116 cells that acquired damage during mitotic arrest, we did observe subsequent death in a fraction of cells. Given that γ-H2AX foci increased with time of mitotic arrest, and that the cytotoxicity of DNA damage is dose dependent, a third implication of our findings is that extensive DNA damage imposed by prolonged or complete inhibition of mitosis may promote cell death or growth arrest. In this way, our data support and extend the idea offered by Wong and Stearns (5) that the growth arrest following pharmacologic induction of mitotic arrest (formerly known as the “tetraploidy checkpoint”) may, at least in some contexts, be a response to DNA damage. Moreover, this consequence may have clinical implications: given the variable degree of γ-H2AX formed on drug-induced arrest in different cancer cell lines, it is plausible that the sensitivity of a tumor to antimitotic chemotherapy could be influenced by its propensity to sustain DNA damage during mitotic arrest. By extension, efforts to identify markers of this propensity might aid attempts to tailor antimitotic chemotherapy to susceptible tumors, as is currently being tried with markers of numerical chromosomal instability (19).
Lastly, our findings do not support a recent report that microtubule-destabilizing agents elicit DNA breaks during G1 phase, and not mitosis (20). Although we do corroborate these authors’ finding that such agents induce breaks, our data indicate that these breaks arise on mitotic chromosomes. This explains why even cells that divide following mitotic arrest possess DNA damage, and it indicates that some yet undefined property of mitosis itself commonly confers vulnerability to DNA damage in human cancer cells.
Supplementary Material
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Acknowledgments
Grant support : NIH grants DK52230, DK64399, and CA84197 (V.W. Yang).
We thank D. Pallas and J. Lee for discussion and assistance with live-cell microscopy and P. Doetsch, D. Jones, A. Corbett, H. Bastians, G. Davis, B. Craige, and M. Wiltenburg for discussion and support.
References
- 1.Rieder CL, Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell. 2004;7:637–51. doi: 10.1016/j.devcel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Dalton WB, Yang VW. Mitotic origins of chromosomal instability in colorectal cancer. Curr Colorectal Cancer Rep. 2007;3:59–64. doi: 10.1007/s11888-007-0001-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rajagopalan H, Jallepalli PV, Rago C, et al. Inactivation of hCDC4 can cause chromosomal instability. Nature. 2004;428:77–81. doi: 10.1038/nature02313. [DOI] [PubMed] [Google Scholar]
- 4.Sotillo R, Hernando E, Diaz-Rodriguez E, et al. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong C, Stearns T. Mammalian cells lack checkpoints for tetraploidy, aberrant centrosome number, and cytokinesis failure. BMC Cell Biol. 2005;6:6. doi: 10.1186/1471-2121-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoon HS, Ghaleb AM, Nandan MO, Hisamuddin IM, Dalton WB, Yang VW. Kruppel-like factor 4 prevents centrosome amplification following γ-irradiation-induced DNA damage. Oncogene. 2005;24:4017–25. doi: 10.1038/sj.onc.1208576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Savage JR. Classification and relationships of induced chromosomal structual changes. J Med Genet. 1976;13:103–22. doi: 10.1136/jmg.13.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang X, Tran T, Zhang L, Hatcher R, Zhang P. DNA damage-induced mitotic catastrophe is mediated by the Chk1-dependent mitotic exit DNA damage checkpoint. Proc Natl Acad Sci U S A. 2005;102:1065–70. doi: 10.1073/pnas.0409130102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu C, Zhu F, Cho YY, et al. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol Cell. 2006;23:121–32. doi: 10.1016/j.molcel.2006.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caserta TM, Smith AN, Gultice AD, Reedy MA, Brown TL. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis. 2003;8:345–52. doi: 10.1023/a:1024116916932. [DOI] [PubMed] [Google Scholar]
- 11.Zhuanzi W, Wenjian L, Dejuan Z, Wei W, Xigang J, Qingxiang G. Chromatid-type aberrations following irradiation in G0 lymphocytes with heavy ions. Mutat Res. 2007;617:98–103. doi: 10.1016/j.mrfmmm.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 12.Schaar BT, Chan GK, Maddox P, Salmon ED, Yen TJ. CENP-E function at kinetochores is essential for chromosome alignment. J Cell Biol. 1997;139:1373–82. doi: 10.1083/jcb.139.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanudji M, Shoemaker J, L’Italien L, Russell L, Chin G, Schebye XM. Gene silencing of CENP-E by small interfering RNA in HeLa cells leads to missegregation of chromosomes after a mitotic delay. Mol Biol Cell. 2004;15:3771–81. doi: 10.1091/mbc.E03-07-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yao X, Abrieu A, Zheng Y, Sullivan KF, Cleveland DW. CENP-E forms a link between attachment of spindle microtubules to kinetochores and the mitotic checkpoint. Nat Cell Biol. 2000;2:484–91. doi: 10.1038/35019518. [DOI] [PubMed] [Google Scholar]
- 15.Sluder G, Thompson EA, Miller FJ, Hayes J, Rieder CL. The checkpoint control for anaphase onset does not monitor excess numbers of spindle poles or bipolar spindle symmetry. J Cell Sci. 1997;110:421–9. doi: 10.1242/jcs.110.4.421. [DOI] [PubMed] [Google Scholar]
- 16.Stewenius Y, Gorunova L, Jonson T, et al. Structural and numerical chromosome changes in colon cancer develop through telomere-mediated anaphase bridges, not through mitotic multipolarity. Proc Natl Acad Sci U S A. 2005;102:5541–6. doi: 10.1073/pnas.0408454102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Gent DC, Hoeijmakers JH, Kanaar R. Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet. 2001;2:196–206. doi: 10.1038/35056049. [DOI] [PubMed] [Google Scholar]
- 18.Therman E, Kuhn EM. Mitotic modifications and aberrations in cancer. Crit Rev Oncog. 1989;1:293–305. [PubMed] [Google Scholar]
- 19.Swanton C, Marani M, Pardo O, et al. Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer Cell. 2007;11:498–512. doi: 10.1016/j.ccr.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 20.Quignon F, Rozier L, Lachages AM, Bieth A, Simili M, Debatisse M. Sustained mitotic block elicits DNA breaks: one-step alteration of ploidy and chromosome integrity in mammalian cells. Oncogene. 2007;26:165–72. doi: 10.1038/sj.onc.1209787. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).